Drug Discovery and Development for Tropical Diseases (TDs)
Share This Topical Collection
Editor
 Dr. Christophe Dardonville
Dr. Christophe Dardonville
Dr. Christophe Dardonville
E-Mail
Website
Collection Editor
1. Instituto de Química Médica—CSIC, Madrid, Spain
2. Medicinal Chemistry Institute—Spanish Council for Scientific Research, Madrid, Spain
Interests: medicinal chemistry; design and synthesis of antiparasitic agents for neglected tropical diseases (trypanosomiasis, leishmaniasis, malaria); cationic compounds; DNA binding study; pKa measurement
Special Issues, Collections and Topics in MDPI journals
Topical Collection Information
Dear Colleagues,
Tropical diseases (TDs) cause immense human suffering and death. They represent a huge socioeconomic and life-threatening burden in less-developed countries. In the last 20 years, a great effort has been made by the international scientific community to tackle this problem and discover new safe and effective drugs against TDs, including the most neglected tropical diseases (NTDs) (https://www.who.int/teams/control-of-neglected-tropical-diseases). In this this Special Issue, we would like to provide a picture of the recent discoveries in the field of drug discovery for TDs. We will acknowledge research papers which topic includes, but is not restricted to, the design, synthesis, in vitro/in vivo evaluation, and development of new compounds for TDs, including promising chemotherapeutic targets to combat these diseases. Review articles outlining recent developments in the field are also welcome.
This collection covers (but is not restricted to) the Tropical Diseases listed below:
- Buruli ulcer
- Chagas disease (American trypanosomiasis)
- Dengue
- Dracunculiasis (Guinea-worm disease)
- Echinococcosis
- Foodborne trematode infections
- Human African trypanosomiasis (sleeping sickness)
- Leishmaniasis·
- Leprosy (Hansen's disease)
- Lymphatic filariasis (Elephantiasis)
- Malaria
- Mycetoma, chromoblastomycosis, and other deep mycoses
- Onchocerciasis (river blindness)
- Rabies
- Scabies and other ectoparasitoses
- Schistosomiasis (Bilharzia)
- Soil-transmitted helminthiases
- Snakebite envenoming
- Taeniasis and cisticercosis
- Trachoma
- Yaws (Endemic treponematoses)
Dr. Christophe Dardonville
Collection Editor
Manuscript Submission Information
Manuscripts should be submitted online at www.mdpi.com by registering and logging in to this website. Once you are registered, click here to go to the submission form. Manuscripts can be submitted until the deadline. All submissions that pass pre-check are peer-reviewed. Accepted papers will be published continuously in the journal (as soon as accepted) and will be listed together on the collection website. Research articles, review articles as well as short communications are invited. For planned papers, a title and short abstract (about 100 words) can be sent to the Editorial Office for announcement on this website.
Submitted manuscripts should not have been published previously, nor be under consideration for publication elsewhere (except conference proceedings papers). All manuscripts are thoroughly refereed through a single-blind peer-review process. A guide for authors and other relevant information for submission of manuscripts is available on the Instructions for Authors page. Pharmaceuticals is an international peer-reviewed open access monthly journal published by MDPI.
Please visit the Instructions for Authors page before submitting a manuscript.
The Article Processing Charge (APC) for publication in this open access journal is 2900 CHF (Swiss Francs).
Submitted papers should be well formatted and use good English. Authors may use MDPI's
English editing service prior to publication or during author revisions.
Keywords
- Tropical diseases (TDs)
- Neglected tropical diseases (NTDs)
- Drug discovery
- Medicinal chemistry
- Drug synthesis
- SAR studies
- in vitro/in vivo activity
- Chemotherapeutic targets
Published Papers (22 papers)
Open AccessCommunication
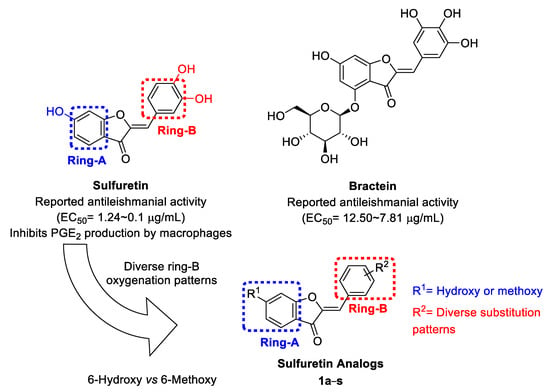
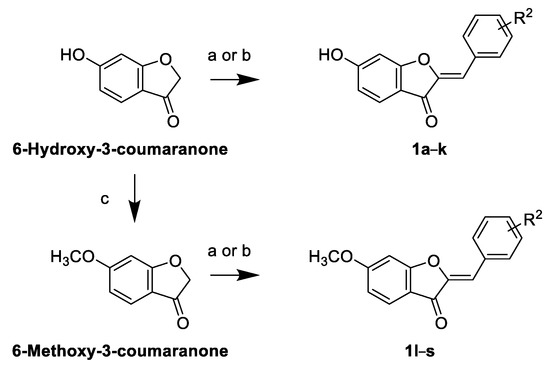
Design, Rational Repurposing, Synthesis, In Vitro Evaluation, Homology Modeling and In Silico Study of Sulfuretin Analogs as Potential Antileishmanial Hit Compounds
by
Ahmed H.E. Hassan, Trong-Nhat Phan, Yeonwoo Choi, Suyeon Moon, Joo Hwan No and Yong Sup Lee
Cited by 13 | Viewed by 1783
Abstract
Direct growth inhibition of infectious organisms coupled with immunomodulation to counteract the immunosuppressive environment might be a beneficial therapeutic approach. Herein, a library of sulfuretin analogs were developed with potential capabilities to inhibit production of the immunosuppressive PGE
2 and elicit direct growth
[...] Read more.
Direct growth inhibition of infectious organisms coupled with immunomodulation to counteract the immunosuppressive environment might be a beneficial therapeutic approach. Herein, a library of sulfuretin analogs were developed with potential capabilities to inhibit production of the immunosuppressive PGE
2 and elicit direct growth inhibition against
Leishmania donovani; the major causative agent of the fatal visceral leishmaniasis. Amongst explored library members bearing diverse methoxy and/or hydroxy substitution patterns at rings B and A, analog
1i retaining the C6-hydroxy moiety at ring-A, but possessing methoxy moieties in place of the polar dihydroxy moieties of sulfuretin ring-B, as well as analog
1q retaining the sulfuretin′s polar dihydroxy moieties at ring-B, but incorporating a C6-methoxy moiety instead of the C6-hydroxy moiety at ring-A, were the most promising hit compounds. Cytotoxicity evaluation suggested that analog
1i possesses a safety profile inducing the death of the parasite rather than host cells. In silico simulation provided insights into their possible binding with
Leishmania donovani fumarate reductase. The current investigation presents sulfuretin analogs
1i and
1q as potential hit compounds for further development of multifunctional therapeutic agents against visceral leishmaniasis.
Full article
►▼
Show Figures
Open AccessArticle
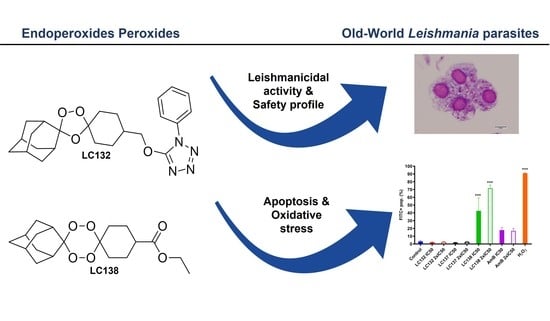
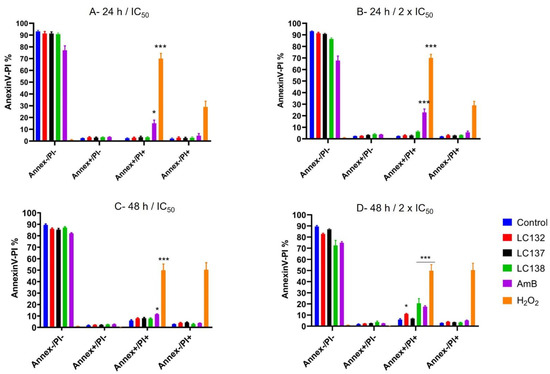
1,2,4-Trioxolane and 1,2,4,5-Tetraoxane Endoperoxides against Old-World Leishmania Parasites: In Vitro Activity and Mode of Action
by
Andreia Mendes, Ana Armada, Lília I. L. Cabral, Patrícia S. M. Amado, Lenea Campino, Maria L. S. Cristiano and Sofia Cortes
Cited by 5 | Viewed by 2361
Abstract
Leishmaniasis remains one of the ten Neglected Tropical Diseases with significant morbidity and mortality in humans. Current treatment of visceral leishmaniasis is difficult due to a lack of effective, non-toxic, and non-extensive medications. This study aimed to evaluate the selectivity of 12 synthetic
[...] Read more.
Leishmaniasis remains one of the ten Neglected Tropical Diseases with significant morbidity and mortality in humans. Current treatment of visceral leishmaniasis is difficult due to a lack of effective, non-toxic, and non-extensive medications. This study aimed to evaluate the selectivity of 12 synthetic endoperoxides (1,2,4-trioxolanes; 1,2,4,5-tetraoxanes) and uncover their biochemical effects on
Leishmania parasites responsible for visceral leishmaniasis. The compounds were screened for in vitro activity against
L. infantum and
L. donovani and for cytotoxicity in two monocytic cell lines (J774A.1 and THP-1) using the methyl thiazol tetrazolium assay. Reactive oxygen species formation, apoptosis, and mitochondrial impairment were measured by flow cytometry. The compounds exhibited fair to moderate anti-proliferative activity against promastigotes of the 2
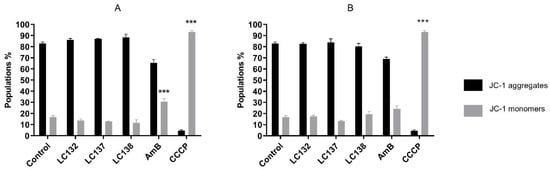
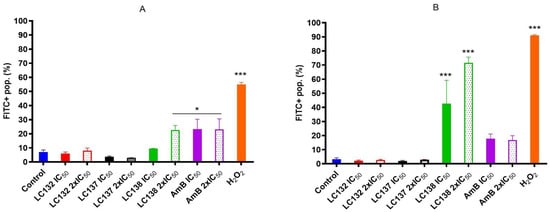
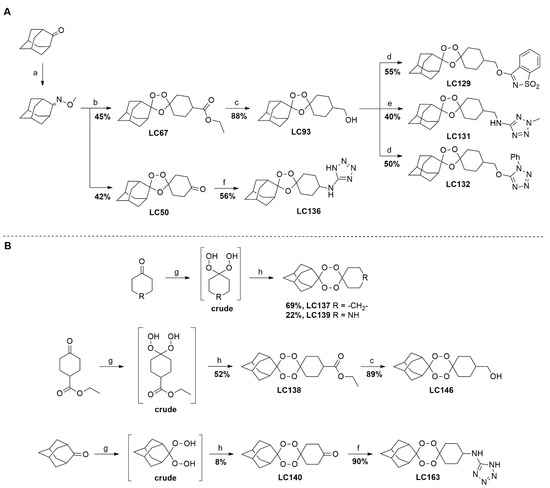
Leishmania species, with IC
50 values ranging from 13.0 ± 1.7 µM to 793.0 ± 37.2 µM. Tetraoxanes LC132 and LC138 demonstrated good leishmanicidal activity on
L. infantum amastigotes (IC
50 13.2 ± 5.2 and 23.9 ± 2.7 µM) with low cytotoxicity in mammalian cells (SIs 22.1 and 118.6), indicating selectivity towards the parasite. Furthermore, LC138 was able to induce late apoptosis and dose-dependent oxidative stress without affecting mithocondria. Compounds LC132 and LC138 can be further explored as potential antileishmanial chemotypes.
Full article
►▼
Show Figures
Open AccessArticle
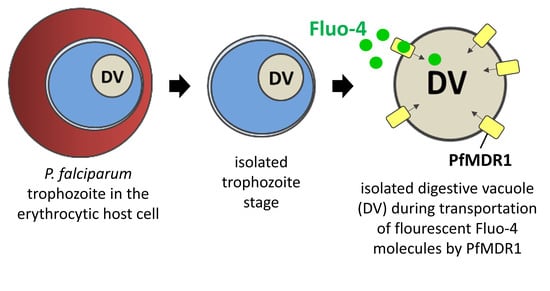
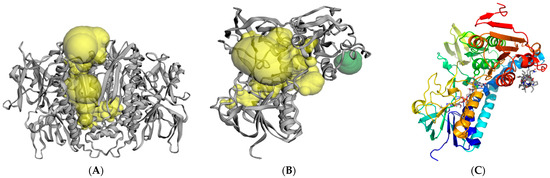
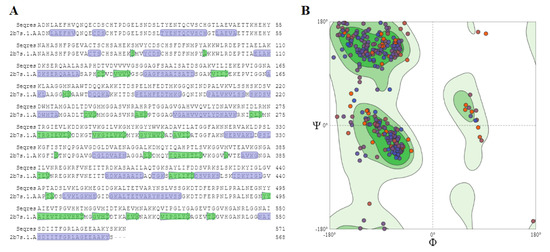
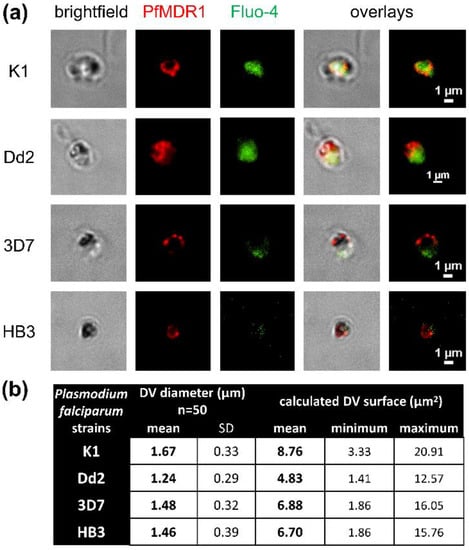
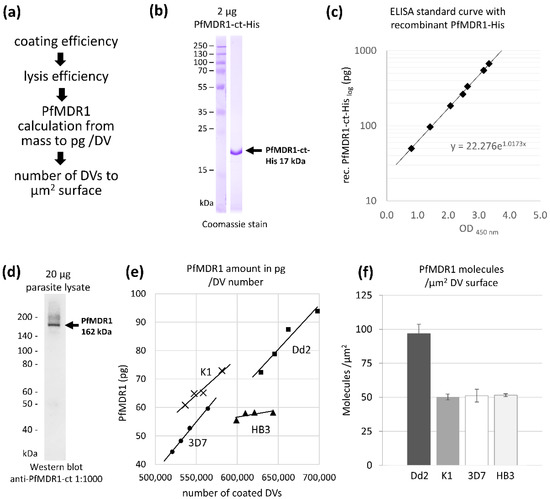
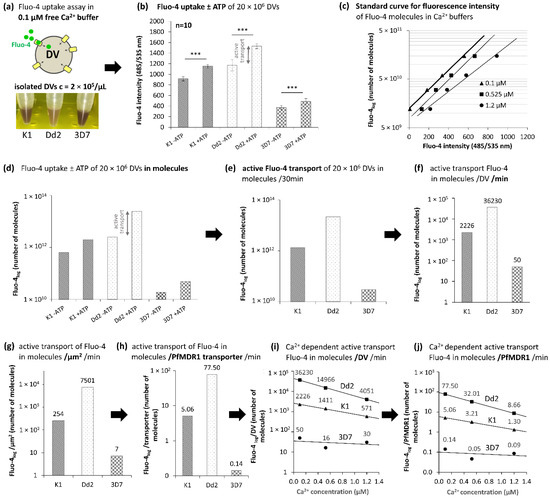
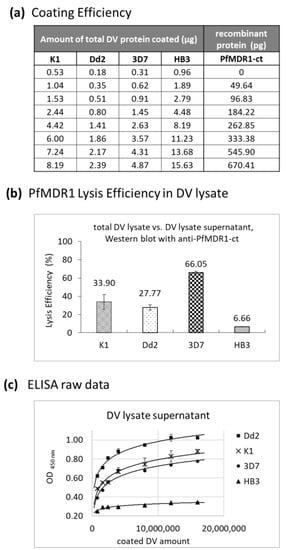
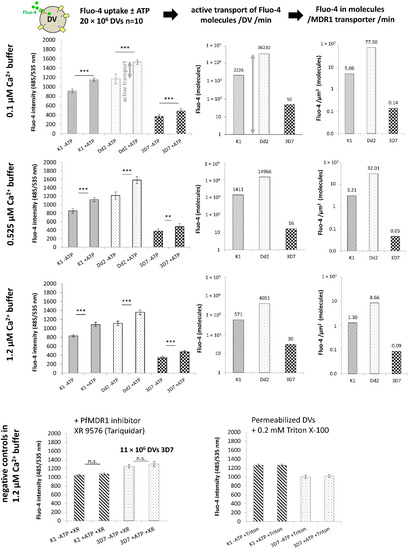
PfMDR1 Transport Rates Assessed in Intact Isolated Plasmodium falciparum Digestive Vacuoles Reflect Functional Drug Resistance Relationship with pfmdr1 Mutations
by
Nina Simon, Cornelia Voigtländer, Barbara Kappes, Petra Rohrbach and Oliver Friedrich
Cited by 3 | Viewed by 2081
Abstract
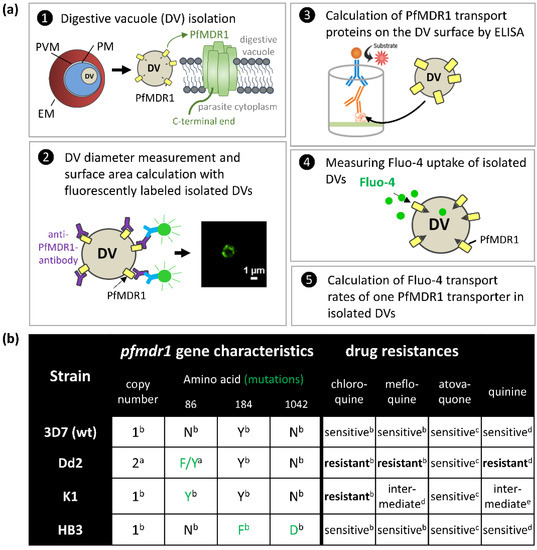
Drug resistance often emerges from mutations in solute transporters. Single amino acid exchanges may alter functionality of transporters with ‘de novo’ ability to transport drugs away from their site of action. The PfMDR1 transporter (or P-glycoprotein 1) is located in the membrane of
[...] Read more.
Drug resistance often emerges from mutations in solute transporters. Single amino acid exchanges may alter functionality of transporters with ‘de novo’ ability to transport drugs away from their site of action. The PfMDR1 transporter (or P-glycoprotein 1) is located in the membrane of the digestive vacuole (DV), functions as an ATP-dependent pump, and transports substrates into the DV. In this study, four strains of
Plasmodium falciparum, carrying various
pfmdr1 gene mutations, were analysed for their transport characteristics of Fluo-4 in isolated DVs of parasites. To obtain quantitative estimates for PfMDR1 DV surface expression, PfMDR1 protein amounts on each strain’s DV membrane were evaluated by quantitative ELISA. Fluo-4, acting as a substrate for PfMDR1, was applied in DV uptake assays (‘reverse Ca
2+ imaging’). Viable DVs were isolated from trophozoite stages with preserved PfMDR1 activity. This newly developed assay enabled us to measure the number of Fluo-4 molecules actively transported into isolated DVs per PfMDR1 molecule. The drug-resistant strain Dd2 presented the highest transport rates, followed by K1 and the drug-sensitive strain 3D7, compatible with their copy numbers. With this assay, an evaluation of the probability of resistance formation for newly developed drugs can be implemented in early stages of drug development.
Full article
►▼
Show Figures
Open AccessArticle
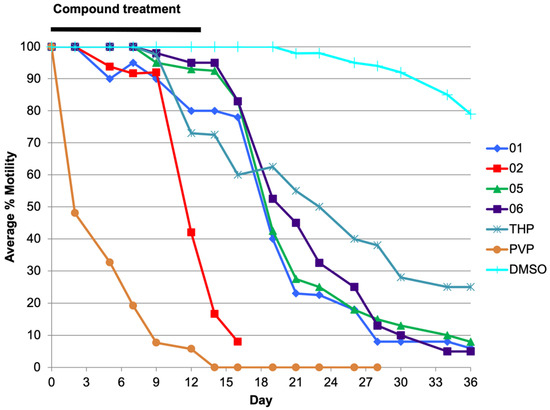
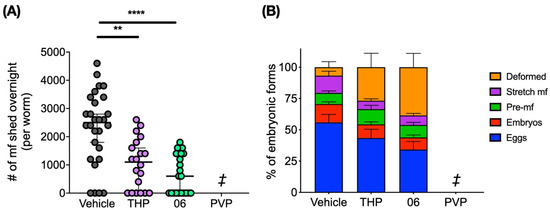
Pyrvinium Pamoate and Structural Analogs Are Early Macrofilaricide Leads
by
Emma L. Gunderson, Clifford Bryant, Christina A. Bulman, Chelsea Fischer, Mona Luo, Ian Vogel, Kee-Chong Lim, Shabnam Jawahar, Nancy Tricoche, Denis Voronin, Christopher Corbo, Rene B. Ayiseh, Faustin P. T. Manfo, Glory E. Mbah, Fidelis Cho-Ngwa, Brenda Beerntsen, Adam R. Renslo, Sara Lustigman and Judy A. Sakanari
Cited by 3 | Viewed by 2241
Abstract
Onchocerciasis and lymphatic filariasis are neglected tropical diseases caused by infection with filarial worms. Annual or biannual mass drug administration with microfilaricidal drugs that kill the microfilarial stages of the parasites has helped reduce infection rates and thus prevent transmission of both infections.
[...] Read more.
Onchocerciasis and lymphatic filariasis are neglected tropical diseases caused by infection with filarial worms. Annual or biannual mass drug administration with microfilaricidal drugs that kill the microfilarial stages of the parasites has helped reduce infection rates and thus prevent transmission of both infections. However, success depends on high population coverage that is maintained for the duration of the adult worm’s lifespan. Given that these filarial worms can live up to 14 years in their human hosts, a macrofilaricidal drug would vastly accelerate elimination efforts. Here, we have evaluated the repurposed drug pyrvinium pamoate as well as newly synthesized analogs of pyrvinium for their efficacy against filarial worms in vitro and in vivo. We found that pyrvinium pamoate, tetrahydropyrvinium and one of the analogs were highly potent in inhibiting worms in in vitro whole-worm screening assays, and that all three compounds reduced female worm fecundity and inhibited embryogenesis in the
Brugia pahangi-gerbil in vivo model of infection.
Full article
►▼
Show Figures
Open AccessEditor’s ChoiceReview
Laboratory Selection of Trypanosomatid Pathogens for Drug Resistance
by
Sabina Beilstein, Radhia El Phil, Suzanne Sherihan Sahraoui, Leonardo Scapozza, Marcel Kaiser and Pascal Mäser
Cited by 1 | Viewed by 2868
Abstract
The selection of parasites for drug resistance in the laboratory is an approach frequently used to investigate the mode of drug action, estimate the risk of emergence of drug resistance, or develop molecular markers for drug resistance. Here, we focused on the How
[...] Read more.
The selection of parasites for drug resistance in the laboratory is an approach frequently used to investigate the mode of drug action, estimate the risk of emergence of drug resistance, or develop molecular markers for drug resistance. Here, we focused on the How rather than the Why of laboratory selection, discussing different experimental set-ups based on research examples with
Trypanosoma brucei,
Trypanosoma cruzi, and
Leishmania spp. The trypanosomatids are particularly well-suited to illustrate different strategies of selecting for drug resistance, since it was with African trypanosomes that Paul Ehrlich performed such an experiment for the first time, more than a century ago. While breakthroughs in reverse genetics and genome editing have greatly facilitated the identification and validation of candidate resistance mutations in the trypanosomatids, the forward selection of drug-resistant mutants still relies on standard in vivo models and in vitro culture systems. Critical questions are: is selection for drug resistance performed in vivo or in vitro? With the mammalian or with the insect stages of the parasites? Under steady pressure or by sudden shock? Is a mutagen used? While there is no bona fide best approach, we think that a methodical consideration of these questions provides a helpful framework for selection of parasites for drug resistance in the laboratory.
Full article
Open AccessReview
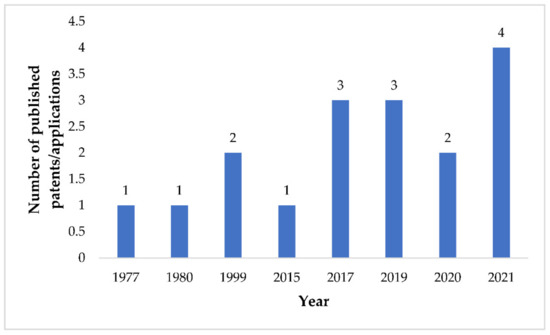
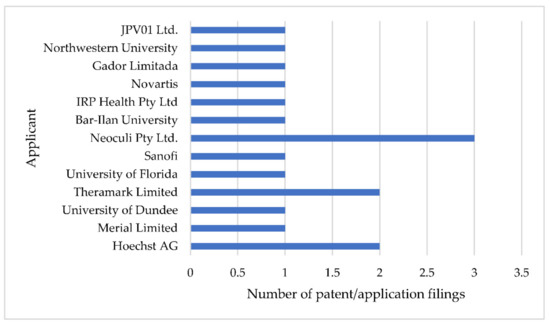
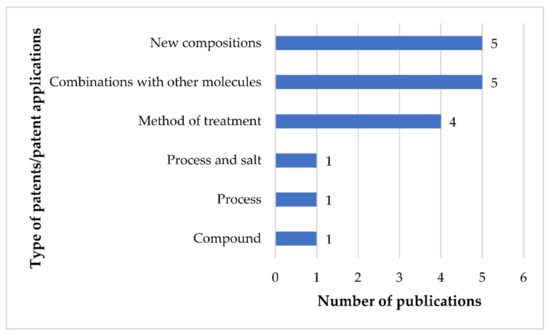
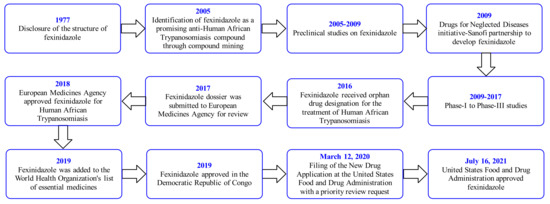
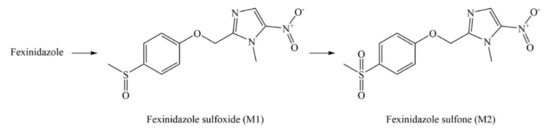
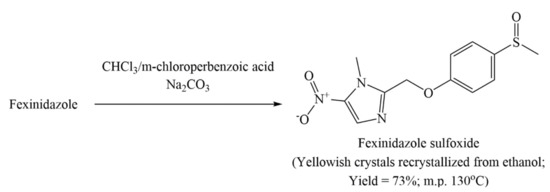
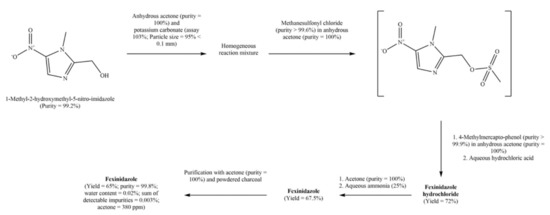
Discovery, Development, Inventions and Patent Review of Fexinidazole: The First All-Oral Therapy for Human African Trypanosomiasis
by
Mohd Imran, Shah Alam Khan, Mohammed Kanan Alshammari, Ashwaq Muiedh Alqahtani, Turkiah Abdullah Alanazi, Mehnaz Kamal, Talha Jawaid, Mohammed M. Ghoneim, Sultan Alshehri and Faiyaz Shakeel
Cited by 19 | Viewed by 4384
Abstract
Human African trypanosomiasis (HAT or ‘sleeping sickness’) is a neglected tropical disease. If untreated, it is always fatal and leads to death. A few treatments are available for HAT, but most of them require a skilled professional, which increases the financial burden on
[...] Read more.
Human African trypanosomiasis (HAT or ‘sleeping sickness’) is a neglected tropical disease. If untreated, it is always fatal and leads to death. A few treatments are available for HAT, but most of them require a skilled professional, which increases the financial burden on the patient. Recently, fexinidazole (FEX) has been approved by the European Medicine Agency (EMA) and the United States Food and Drug Administration (USFDA) as the first all-oral therapy for the treatment of stage-1 (hemolymphatic) as well as stage-2 (meningoencephalitic) of HAT. Before the FEX approval, there were separate treatments for stage-1 and stage-2 of HAT. This study reviews the discovery, development timeline, inventions, and patent literature of FEX. It was first approved by EMA and USFDA in 2018 and 2021, respectively. FEX was also added to the World Health Organization’s list of essential drugs in 2019. The patent literature search revealed many types of patents/patent applications (compound, salt, process, method of treatment, drug combinations, and compositions) related to FEX, which have been summarized in this article. The authors foresee a great scope to develop more inventions based on FEX (novel salts, polymorphs, drug conjugates, cyclodextrin complex, etc.) for the treatment of many protozoal diseases (Leishmaniasis and Chagas disease), inflammatory diseases, and other microbial infections. New combinations of FEX with other treatments of HAT may also provide fruitful results. This review might be useful to the scientists working on the HAT and other neglected diseases to develop novel inventions and innovations of therapeutic relevance.
Full article
►▼
Show Figures
Open AccessArticle
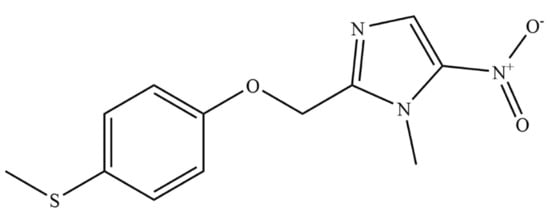
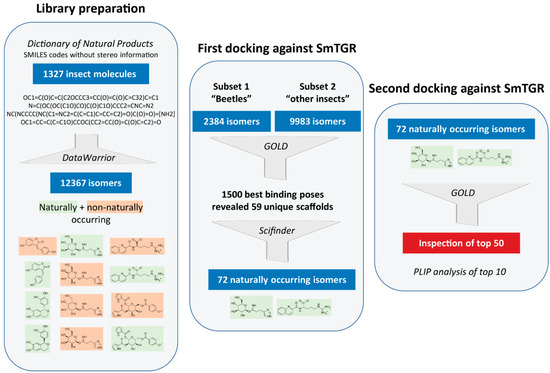
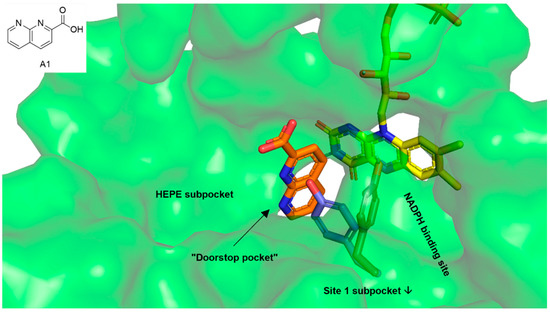
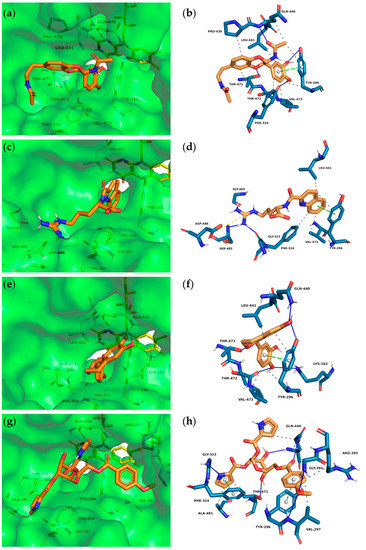
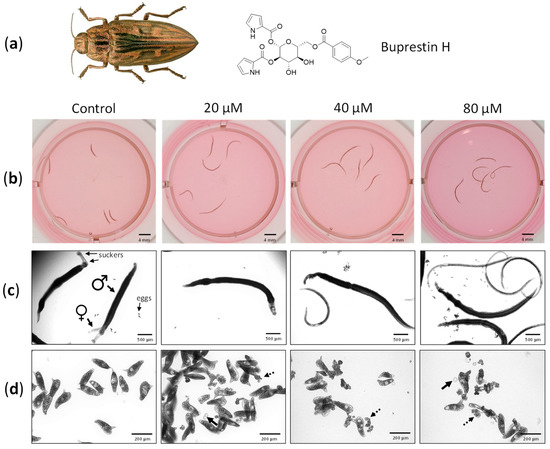
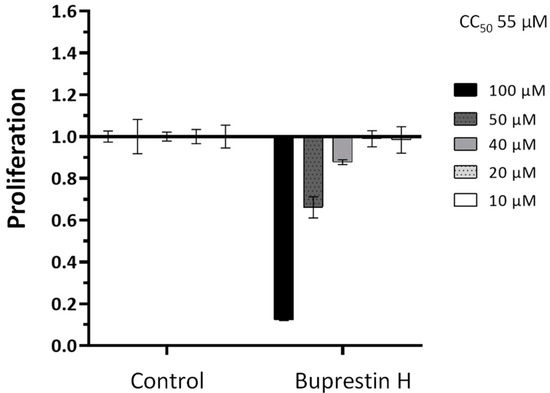
First In Silico Screening of Insect Molecules for Identification of Novel Anti-Parasitic Compounds
by
Tom L. Gallinger, Samuel Y. Aboagye, Wiebke Obermann, Michael Weiss, Arnold Grünweller, Carlo Unverzagt, David L. Williams, Martin Schlitzer and Simone Haeberlein
Cited by 8 | Viewed by 3080
Abstract
Schistosomiasis is a neglected tropical disease caused by blood flukes of the genus
Schistosoma. In silico screenings of compounds for the identification of novel anti-parasitic drug candidates have received considerable attention in recent years, including the screening of natural compounds. For the
[...] Read more.
Schistosomiasis is a neglected tropical disease caused by blood flukes of the genus
Schistosoma. In silico screenings of compounds for the identification of novel anti-parasitic drug candidates have received considerable attention in recent years, including the screening of natural compounds. For the first time, we investigated molecules from insects, a rather neglected source in drug discovery, in an in silico screening approach to find novel antischistosomal compounds. Based on the Dictionary of Natural Products (DNP), we created a library of 1327 insect compounds suitable for molecular docking. A structure-based virtual screening against the crystal structure of a known druggable target in
Schistosoma mansoni, the thioredoxin glutathione reductase (SmTGR), was performed. The top ten compounds predominantly originated from beetles and were predicted to interact particularly with amino acids in the doorstop pocket of SmTGR. For one compound from a jewel beetle, buprestin H, we tested and confirmed antischistosomal activity against adult and juvenile parasites in vitro. At concentrations with anti-parasitic activity, we could also exclude any unspecific cytotoxic activity against human HepG2 cells. This study highlights the potential of insect molecules for the identification of novel antischistosomal compounds. Our library of insect-derived molecules could serve not only as basis for future in silico screenings against additional target proteins of schistosomes, but also of other parasites.
Full article
►▼
Show Figures
Open AccessReview
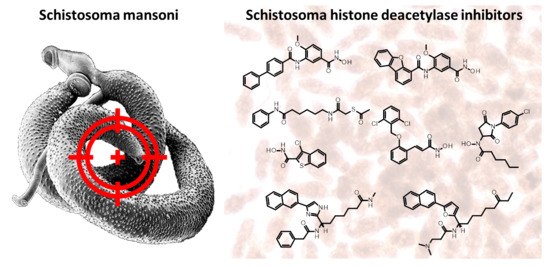
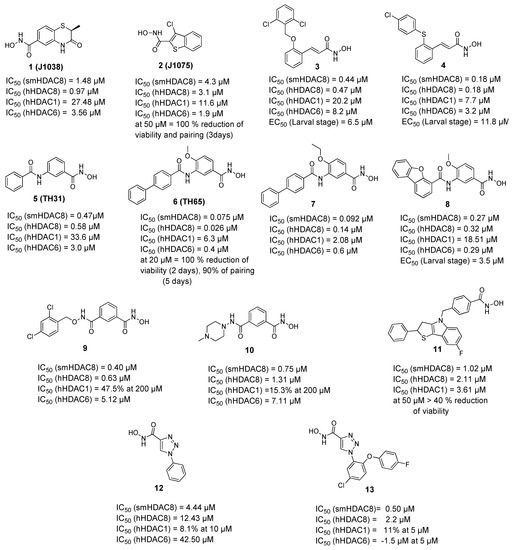
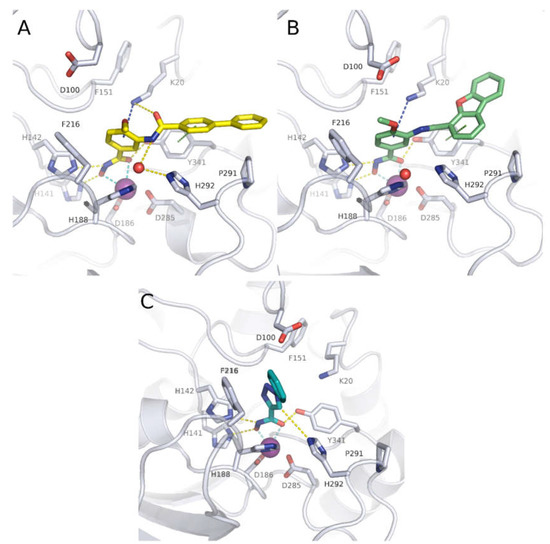
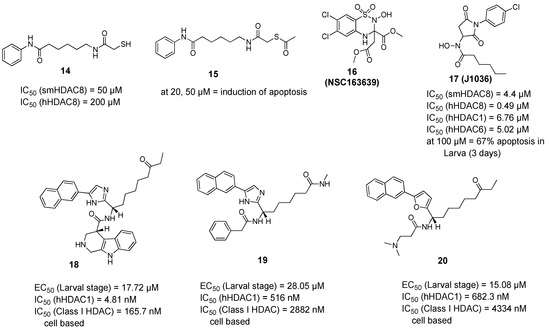
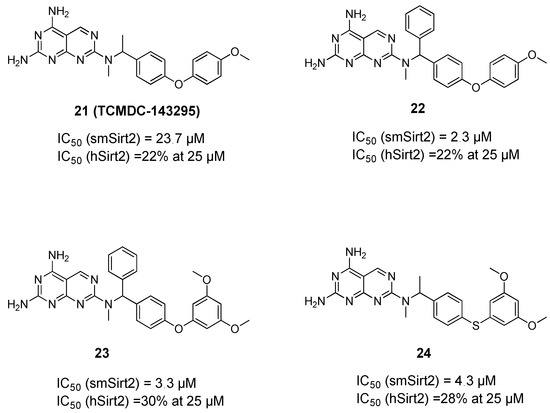
Histone Deacetylase (HDAC) Inhibitors for the Treatment of Schistosomiasis
by
Ehab Ghazy, Mohamed Abdelsalam, Dina Robaa, Raymond J. Pierce and Wolfgang Sippl
Cited by 11 | Viewed by 3395
Abstract
Schistosomiasis is a major neglected parasitic disease that affects more than 240 million people worldwide and for which the control strategy consists of mass treatment with the only available drug, praziquantel. Schistosomes display morphologically distinct stages during their life cycle and the transformations
[...] Read more.
Schistosomiasis is a major neglected parasitic disease that affects more than 240 million people worldwide and for which the control strategy consists of mass treatment with the only available drug, praziquantel. Schistosomes display morphologically distinct stages during their life cycle and the transformations between stages are controlled by epigenetic mechanisms. The targeting of epigenetic actors might therefore represent the parasites’ Achilles’ heel. Specifically, histone deacetylases have been recently characterized as drug targets for the treatment of schistosomiasis. This review focuses on the recent development of inhibitors for schistosome histone deacetylases. In particular, advances in the development of inhibitors of
Schistosoma mansoni histone deacetylase 8 have indicated that targeting this enzyme is a promising approach for the treatment of this infection.
Full article
►▼
Show Figures
Open AccessArticle
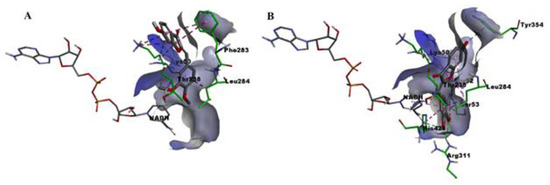
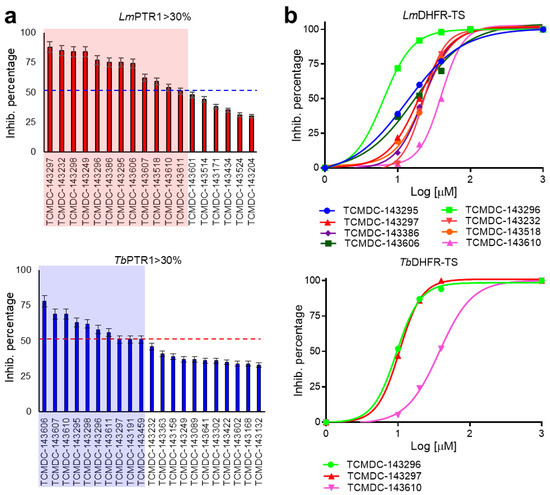
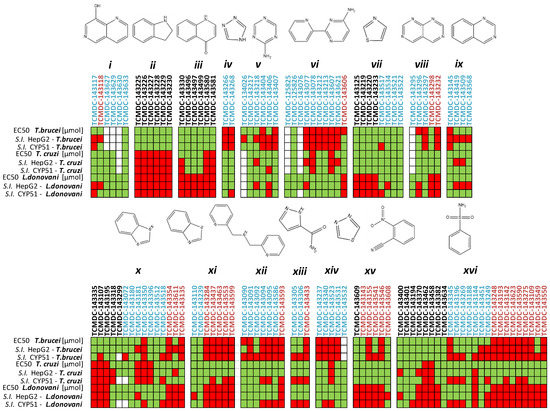
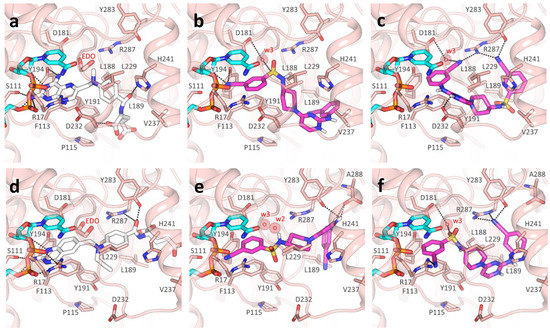
Repurposing the Trypanosomatidic GSK Kinetobox for the Inhibition of Parasitic Pteridine and Dihydrofolate Reductases
by
Matteo Santucci, Rosaria Luciani, Eleonora Gianquinto, Cecilia Pozzi, Flavio di Pisa, Lucia dello Iacono, Giacomo Landi, Lorenzo Tagliazucchi, Stefano Mangani, Francesca Spyrakis and Maria Paola Costi
Cited by 3 | Viewed by 2657
Abstract
Three open-source anti-kinetoplastid chemical boxes derived from a whole-cell phenotypic screening by GlaxoSmithKline (Tres Cantos Anti-Kinetoplastid Screening, TCAKS) were exploited for the discovery of a novel core structure inspiring new treatments of parasitic diseases targeting the trypansosmatidic pteridine reductase 1 (PTR1) and dihydrofolate
[...] Read more.
Three open-source anti-kinetoplastid chemical boxes derived from a whole-cell phenotypic screening by GlaxoSmithKline (Tres Cantos Anti-Kinetoplastid Screening, TCAKS) were exploited for the discovery of a novel core structure inspiring new treatments of parasitic diseases targeting the trypansosmatidic pteridine reductase 1 (PTR1) and dihydrofolate reductase (DHFR) enzymes. In total, 592 compounds were tested through medium-throughput screening assays. A subset of 14 compounds successfully inhibited the enzyme activity in the low micromolar range of at least one of the enzymes from both
Trypanosoma brucei and
Lesihmania major parasites (pan-inhibitors), or from both PTR1 and DHFR-TS of the same parasite (dual inhibitors). Molecular docking studies of the protein–ligand interaction focused on new scaffolds not reproducing the well-known antifolate core clearly explaining the experimental data. TCMDC-143249, classified as a benzenesulfonamide derivative by the QikProp descriptor tool, showed selective inhibition of PTR1 and growth inhibition of the kinetoplastid parasites in the 5 μM range. In our work, we enlarged the biological profile of the GSK Kinetobox and identified new core structures inhibiting selectively PTR1, effective against the kinetoplastid infectious protozoans. In perspective, we foresee the development of selective PTR1 and DHFR inhibitors for studies of drug combinations.
Full article
►▼
Show Figures
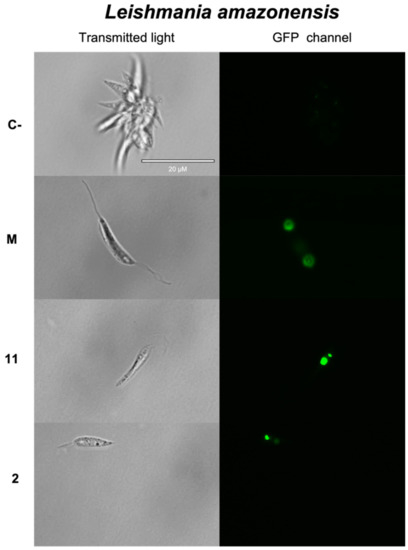
Open AccessArticle
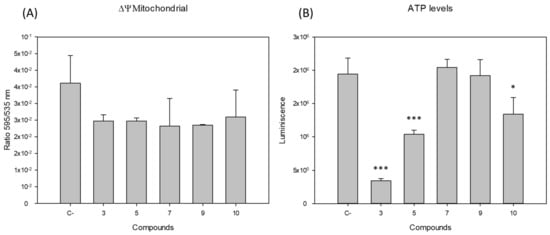
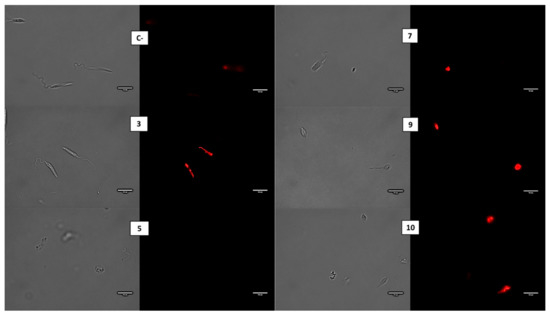
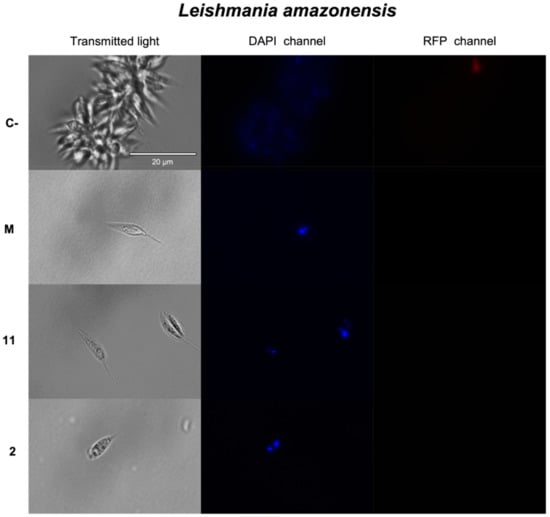
Discovery of New Chemical Tools against Leishmania amazonensis via the MMV Pathogen Box
by
Atteneri López-Arencibia, Ines Sifaoui, María Reyes-Batlle, Carlos J. Bethencourt-Estrella, Desirée San Nicolás-Hernández, Jacob Lorenzo-Morales and José E. Piñero
Cited by 5 | Viewed by 1459
Abstract
The protozoan parasite
Leishmania causes a spectrum of diseases and there are over 1 million infections each year. Current treatments are toxic, expensive, and difficult to administer, and resistance to them is emerging. In this study, we screened the antileishmanial activity of the
[...] Read more.
The protozoan parasite
Leishmania causes a spectrum of diseases and there are over 1 million infections each year. Current treatments are toxic, expensive, and difficult to administer, and resistance to them is emerging. In this study, we screened the antileishmanial activity of the Pathogen Box compounds from the Medicine for Malaria Venture against
Leishmania amazonensis, and compared their structures and cytotoxicity. The compounds MMV676388 (3), MMV690103 (5), MMV022029 (7), MMV022478 (9) and MMV021013 (10) exerted a significant dose-dependent inhibition effect on the proliferation of
L. amazonensis promastigotes and intracellular amastigotes. Moreover, studies on the mechanism of cell death showed that compounds 3 and 5 induced an apoptotic process while the compounds 7, 9 and 10 seem to induce an autophagic mechanism. The present findings underline the potential of these five molecules as novel therapeutic leishmanicidal agents.
Full article
►▼
Show Figures
Open AccessArticle
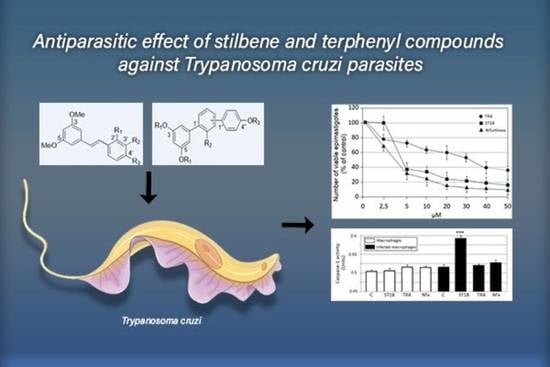
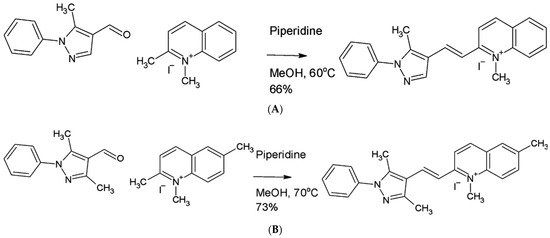
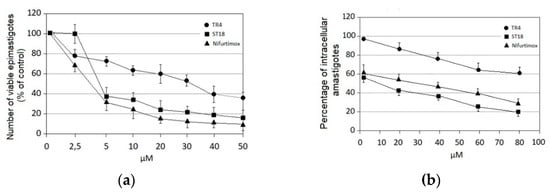
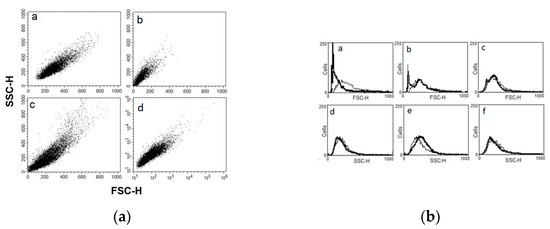
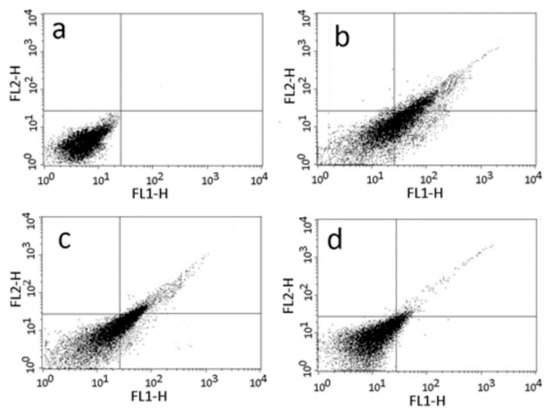
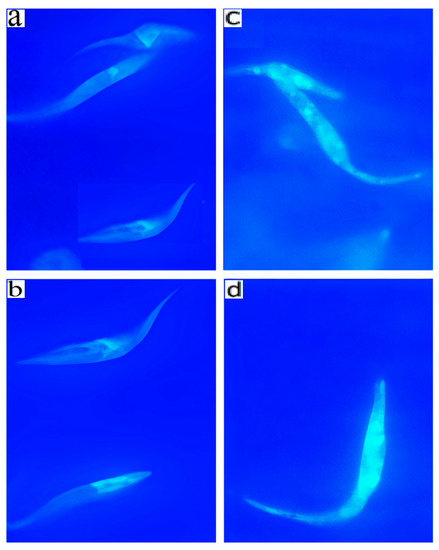
Antiparasitic Effect of Stilbene and Terphenyl Compounds against Trypanosoma cruzi Parasites
by
Federica Bruno, Germano Castelli, Fabrizio Vitale, Simone Catanzaro, Valeria Vitale Badaco, Marinella Roberti, Claudia Colomba, Antonio Cascio and Manlio Tolomeo
Cited by 3 | Viewed by 1826
Abstract
Background: Chagas disease, also known as American trypanosomiasis, is a potentially life-threatening illness caused by the protozoan parasite
Trypanosoma cruzi. No progress in the treatment of this pathology has been made since Nifurtimox was introduced more than fifty years ago, and this
[...] Read more.
Background: Chagas disease, also known as American trypanosomiasis, is a potentially life-threatening illness caused by the protozoan parasite
Trypanosoma cruzi. No progress in the treatment of this pathology has been made since Nifurtimox was introduced more than fifty years ago, and this drug is considered very aggressive and may cause several adverse effects. This drug currently has severe limitations, including a high frequency of undesirable side effects and limited efficacy and availability, so research to discover new drugs for the treatment of Chagas disease is imperative. Many drugs available on the market are natural products as found in nature or compounds designed based on the structure and activity of these natural products. Methods: This study evaluated the in vitro antiparasitic activity of a series of previously synthesized stilbene and terphenyl compounds in
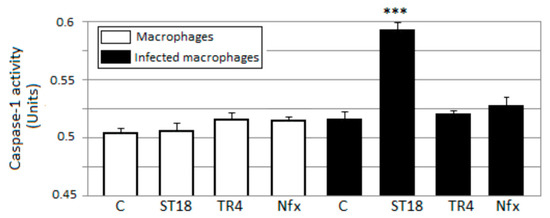
T. cruzi epimastigotes and intracellular amastigotes. The action of the most selective compounds was investigated by flow cytometric analysis to evaluate the mechanism of cell death. The ability to induce apoptosis or caspase-1 inflammasomes was assayed in macrophages infected with
T. cruzi after treatment, comparing it with that of Nifurtimox. Results: The stilbene ST18 was the most potent compound of the series. It was slightly less active than Nifurtimox in epimastigotes but most active in intracellular amastigotes. Compared to Nifurtimox, it was markedly less cytotoxic when tested in vitro on normal cells. ST18 was able to induce a marked increase in parasites positive for Annexin V and monodansylcadaverine. Moreover, ST18 induced the activation, in infected macrophages, of caspase-1, a conserved enzyme that plays a major role in controlling parasitemia, host survival and the onset of the adaptive immune response in Trypanosoma infection. Conclusions: The antiparasitic activity of ST18 together with its ability to activate caspase-1 in infected macrophages and its low toxicity toward normal cells makes this compound interesting for further clinical investigation.
Full article
►▼
Show Figures
Open AccessReview
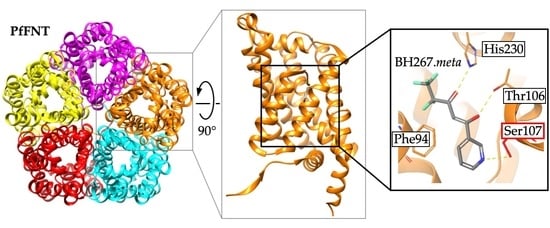
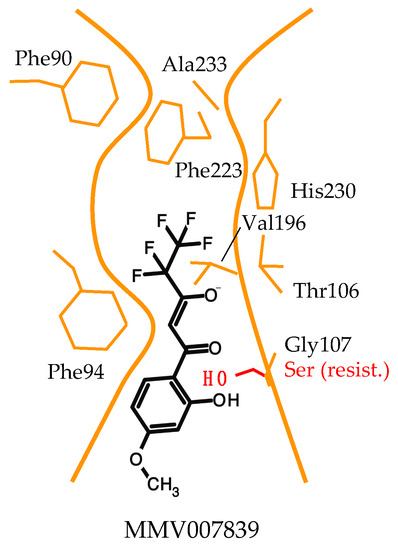
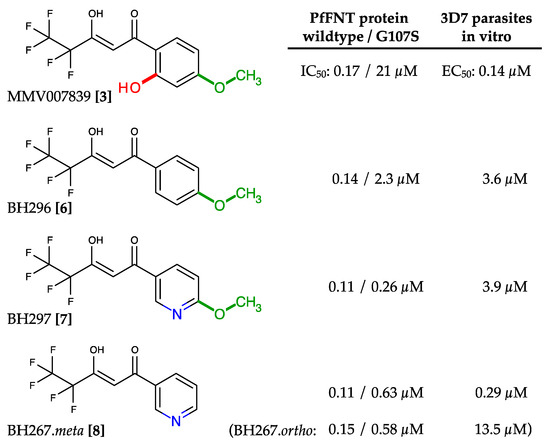
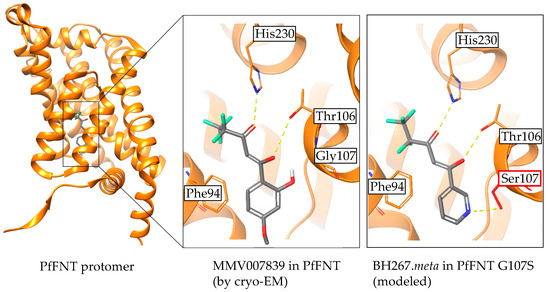
Discovery and Development of Inhibitors of the Plasmodial FNT-Type Lactate Transporter as Novel Antimalarials
by
Cornelius Nerlich, Nathan H. Epalle, Philip Seick and Eric Beitz
Cited by 5 | Viewed by 2299
Abstract
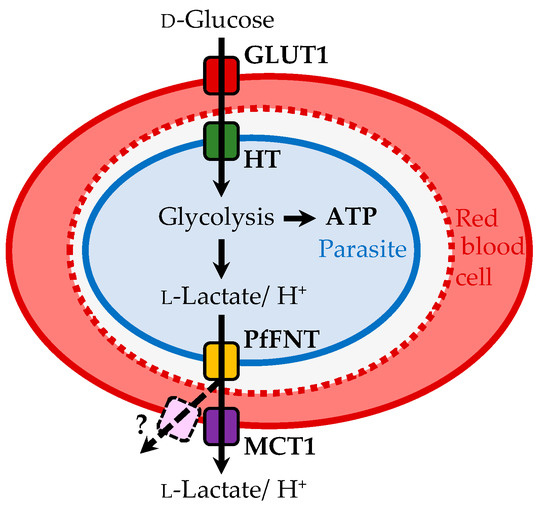
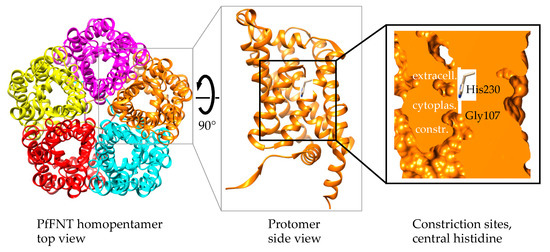
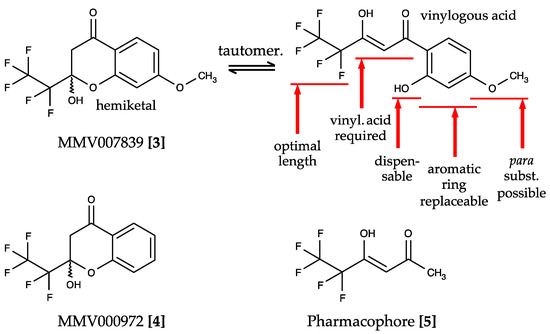
Plasmodium spp. malaria parasites in the blood stage draw energy from anaerobic glycolysis when multiplying in erythrocytes. They tap the ample glucose supply of the infected host using the erythrocyte glucose transporter 1, GLUT1, and a hexose transporter, HT, of the parasite’s plasma
[...] Read more.
Plasmodium spp. malaria parasites in the blood stage draw energy from anaerobic glycolysis when multiplying in erythrocytes. They tap the ample glucose supply of the infected host using the erythrocyte glucose transporter 1, GLUT1, and a hexose transporter, HT, of the parasite’s plasma membrane. Per glucose molecule, two lactate anions and two protons are generated as waste that need to be released rapidly from the parasite to prevent blockage of the energy metabolism and acidification of the cytoplasm. Recently, the missing
Plasmodium lactate/H
+ cotransporter was identified as a member of the exclusively microbial formate–nitrite transporter family, FNT. Screening of an antimalarial compound selection with unknown targets led to the discovery of specific and potent FNT-inhibitors, i.e., pentafluoro-3-hydroxy-pent-2-en-1-ones. Here, we summarize the discovery and further development of this novel class of antimalarials, their modes of binding and action, circumvention of a putative resistance mutation of the FNT target protein, and suitability for in vivo studies using animal malaria models.
Full article
►▼
Show Figures
Open AccessArticle
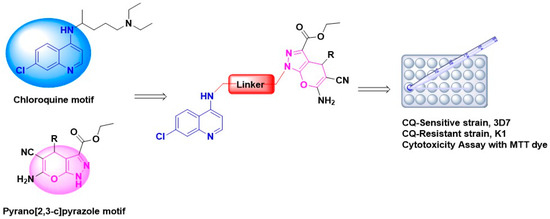
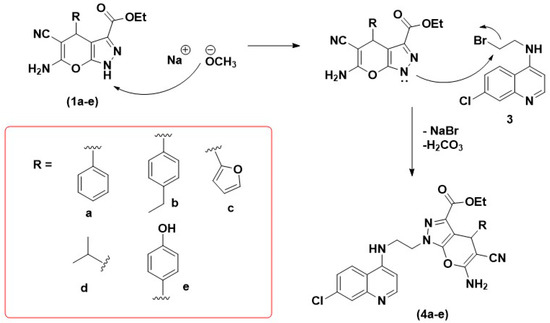
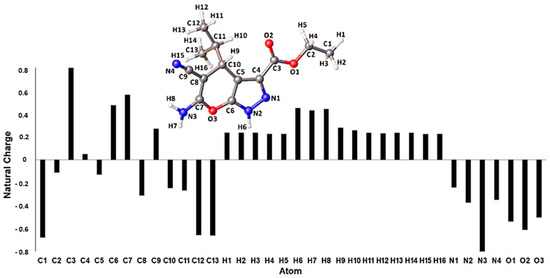
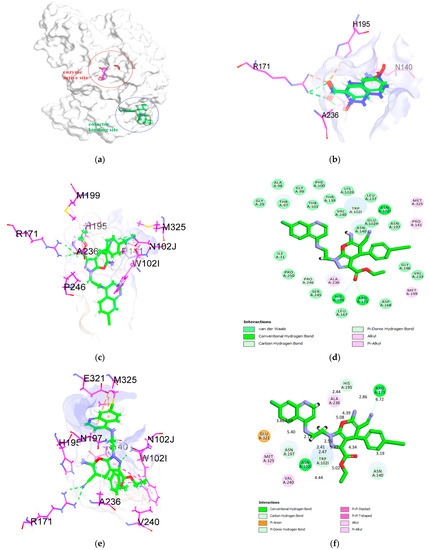
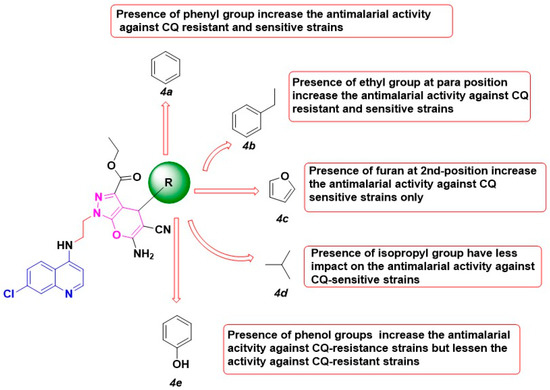
Synthesis, Molecular Docking, and Antimalarial Activity of Hybrid 4-Aminoquinoline-pyrano[2,3-c]pyrazole Derivatives
by
Mohd Asyraf Shamsuddin, Amatul Hamizah Ali, Nur Hanis Zakaria, Mohd Fazli Mohammat, Ahmad Sazali Hamzah, Zurina Shaameri, Kok Wai Lam, Wun Fui Mark-Lee, Hani Kartini Agustar, Mohd Ridzuan Mohd Abd Razak, Jalifah Latip and Nurul Izzaty Hassan
Cited by 14 | Viewed by 2910
Abstract
Widespread resistance of
Plasmodium falciparum to current artemisinin-based combination therapies necessitate the discovery of new medicines. Pharmacophoric hybridization has become an alternative for drug resistance that lowers the risk of drug–drug adverse interactions. In this study, we synthesized a new series of hybrids
[...] Read more.
Widespread resistance of
Plasmodium falciparum to current artemisinin-based combination therapies necessitate the discovery of new medicines. Pharmacophoric hybridization has become an alternative for drug resistance that lowers the risk of drug–drug adverse interactions. In this study, we synthesized a new series of hybrids by covalently linking the scaffolds of pyrano[2,3-c]pyrazole with 4-aminoquinoline via an ethyl linker. All synthesized hybrid molecules were evaluated through in vitro screenings against chloroquine-resistant (K1) and -sensitive (3D7)
P. falciparum strains, respectively. Data from in vitro assessments showed that hybrid
4b displayed significant antiplasmodial activities against the 3D7 strain (EC
50 = 0.0130 ± 0.0002 μM) and the K1 strain (EC
50 = 0.02 ± 0.01 μM), with low cytotoxic effect against Vero mammalian cells. The high selectivity index value on the 3D7 strain (SI > 1000) and the K1 strain (SI > 800) and the low resistance index value from compound
4b suggested that the pharmacological effects of this compound were due to selective inhibition on the 3D7 and K1 strains. Molecular docking analysis also showed that
4b recorded the highest binding energy on
P. falciparum lactate dehydrogenase. Thus,
P. falciparum lactate dehydrogenase is considered a potential molecular target for the synthesized compound.
Full article
►▼
Show Figures
Open AccessArticle
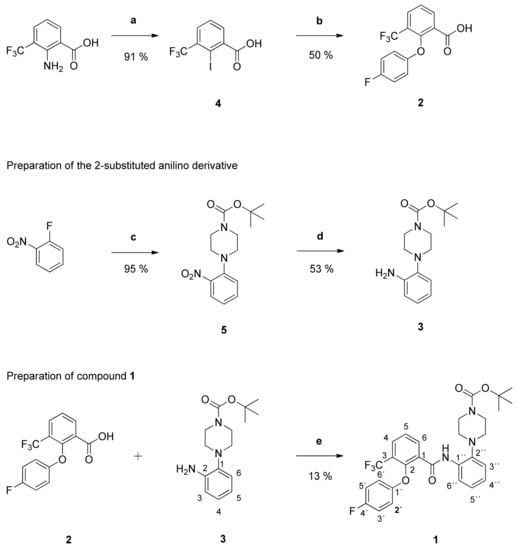
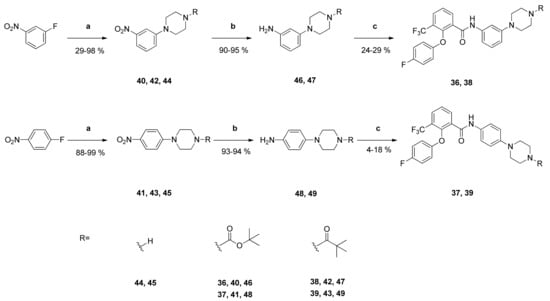
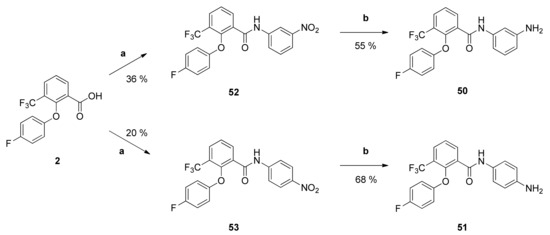
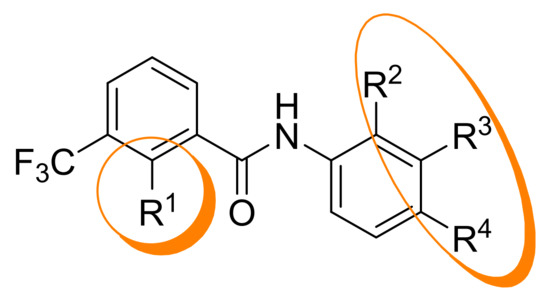
Synthesis and Structure-Activity Relationships of New 2-Phenoxybenzamides with Antiplasmodial Activity
by
Theresa Hermann, Patrick Hochegger, Johanna Dolensky, Werner Seebacher, Eva-Maria Pferschy-Wenzig, Robert Saf, Marcel Kaiser, Pascal Mäser and Robert Weis
Cited by 1 | Viewed by 2199
Abstract
The 2-phenoxybenzamide
1 from the Medicines for Malaria Venture Malaria Box Project has shown promising multi-stage activity against different strains of
P. falciparum. It was successfully synthesized via a retrosynthetic approach. Subsequently, twenty-one new derivatives were prepared and tested for their in
[...] Read more.
The 2-phenoxybenzamide
1 from the Medicines for Malaria Venture Malaria Box Project has shown promising multi-stage activity against different strains of
P. falciparum. It was successfully synthesized via a retrosynthetic approach. Subsequently, twenty-one new derivatives were prepared and tested for their in vitro activity against blood stages of the NF54 strain of
P. falciparum. Several insights into structure-activity relationships were revealed. The antiplasmodial activity and cytotoxicity of compounds strongly depended on the substitution pattern of the anilino partial structure as well as on the size of substituents. The diaryl ether partial structure had further impacts on the activity. Additionally, several physicochemical and pharmacokinetic parameters were calculated (log P, log D
7.
4 and ligand efficiency) or determined experimentally (passive permeability and CYP3A4 inhibition). The
tert-butyl-4-{4-[2-(4-fluorophenoxy)-3-(trifluoromethyl)benzamido]phenyl}piperazine-1-carboxylate possesses high antiplasmodial activity against
P. falciparum NF54 (
PfNF54 IC
50 = 0.2690 µM) and very low cytotoxicity (L-6 cells IC
50 = 124.0 µM) resulting in an excellent selectivity index of 460. Compared to the lead structure
1 the antiplasmodial activity was improved as well as the physicochemical and some pharmacokinetic parameters.
Full article
►▼
Show Figures
Open AccessArticle
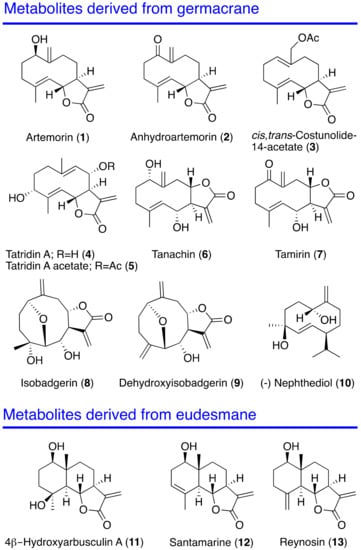
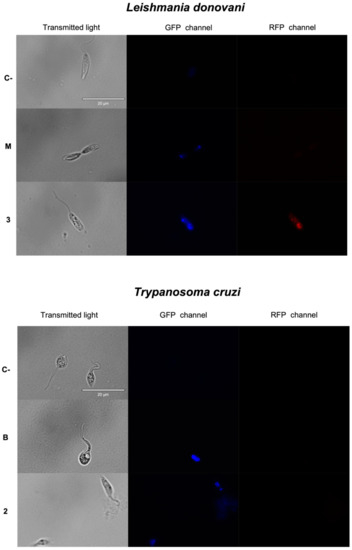
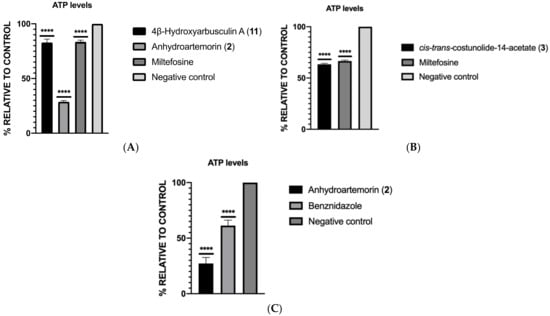
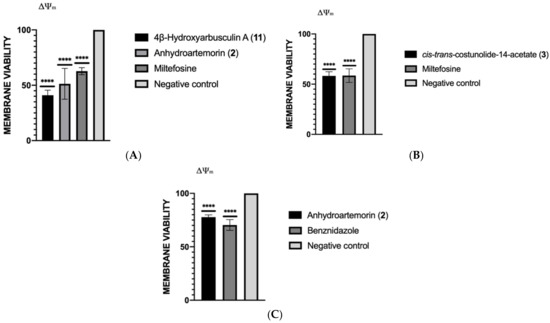
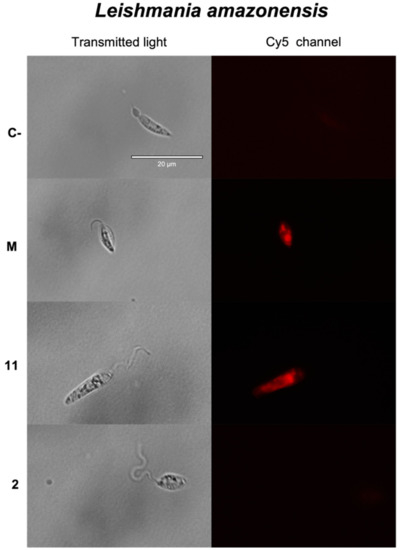
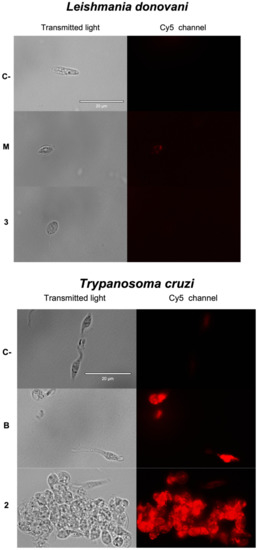
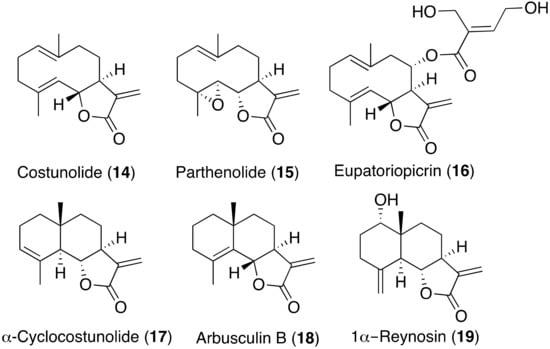
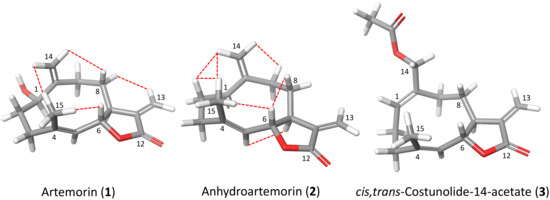
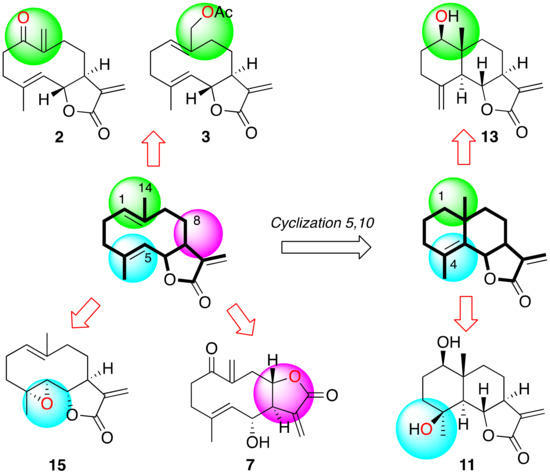
Antikinetoplastid Activity of Sesquiterpenes Isolated from the Zoanthid Palythoa aff. clavata
by
Carlos J. Bethencourt-Estrella, Nathalia Nocchi, Atteneri López-Arencibia, Desirée San Nicolás-Hernández, María L. Souto, Blanca Suárez-Gómez, Ana R. Díaz-Marrero, José J. Fernández, Jacob Lorenzo-Morales and José E. Piñero
Cited by 8 | Viewed by 1768
Abstract
Leishmaniasis and Chagas disease are neglected tropical diseases that cause problems in developing countries. The causative agents,
Leishmania spp. and
Trypanosoma cruzi, produce a clinical picture that can be fatal for the patient, such as Chagas heart disease, visceral leishmaniasis and megacolon,
[...] Read more.
Leishmaniasis and Chagas disease are neglected tropical diseases that cause problems in developing countries. The causative agents,
Leishmania spp. and
Trypanosoma cruzi, produce a clinical picture that can be fatal for the patient, such as Chagas heart disease, visceral leishmaniasis and megacolon, among others. Current treatments for these diseases are not very effective and highly toxic, since they require very prolonged treatments. The development of innovative, effective and safe drugs to fight infections caused by these parasites remains a challenge. For this reason, in recent years, there has been an increase in the search for new therapies. In this study, the antikinetoplastid activity of 13 sesquiterpene lactones obtained from
Palythoa aff. clavata was screened against
L. amazonensis,
L. donovani and
T. cruzi. The results revealed that the sesquiterpene lactones anhydroartemorin (
2),
cis,trans-costunolide-14-acetate (
3) and 4-hydroxyarbusculin A (
11) were the most selective against the kinetoplastid species studied. These molecules seem to induce the mechanisms involved in an apoptotic-like death or programmed cell death (PCD) in the kinetoplastids, and since they do not cause necrosis, the inflammatory events associated with this type of cell death will not be triggered.
Full article
►▼
Show Figures
Open AccessArticle
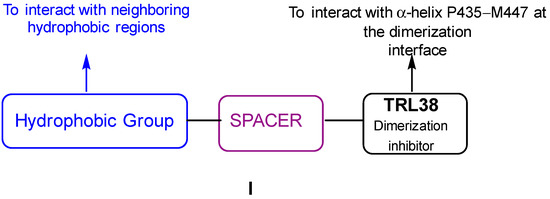
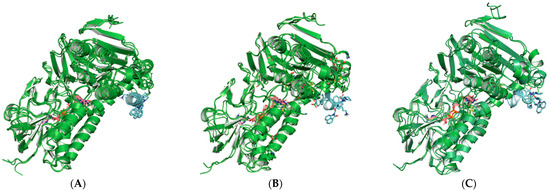
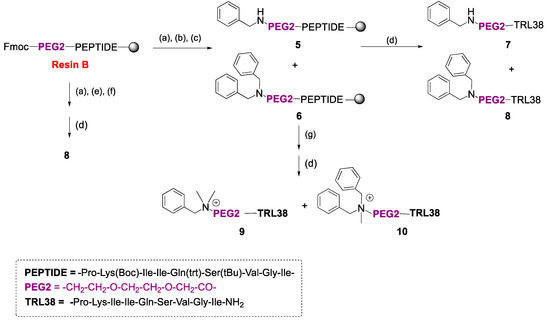
Small Molecule–Peptide Conjugates as Dimerization Inhibitors of Leishmania infantum Trypanothione Disulfide Reductase
by
Alejandro Revuelto, Isabel López-Martín, Héctor de Lucio, Juan Carlos García-Soriano, Nicola Zanda, Sonia de Castro, Federico Gago, Antonio Jiménez-Ruiz, Sonsoles Velázquez and María-José Camarasa
Viewed by 2946
Abstract
Trypanothione disulfide reductase (TryR) is an essential homodimeric enzyme of trypanosomatid parasites that has been validated as a drug target to fight human infections. Using peptides and peptidomimetics, we previously obtained proof of concept that disrupting protein–protein interactions at the dimer interface of
[...] Read more.
Trypanothione disulfide reductase (TryR) is an essential homodimeric enzyme of trypanosomatid parasites that has been validated as a drug target to fight human infections. Using peptides and peptidomimetics, we previously obtained proof of concept that disrupting protein–protein interactions at the dimer interface of
Leishmania infantum TryR
(LiTryR
) offered an innovative and so far unexploited opportunity for the development of novel antileishmanial agents. Now, we show that linking our previous peptide prototype
TRL38 to selected hydrophobic moieties provides a novel series of small-molecule–peptide conjugates that behave as good inhibitors of both
LiTryR activity and dimerization.
Full article
►▼
Show Figures
Open AccessArticle
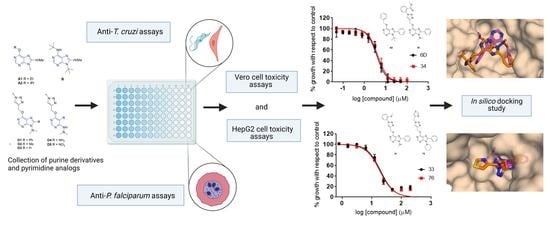
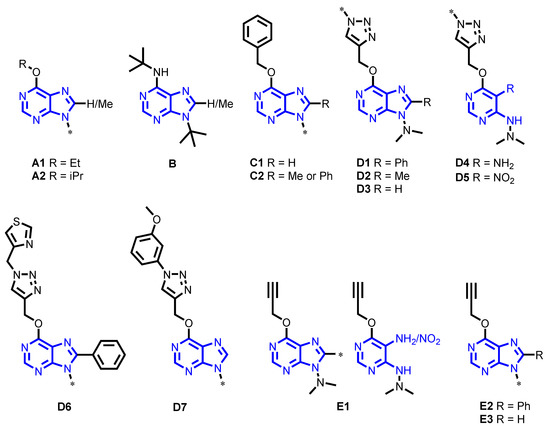
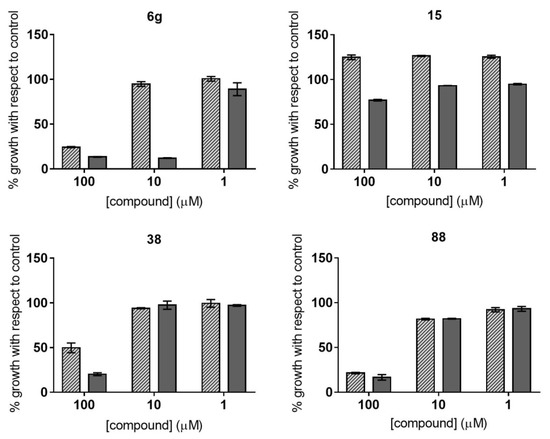
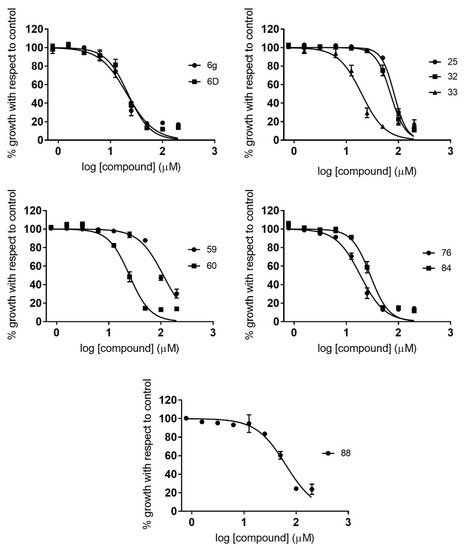
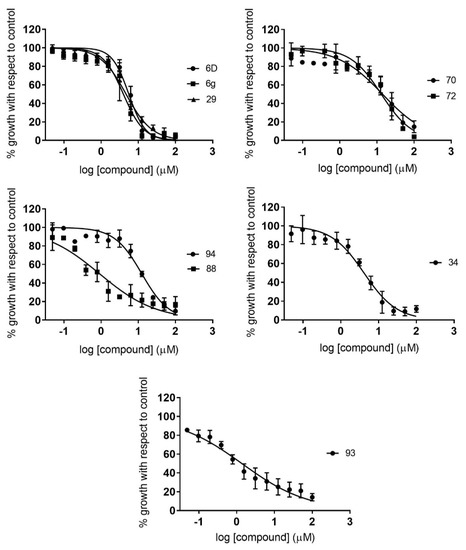
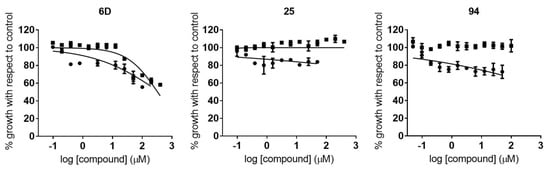
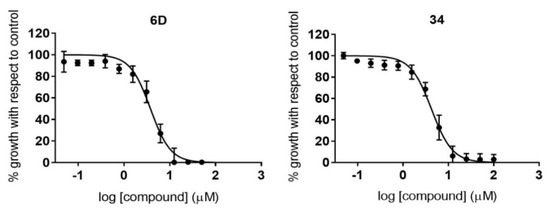
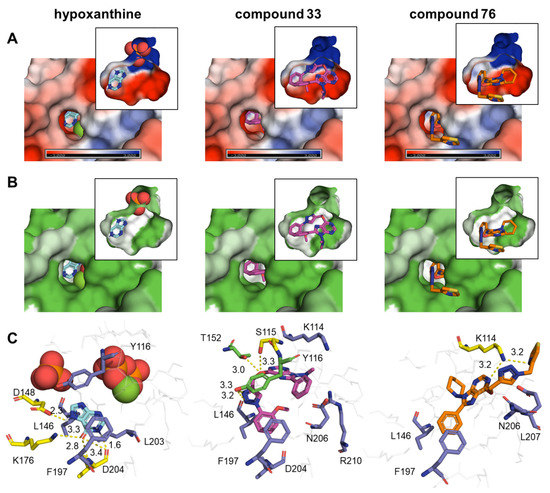
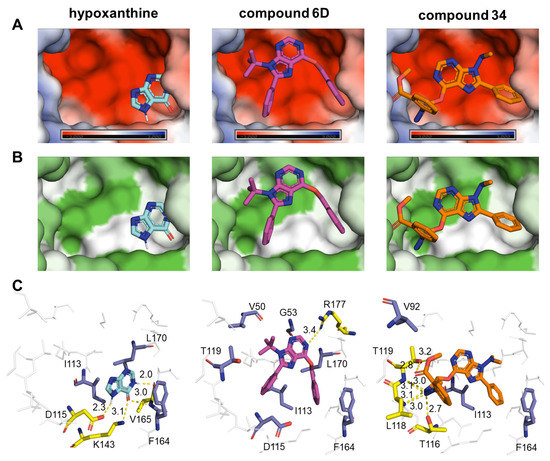
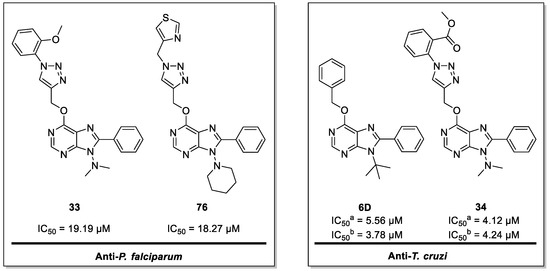
Novel Purine Chemotypes with Activity against Plasmodium falciparum and Trypanosoma cruzi
by
Nieves Martinez-Peinado, Álvaro Lorente-Macías, Alejandro García-Salguero, Nuria Cortes-Serra, Ángel Fenollar-Collado, Albert Ros-Lucas, Joaquim Gascon, Maria-Jesus Pinazo, Ignacio J. Molina, Asier Unciti-Broceta, Juan J. Díaz-Mochón, María J. Pineda de las Infantas y Villatoro, Luis Izquierdo and Julio Alonso-Padilla
Cited by 6 | Viewed by 3692
Abstract
Malaria and Chagas disease, caused by
Plasmodium spp. and
Trypanosoma cruzi parasites, remain important global health problems. Available treatments for those diseases present several limitations, such as lack of efficacy, toxic side effects, and drug resistance. Thus, new drugs are urgently needed. The
[...] Read more.
Malaria and Chagas disease, caused by
Plasmodium spp. and
Trypanosoma cruzi parasites, remain important global health problems. Available treatments for those diseases present several limitations, such as lack of efficacy, toxic side effects, and drug resistance. Thus, new drugs are urgently needed. The discovery of new drugs may be benefited by considering the significant biological differences between hosts and parasites. One of the most striking differences is found in the purine metabolism, because most of the parasites are incapable of de novo purine biosynthesis. Herein, we have analyzed the in vitro anti-
P. falciparum and anti-
T. cruzi activity of a collection of 81 purine derivatives and pyrimidine analogs. We firstly used a primary screening at three fixed concentrations (100, 10, and 1 µM) and progressed those compounds that kept the growth of the parasites < 30% at 100 µM to dose–response assays. Then, we performed two different cytotoxicity assays on Vero cells and human HepG2 cells. Finally, compounds specifically active against
T. cruzi were tested against intracellular amastigote forms. Purines
33 (IC
50 = 19.19 µM) and
76 (IC
50 = 18.27 µM) were the most potent against
P. falciparum. On the other hand,
6D (IC
50 = 3.78 µM) and
34 (IC
50 = 4.24 µM) were identified as hit purines against
T. cruzi amastigotes. Moreover, an in silico docking study revealed that
P. falciparum and
T. cruzi hypoxanthine guanine phosphoribosyltransferase enzymes could be the potential targets of those compounds. Our study identified two novel, purine-based chemotypes that could be further optimized to generate potent and diversified anti-parasitic drugs against both parasites.
Full article
►▼
Show Figures
Open AccessArticle
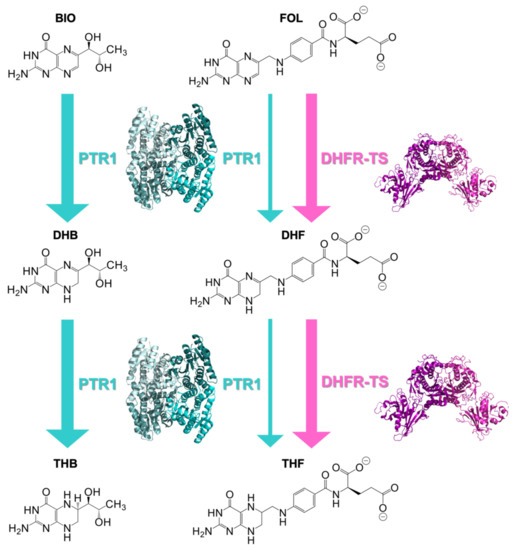
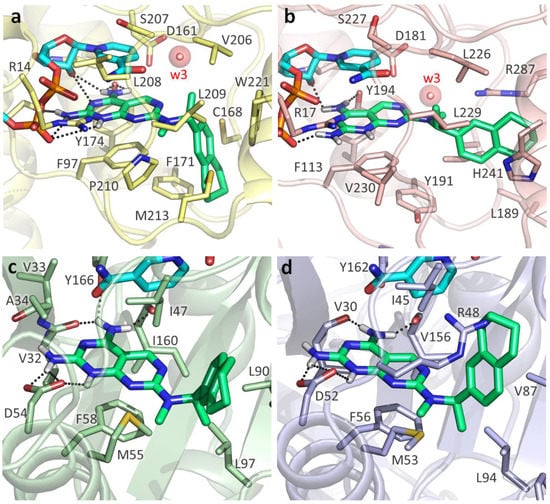
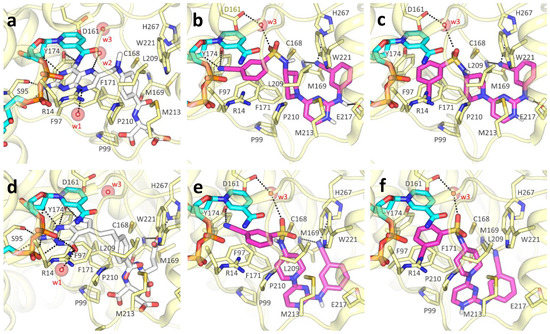
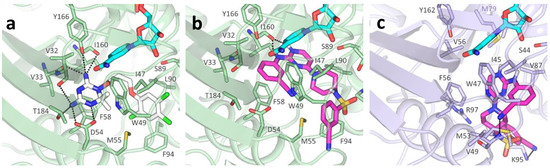
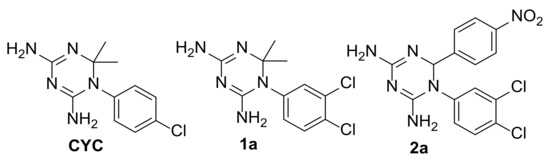
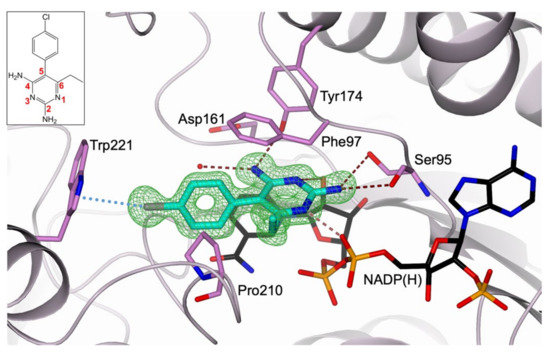
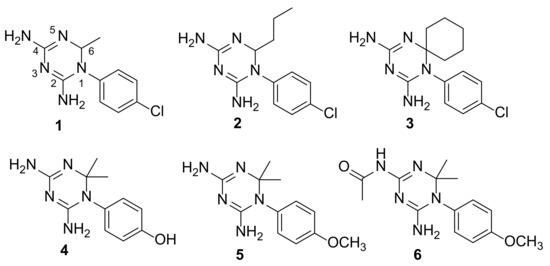
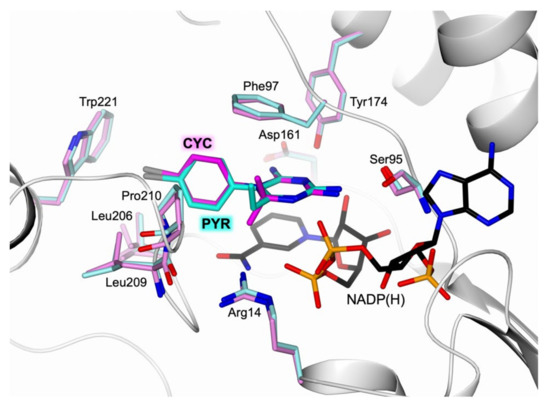
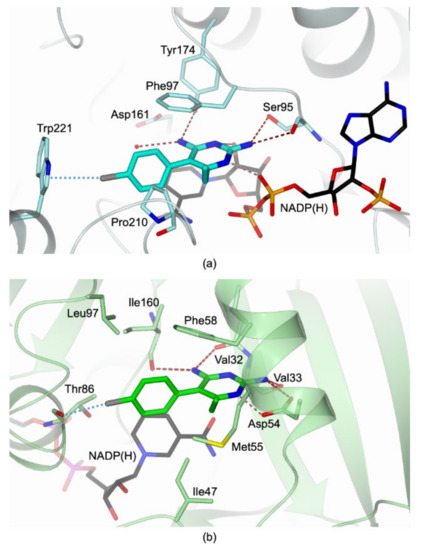
Evidence of Pyrimethamine and Cycloguanil Analogues as Dual Inhibitors of Trypanosoma brucei Pteridine Reductase and Dihydrofolate Reductase
by
Giusy Tassone, Giacomo Landi, Pasquale Linciano, Valeria Francesconi, Michele Tonelli, Lorenzo Tagliazucchi, Maria Paola Costi, Stefano Mangani and Cecilia Pozzi
Cited by 10 | Viewed by 3306
Abstract
Trypanosoma and
Leishmania parasites are the etiological agents of various threatening neglected tropical diseases (NTDs), including human African trypanosomiasis (HAT), Chagas disease, and various types of leishmaniasis. Recently, meaningful progresses in the treatment of HAT, due to
Trypanosoma brucei (
Tb), have
[...] Read more.
Trypanosoma and
Leishmania parasites are the etiological agents of various threatening neglected tropical diseases (NTDs), including human African trypanosomiasis (HAT), Chagas disease, and various types of leishmaniasis. Recently, meaningful progresses in the treatment of HAT, due to
Trypanosoma brucei (
Tb), have been achieved by the introduction of fexinidazole and the combination therapy eflornithine–nifurtimox. Nevertheless, due to drug resistance issues and the exitance of animal reservoirs, the development of new NTD treatments is still required. For this purpose, we explored the combined targeting of two key folate enzymes, dihydrofolate reductase (DHFR) and pteridine reductase 1 (PTR1). We formerly showed that the
TbDHFR inhibitor cycloguanil (CYC) also targets
TbPTR1, although with reduced affinity. Here, we explored a small library of CYC analogues to understand how their substitution pattern affects the inhibition of both
TbPTR1 and
TbDHFR. Some novel structural features responsible for an improved, but preferential, ability of CYC analogues to target
TbPTR1 were disclosed. Furthermore, we showed that the known drug pyrimethamine (PYR) effectively targets both enzymes, also unveiling its binding mode to
TbPTR1. The structural comparison between PYR and CYC binding modes to
TbPTR1 and
TbDHFR provided key insights for the future design of dual inhibitors for HAT therapy.
Full article
►▼
Show Figures
Open AccessArticle
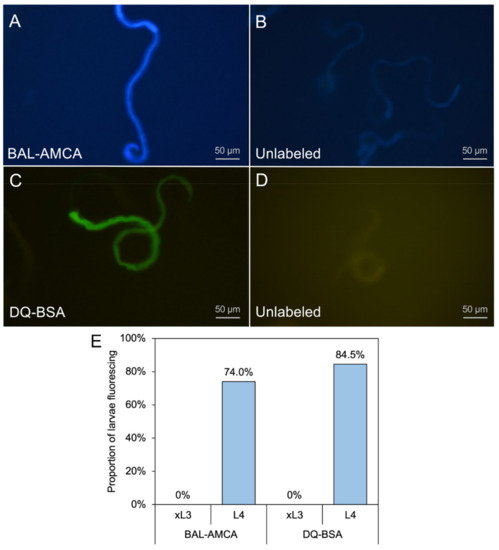
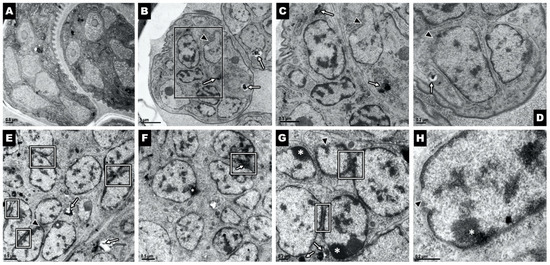
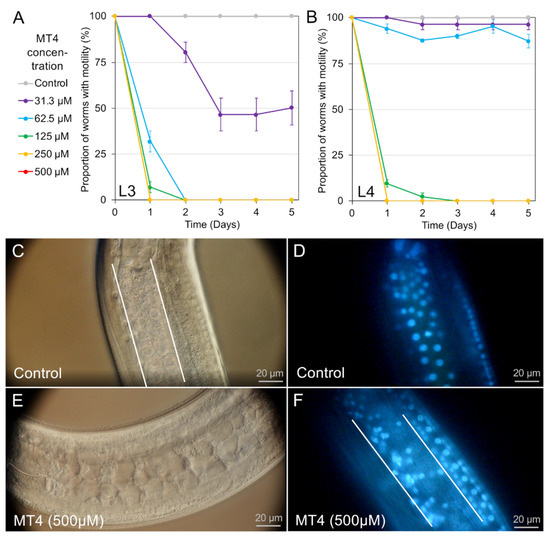
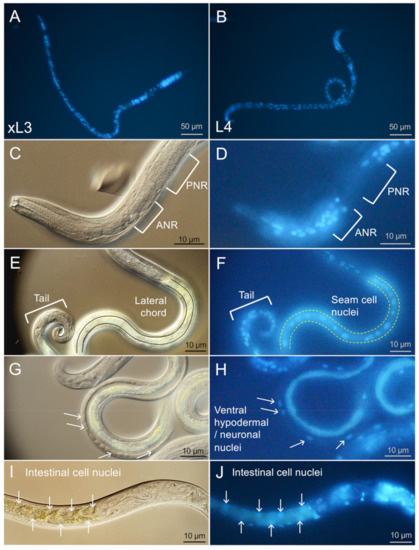
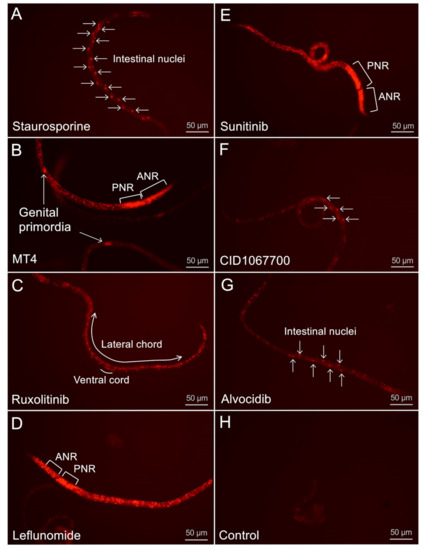
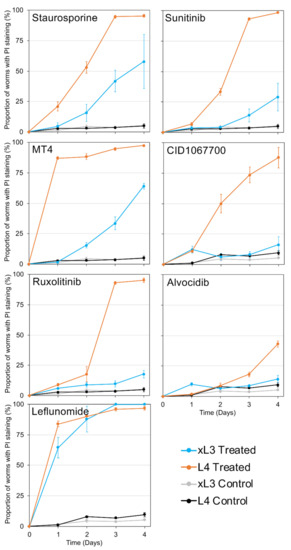
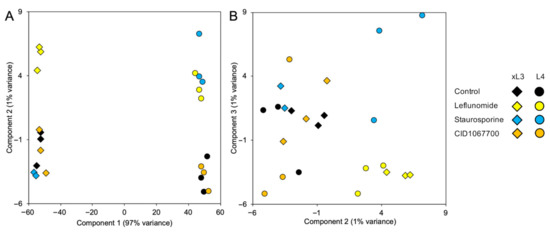
Cell Death and Transcriptional Responses Induced in Larvae of the Nematode Haemonchus contortus by Toxins/Toxicants with Broad Phylogenetic Efficacy
by
Douglas P. Jasmer, Bruce A. Rosa and Makedonka Mitreva
Cited by 3 | Viewed by 2517
Abstract
Establishing methods to investigate treatments that induce cell death in parasitic nematodes will promote experimental approaches to elucidate mechanisms and to identify prospective anthelmintics capable of inducing this outcome. Here, we extended recent progress on a method to monitor cell death and to
[...] Read more.
Establishing methods to investigate treatments that induce cell death in parasitic nematodes will promote experimental approaches to elucidate mechanisms and to identify prospective anthelmintics capable of inducing this outcome. Here, we extended recent progress on a method to monitor cell death and to identify small molecule inhibitors in
Ascaris suum to
Haemonchus contortus, a phylogenetically distant parasitic nematode of significance for both human and agricultural animal health. We utilized a diverse group of small molecule inhibitors referred to as nematode intestinal toxins/toxicants (NITs) coupled with motility, cytological and cell death assays to resolve gross effects on motility and individual cells and organ systems of two
H. contortus larval stages in culture. Early transcriptional response evaluation identified NIT-responsive genes and pathways. The scope of death among cells in larvae varied among NITs but shared patterns with
A. suum, despite the approach having some limitations due to characteristics of
H. contortus larvae. Gene response patterns varied among NITs tested and provided information on the cell targets and pathways affected. Experimental NIT assays provide tools capable of inducing cell death in larval stages of parasitic nematodes, and can resolve many individual cells and organ systems in which cell death can be induced.
Full article
►▼
Show Figures
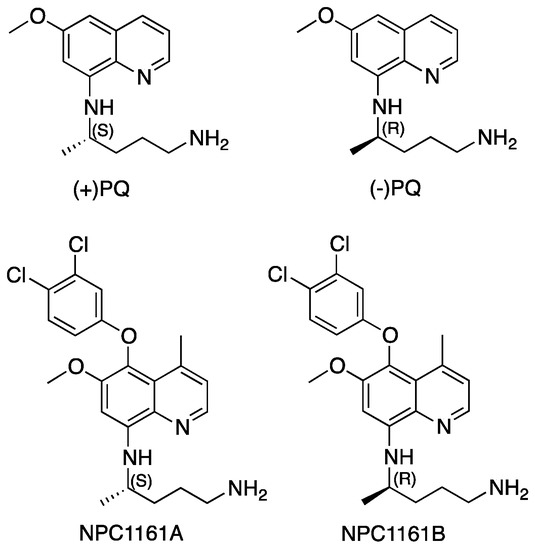
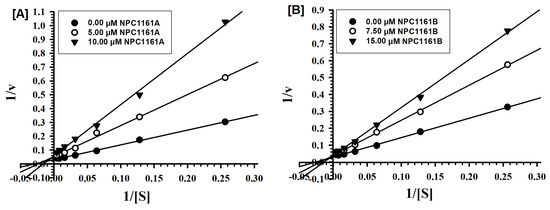
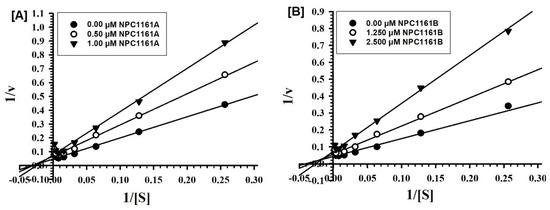
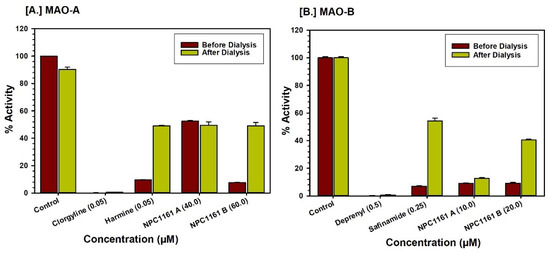
Open AccessArticle
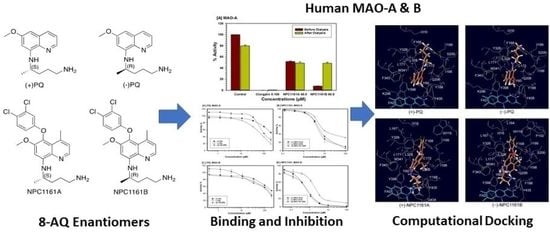
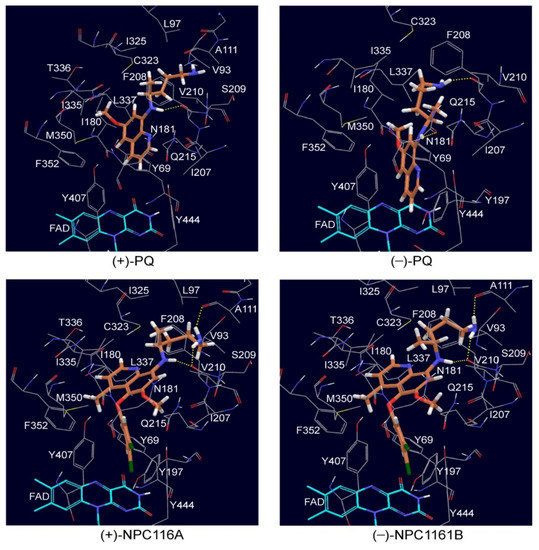
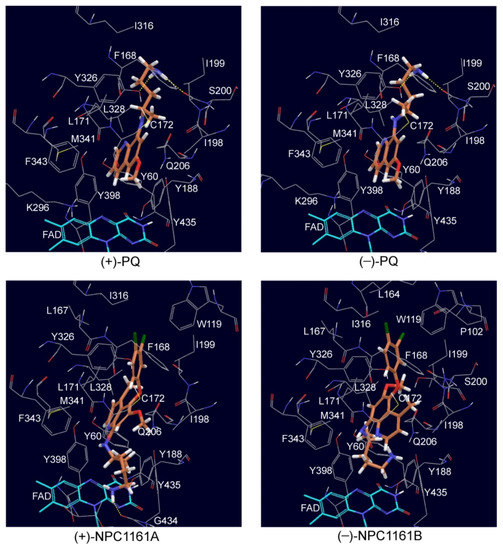
Enantioselective Interactions of Anti-Infective 8-Aminoquinoline Therapeutics with Human Monoamine Oxidases A and B
by
Narayan D. Chaurasiya, Haining Liu, Robert J. Doerksen, N. P. Dhammika Nanayakkara, Larry A. Walker and Babu L. Tekwani
Cited by 4 | Viewed by 2054
Abstract
8-Aminoquinolines (8-AQs) are an important class of anti-infective therapeutics. The monoamine oxidases (MAOs) play a key role in metabolism of 8-AQs. A major role for MAO-A in metabolism of primaquine (PQ), the prototypical 8-AQ antimalarial, has been demonstrated. These investigations were further extended
[...] Read more.
8-Aminoquinolines (8-AQs) are an important class of anti-infective therapeutics. The monoamine oxidases (MAOs) play a key role in metabolism of 8-AQs. A major role for MAO-A in metabolism of primaquine (PQ), the prototypical 8-AQ antimalarial, has been demonstrated. These investigations were further extended to characterize the enantioselective interactions of PQ and NPC1161 (8-[(4-amino-1-methylbutyl) amino]-5-[3, 4-dichlorophenoxy]-6-methoxy-4-methylquinoline) with human MAO-A and -B. NPC1161B, the (
R)-(−) enantiomer with outstanding potential for malaria radical cure, treatment of visceral leishmaniasis and pneumocystis pneumonia infections is poised for clinical development. PQ showed moderate inhibition of human MAO-A and -B. Racemic PQ and (
R)-(−)-PQ both showed marginally greater (1.2- and 1.6-fold, respectively) inhibition of MAO-A as compared to MAO-B. However, (
S)-(+)-PQ showed a reverse selectivity with greater inhibition of MAO-B than MAO-A. Racemic NPC1161 was a strong inhibitor of MAOs with 3.7-fold selectivity against MAO-B compared to MAO-A. The (
S)-(+) enantiomer (NPC1161A) was a better inhibitor of MAO-A and -B compared to the (
R)-(−) enantiomer (NPC1161B), with more than 10-fold selectivity for inhibition of MAO-B over MAO-A. The enantioselective interaction of NPC1161 and strong binding of NPC1161A with MAO-B was further confirmed by enzyme-inhibitor binding and computational docking analyses. Differential interactions of PQ and NPC1161 enantiomers with human MAOs may contribute to the enantioselective pharmacodynamics and toxicity of anti-infective 8-AQs therapeutics.
Full article
►▼
Show Figures
Open AccessArticle
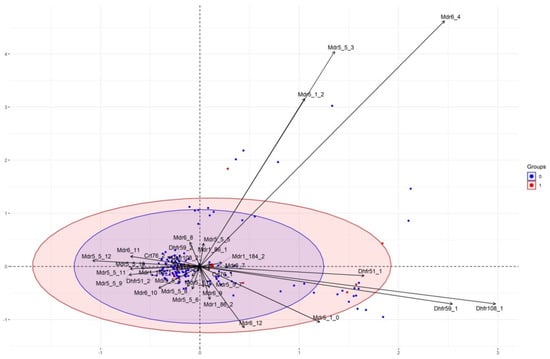
Absence of Association between Methylene Blue Reduced Susceptibility and Polymorphisms in 12 Genes Involved in Antimalarial Drug Resistance in African Plasmodium falciparum
by
Mathieu Gendrot, Océane Delandre, Marie Gladys Robert, Francis Tsombeng Foguim, Nicolas Benoit, Rémy Amalvict, Isabelle Fonta, Joel Mosnier, Marylin Madamet, Bruno Pradines and on behalf of the French National Reference Centre for Imported Malaria Study Group
Cited by 2 | Viewed by 2170
Abstract
Half the human population is exposed to malaria.
Plasmodium falciparum antimalarial drug resistance monitoring and development of new drugs are major issues related to the control of malaria. Methylene blue (MB), the oldest synthetic antimalarial, is again a promising drug after the break
[...] Read more.
Half the human population is exposed to malaria.
Plasmodium falciparum antimalarial drug resistance monitoring and development of new drugs are major issues related to the control of malaria. Methylene blue (MB), the oldest synthetic antimalarial, is again a promising drug after the break of its use as an antimalarial drug for more than 80 years and a potential partner for triple combination. Very few data are available on the involvement of polymorphisms on genes known to be associated with standard antimalarial drugs and parasite in vitro susceptibility to MB (cross-resistance). In this context, MB susceptibility was evaluated against 482 isolates of imported malaria from Africa by HRP2-based ELISA chemosusceptibility assay. A total of 12 genes involved in antimalarial drug resistance (
Pfcrt, Pfdhfr, Pfmdr1, Pfmdr5, Pfmdr6, PfK13, Pfubq, Pfcarl, Pfugt, Pfact, Pfcoronin, and copy number of
Pfpm2) were sequenced by Sanger method and quantitative PCR. On the
Pfmdr1 gene, the mutation 86Y combined with 184F led to more susceptible isolates to MB (8.0 nM vs. 11.6 nM,
p = 0.03). Concerning
Pfmdr6, the isolates bearing 12 Asn repetitions were more susceptible to MB (4.6 nM vs. 11.6 nM,
p = 0.005). None of the polymorphisms previously described as involved in antimalarial drug resistance was shown to be associated with reduced susceptibility to MB. Some genes (particularly
PfK13,
Pfugt,
Pfact,
Pfpm2) did not present enough genetic variability to draw conclusions about their involvement in reduced susceptibility to MB. None of the polymorphisms analyzed by multiple correspondence analysis (MCA) had an impact on the MB susceptibility of the samples successfully included in the analysis. It seems that there is no in vitro cross-resistance between MB and commonly used antimalarial drugs.
Full article
►▼
Show Figures
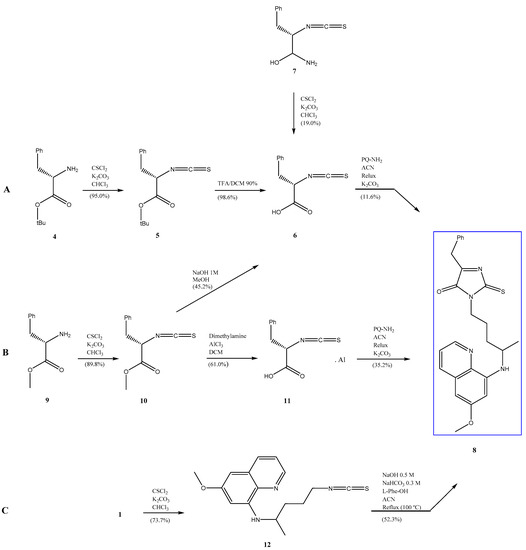
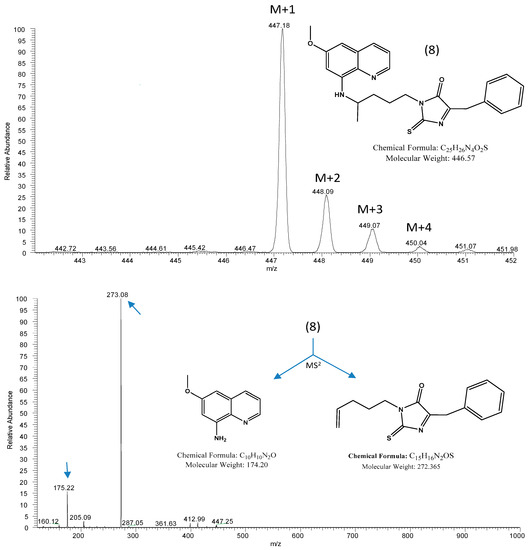
Open AccessArticle
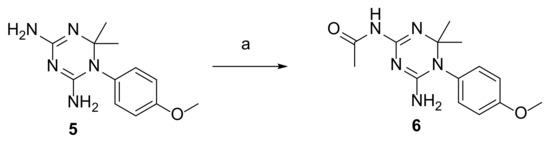
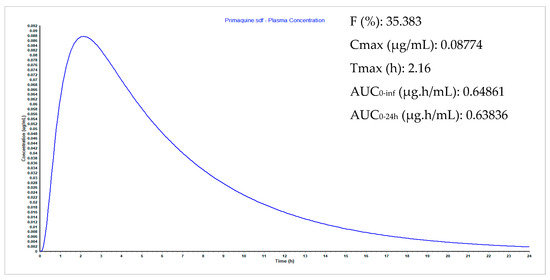
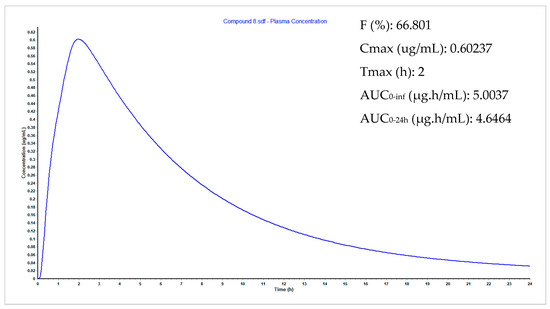
Synthesis, Biological Activity and In Silico Pharmacokinetic Prediction of a New 2-Thioxo-Imidazoldidin-4-One of Primaquine
by
Mariana Pereira, Guy Caljon, Maria João Gouveia, Louis Maes and Nuno Vale
Cited by 2 | Viewed by 2103
Abstract
The discovery of novel antiparasitic drugs for neglected tropical diseases (NTDs) constitutes a global urgency and requires a range of innovative strategies to ensure a sustainable pipeline of lead compounds. Thus far, primaquine (PQ) is the only transmission-blocking antimalarial that is clinically available,
[...] Read more.
The discovery of novel antiparasitic drugs for neglected tropical diseases (NTDs) constitutes a global urgency and requires a range of innovative strategies to ensure a sustainable pipeline of lead compounds. Thus far, primaquine (PQ) is the only transmission-blocking antimalarial that is clinically available, displaying marked activity against gametocytes of all causative species of human malaria (
Plasmodium spp.). Chagas disease, caused by
Trypanosoma cruzi, is another PQ-sensitive illness besides malaria. One of the major drawbacks of PQ is its metabolism into carboxyprimaquine (CPQ), which is less active than the parent drug. In this study, we developed different synthetic pathways to confer N-protection to PQ through introduction of thioxo-imidazolidin-4-one. The introduction of this group prevents the formation of CPQ, counteracting one major drawback of the parent drug. After that, we evaluated the potential biological activity of the novel 2-thioxo-imidazolidin-4-one derivative of PQ, which showed relevant in vitro activity against
Trypanosoma cruzi (IC
50 1.4 μM) compared to PQ (IC
50 1.7 μM) and the reference drug benznidazole (IC
50 1.6 μM). Noting its acceptable pharmacokinetic profile, this PQ conjugate may be a potential scaffold for novel drug exploration against Chagas disease.
Full article
►▼
Show Figures
Planned Papers
The below list represents only planned manuscripts. Some of these
manuscripts have not been received by the Editorial Office yet. Papers
submitted to MDPI journals are subject to peer-review.
Type: Article
Title: New (E)-1-[3-(halophenyl)acryloyl]-piperidin-2-ones and –pyrrolidin-2-ones with activity against protozoal parasites
Author(s): Ibrahim S. Al Nasr, Waleed S. Koko, Tariq A. Khan, Rainer Schobert, Bernhard Biersack
The Corresponding author: Bernhard Biersack
Affiliation(s): Qassim University, KSA; University of Bayreuth, Germany
Abstract: New synthetic cinnamoyl derivatives with variable halogen substituents (F, Cl, and Br) were prepared and analyzed. Their activities against apicomplexan Toxoplasma gondii and kinetoplastid Leishmania major protozoal parasites were evaluated. Structure-activity-relationships were investigated, which revealed notable relations of antiparasitic activity with the applied halogen substituents.
Expected Submission Time: 31st December 2023





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}