
Synthesis, Biological Activity and In Silico Pharmacokinetic Prediction of a New 2-Thioxo-Imidazoldidin-4-One of Primaquine




Abstract
:1. Introduction
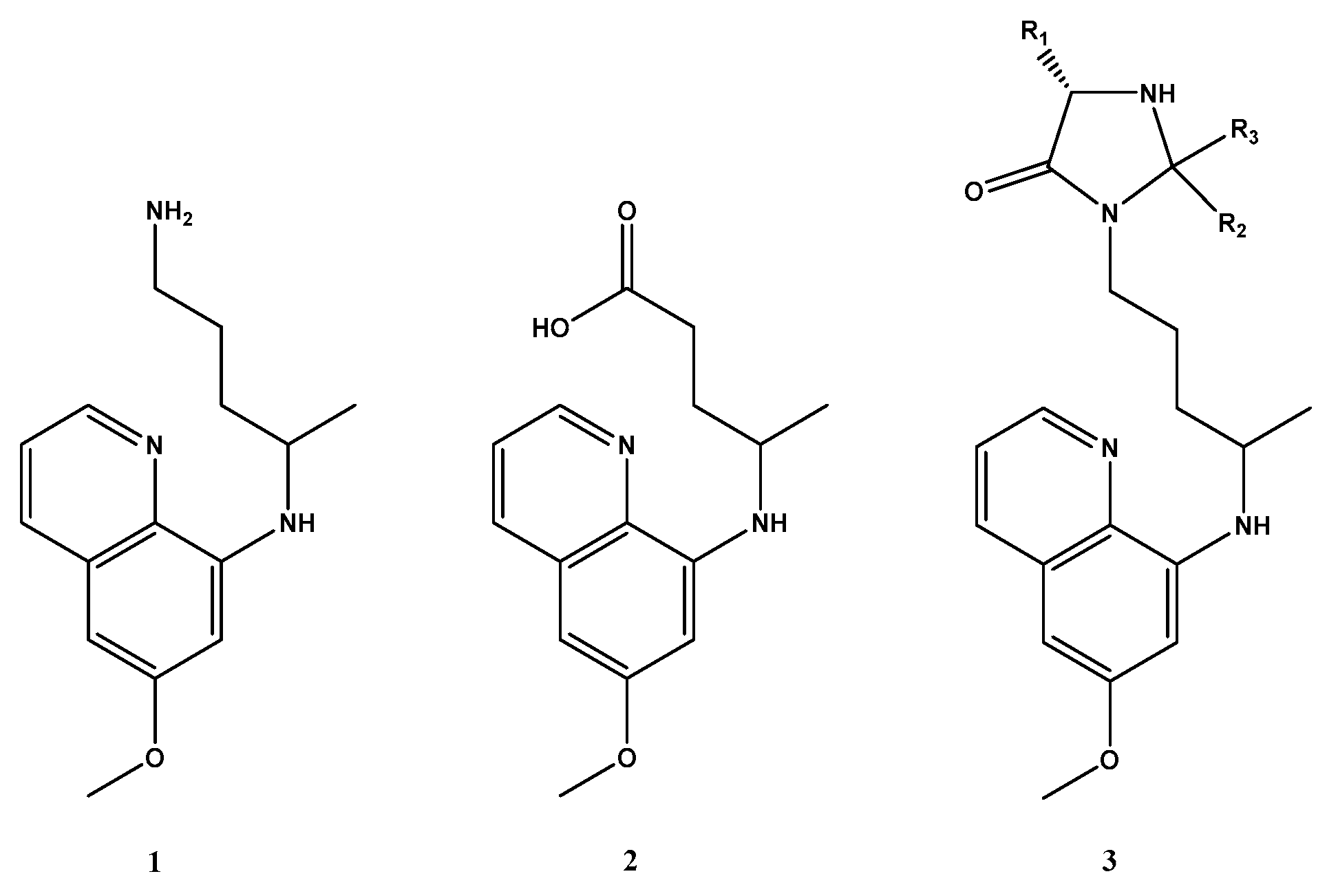
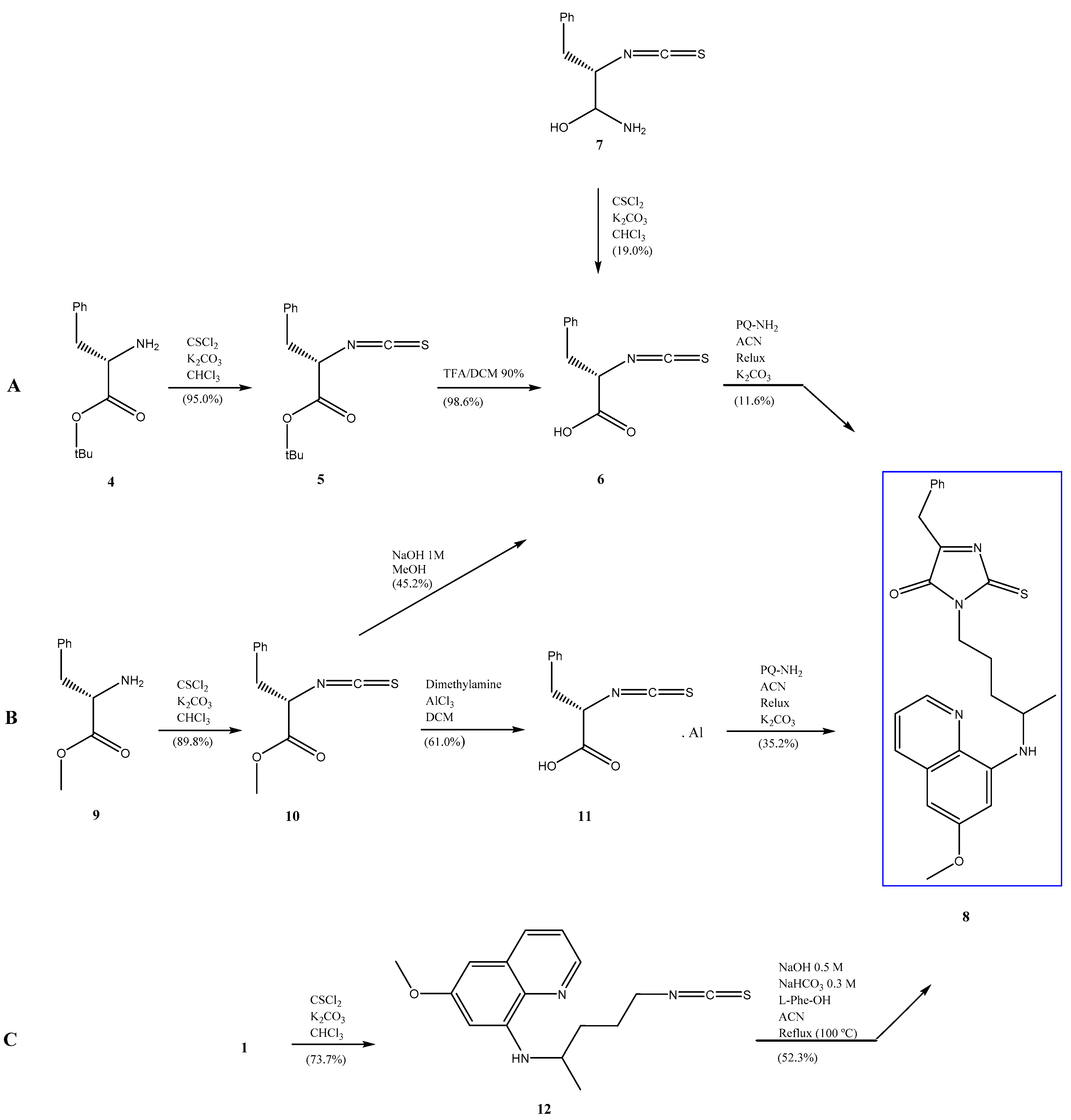
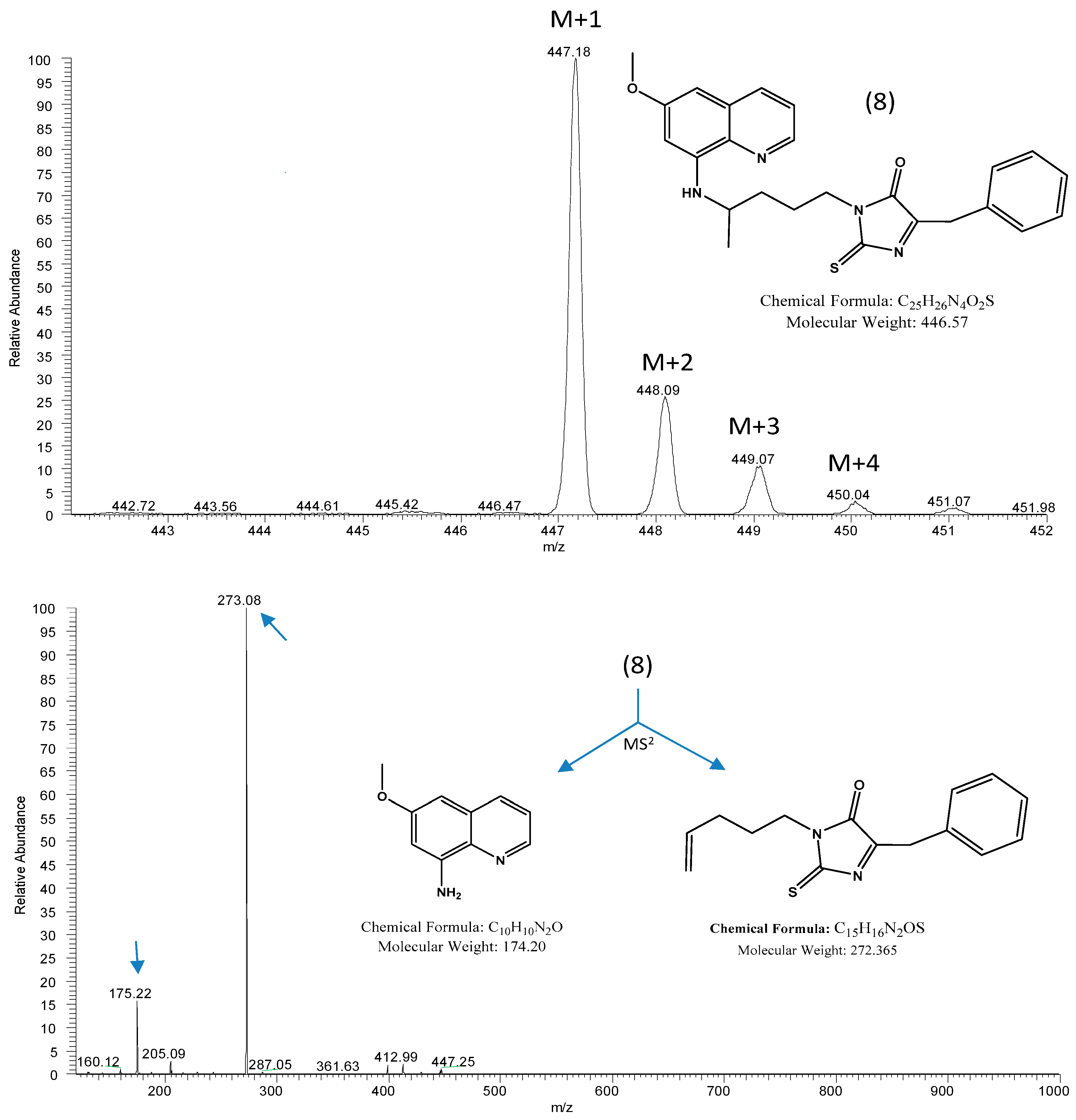
2. Results and Discussion
3. Materials and Methods
3.1. Chemicals
3.2. Hydrolysis in Aqueous Solution
3.3. Biological Assay
3.4. Antitrypanosomal Activity
3.5. Antiplasmodial Activity
3.6. Antileishmanial Activity
3.7. Antibacterial Activity
3.8. Cytotoxicity Assay
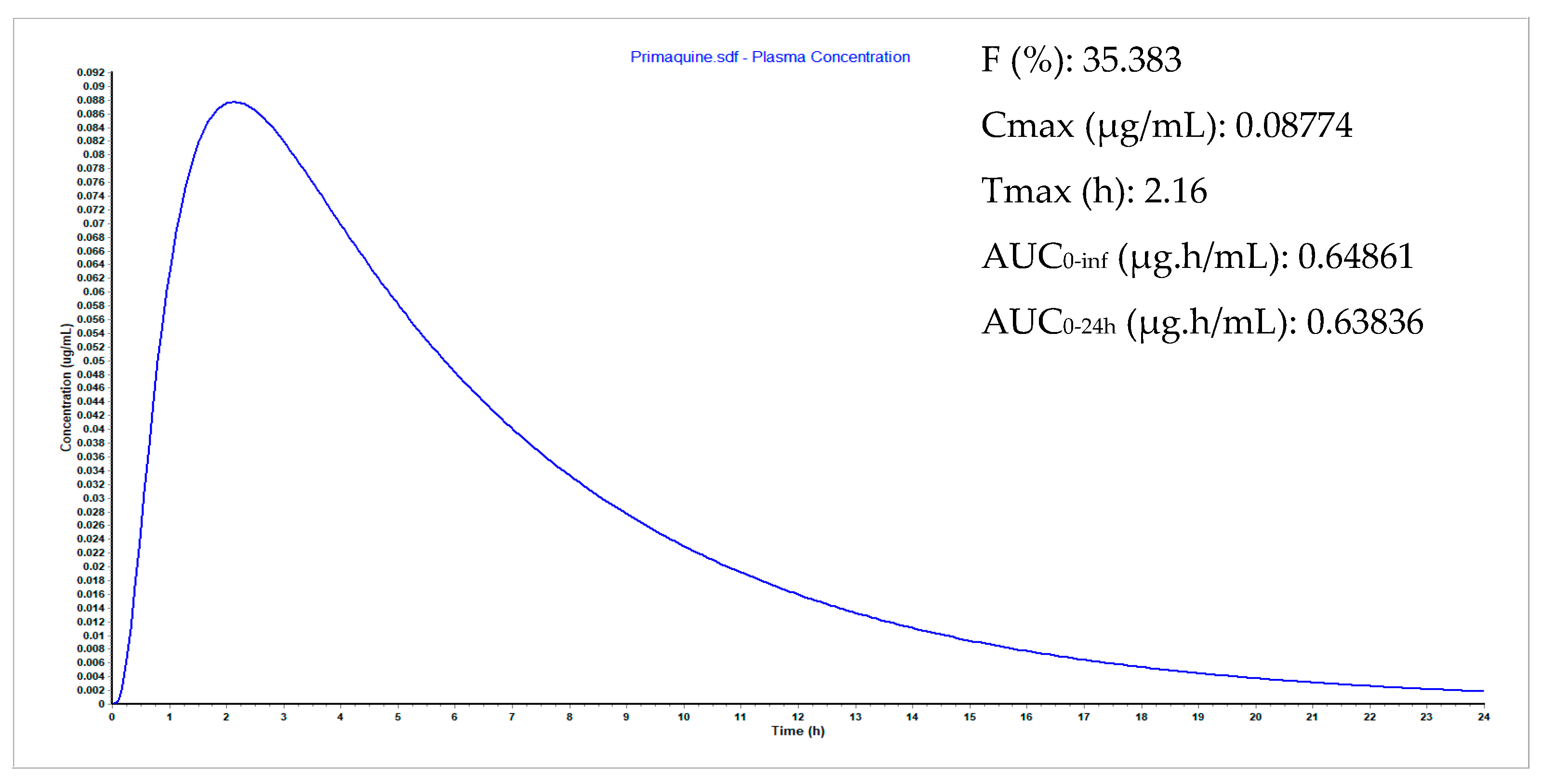
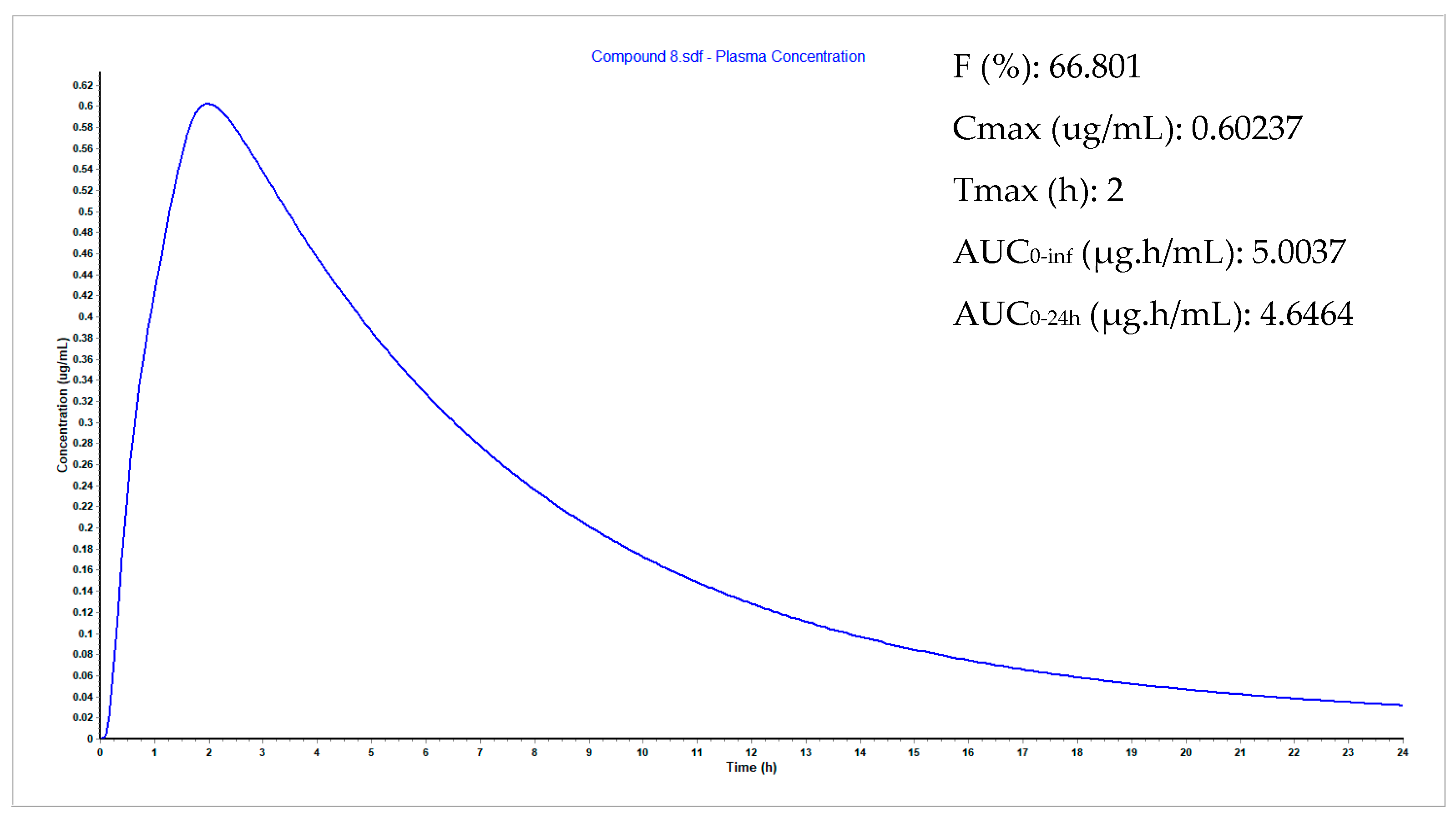
3.9. Plasma Concentration Simulations
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chris, B.; Singh, S.; Sudarshi, D. Neglected tropical diseases, conflict, and the right to health. In The Causes and Impacts of Neglected Tropical and Zoonotic Diseases: Opportunities for Integrated Intervention Strategies; National Academies Press (US): Washington, DC, USA, 2011. [Google Scholar]
- Mitra, A.K.; Mawson, A.R. Neglected tropical diseases: Epidemiology and global burden. Trop. Med. Infect. Dis. 2017, 2, 36. [Google Scholar] [CrossRef] [Green Version]
- Wetsman, N. Turning up the heat on neglected diseases. Nat. Med. 2019, 25, 1632–1633. [Google Scholar] [CrossRef]
- Baird, J.K.; Hoffman, S.L. Primaquine therapy for malaria. Clin. Infect. Dis. 2004, 39, 1336–1345. [Google Scholar] [CrossRef] [Green Version]
- Hill, D.R.; Baird, J.K.; Parise, M.E.; Lewis, L.S.; Ryan, E.T.; Magill, A.J. Primaquine: Report from CDC expert meeting on malaria chemoprophylaxis I. Am. J. Trop. Med. Hyg. 2006, 75, 402–415. [Google Scholar] [CrossRef] [Green Version]
- Vale, N.; Moreira, R.; Gomes, P. Primaquine revisited six decades after its discovery. Eur. J. Med. Chem. 2009, 44, 937–953. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. Guidelines for the Treatment of Malaria; World Health Organization: Geneva, Switzerland, 2015. [Google Scholar]
- Greaves, J.; Evans, D.A.; Gilles, H.M.; Fletcher, K.A.; Bunnag, D.; Harinasuta, T. Plasma kinetics and urinary excretion of primaquine in man. Br. J. Clin. Pharmacol. 1980, 10, 399–404. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.R.; Kuh, H.J.; Kim, M.Y.; Kim, Y.S.; Chung, W.C.; Kim, S.I.; Kang, M.W. Pharmacokinetics of primaquine and carboxyprimaquine in Korean patients with vivax malaria. Arch. Pharm. Res. 2004, 27, 576–580. [Google Scholar] [CrossRef]
- Clark, A.M.; Baker, J.K.; McChesney, J.D. Excretion, distribution, and metabolism of primaquine in rats. J. Pharm. Sci. 1984, 73, 502–506. [Google Scholar] [CrossRef] [PubMed]
- Ashley, E.A.; Recht, J.; White, N.J. Primaquine: The risks and the benefits. Malar. J. 2014, 13, 418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, E.; Zhong, D.; Raya, B.; Pestana, K.; Koepfli, C.; Lee, M.C.; Yewhalaw, D.; Yan, G. Prevalence and distribution of G6PD deficiency: Implication for the use of primaquine in malaria treatment in Ethiopia. Malar. J. 2019, 18, 340. [Google Scholar] [CrossRef]
- Pybus, B.S.; Marcsisin, S.R.; Jin, X.; Deye, G.; Sousa, J.C.; Li, Q.; Caridha, D.; Zeng, Q.; Reichard, G.A.; Ockenhouse, C.; et al. The metabolism of primaquine to its active metabolite is dependent on CYP 2D6. Malar. J. 2013, 12, 212. [Google Scholar] [CrossRef] [Green Version]
- Spring, M.D.; Sousa, J.C.; Li, Q.; Darko, C.A.; Morrison, M.N.; Marcsisin, S.R.; Mills, K.T.; Potter, B.M.; Paolino, K.M.; Twomey, P.S.; et al. Determination of Cytochrome P450 Isoenzyme 2D6 (CYP2D6) Genotypes and Pharmacogenomic Impact on Primaquine Metabolism in an Active-Duty US Military Population. J. Infect. Dis. 2019, 220, 1761–1770. [Google Scholar] [CrossRef] [PubMed]
- Pybus, B.S.; Sousa, J.C.; Jin, X.; Ferguson, J.A.; Christian, R.E.; Barnhart, R.; Vuong, C.; Sciotti, R.J.; Reichard, G.A.; Kozar, M.P.; et al. CYP450 phenotyping and accurate mass identification of metabolites of the 8-aminoquinoline, anti-malarial drug primaquine. Malar. J. 2012, 11, 259. [Google Scholar] [CrossRef] [Green Version]
- Mihaly, G.W.; Ward, S.A.; Edwards, G.; Orme, M.L.; Breckenridge, A.M. Pharmacokinetics of primaquine in man: Identification of the carboxylic acid derivative as a major plasma metabolite. Br. J. Clin. Pharmacol. 1984, 17, 441–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Constantino, L.; Paixão, P.; Moreira, R.; Portela, M.J.; Do Rosario, V.E.; Iley, J. Metabolism of primaquine by liver homogenate fractions. Evidence for monoamine oxidase and cytochrome P450 involvement in the oxidative deamination of primaquine to carboxyprimaquine. Exp. Toxicol. Pathol. 1999, 51, 299–303. [Google Scholar] [CrossRef]
- Camarda, G.; Jirawatcharadech, P.; Priestley, R.S.; Saif, A.; March, S.; Wong, M.H.L.; Leung, S.; Miller, A.B.; Baker, D.A.; Alano, P.; et al. Antimalarial activity of primaquine operates via a two-step biochemical relay. Nat. Commun. 2019, 10, 3226. [Google Scholar] [CrossRef] [PubMed]
- Potter, B.M.J.; Xie, L.H.; Vuong, C.; Zhang, J.; Zhang, P.; Duan, D.; Luong, T.-L.T.; Bandara Herath, H.M.T.; Dhammika Nanayakkara, N.P.; Tekwani, B.L.; et al. Differential CYP 2D6 metabolism alters primaquine pharmacokinetics. Antimicrob. Agents Chemother. 2015, 59, 2380–2387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avula, B.; Tekwani, B.L.; Chaurasiya, N.D.; Fasinu, P.; Dhammika Nanayakkara, N.P.; Bhandara Herath, H.M.T.; Wang, Y.H.; Bae, J.Y.; Khan, S.I.; Elsohly, M.A.; et al. Metabolism of primaquine in normal human volunteers: Investigation of phase I and phase II metabolites from plasma and urine using ultra-high performance liquid chromatography-quadrupole time-of-flight mass spectrometry. Malar. J. 2018, 17, 294. [Google Scholar] [CrossRef] [Green Version]
- Araújo, M.J.; Bom, J.; Capela, R.; Casimiro, C.; Chambel, P.; Gomes, P.; Iley, J.; Lopes, F.; Morais, J.; Moreira, R.; et al. Imidazolidin-4-one derivatives of primaquine as novel transmission-blocking antimalarials. J. Med. Chem. 2005, 48, 888–892. [Google Scholar] [CrossRef] [Green Version]
- Vale, N.; Collins, M.S.; Gut, J.; Ferraz, R.; Rosenthal, P.J.; Cushion, M.T.; Moreira, R.; Gomes, P. Anti-Pneumocystis carinii and antiplasmodial activities of primaquine-derived imidazolidin-4-ones. Bioorg. Med. Chem. Lett. 2008, 18, 485–488. [Google Scholar] [CrossRef] [Green Version]
- Kinnamon, K.E.; Steck, E.A.; Hanson, W.L.; Chapman, W.L., Jr. In search of anti-Trypanosoma cruzi drugs: New leads from a mouse model. J. Med. Chem. 1977, 20, 741–744. [Google Scholar] [CrossRef] [PubMed]
- La-Scalea, M.A.; Chin, C.M.; Cruz, M.L.; Serrano, S.H.; Ferreira, E.I. Dissociation and electrooxidation of primaquine diphosphate as an approach to the study of anti-chagas prodrugs mechanism of action. Bioelectrochemistry 2001, 53, 55–59. [Google Scholar] [CrossRef]
- Caplan, A.; Zink, A. Adverse event management in mass drug administration for neglected tropical diseases. Clin. Ther. 2014, 36, 421–424. [Google Scholar] [CrossRef] [PubMed]
- Mihaly, G.W.; Ward, S.A.; Edwards, G.; Nicholl, D.D.; Orme, M.L.; Breckenridge, A.M. Pharmacokinetics of primaquine in man. I. Studies of the absolute bioavailability and effects of dose size. Br. J. Clin. Pharmacol. 1985, 19, 745–750. [Google Scholar] [CrossRef] [Green Version]
- Toledo, M.J.; Bahia, M.T.; Veloso, V.M.; Carneiro, C.M.; Machado-Coelho, G.L.; Alves, C.F.; Martins, H.R.; Cruz, R.E.; Tafuri, W.L.; Lana, M. Effects of specific treatment on parasitological and histopathological parameters in mice infected with different Trypanosoma cruzi clonal genotypes. J. Antimicrob. Chemother. 2004, 53, 1045–1053. [Google Scholar] [CrossRef] [Green Version]
- Gobeau, N.; Stringer, R.; De Buck, S.; Tuntland, T.; Faller, B. Evaluation of the GastroPlus™ Advanced Compartmental and Transit (ACAT) Model in Early Discovery. Pharm. Res. 2016, 33, 2126–2139. [Google Scholar] [CrossRef] [PubMed]
- Cos, P.; Vlietinck, A.J.; Berghe, D.V.; Maes, L. Anti-infective potential of natural products: How to develop a stronger in vitro ’proof-of-concept’. J. Ethnopharmacol. 2006, 106, 290–302. [Google Scholar] [CrossRef]
- Räz, B.; Iten, M.; Grether-Bühler, Y.; Kaminsky, R.; Brun, R. The Alamar Blue assay to determine drug sensitivity of African trypanosomes (T.b. rhodesiense and T.b. gambiense) in vitro. Acta Trop. 1997, 68, 139–147. [Google Scholar] [CrossRef]
- Gruber, A.; Czejka, M.; Buchner, P.; Kitzmueller, M.; Kirchbaumer Baroian, N.; Dittrich, C.; Sahmanovic Hrgovcic, A. Monitoring of erlotinib in pancreatic cancer patients during long-time administration and comparison to a physiologically based pharmacokinetic model. Cancer Chemother. Pharmacol. 2018, 81, 763–771. [Google Scholar] [CrossRef] [Green Version]
- Duque, M.D.; Silva, D.A.; Issa, M.G.; Porta, V.; Löbenberg, R.; Ferraz, H.G. In silico prediction of plasma concentrations of flucona-zole capsules with different dissolution profiles and bioequivalence study using population simulation. Pharmaceuticals 2019, 11, 215. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, A.; Lapa, R.; Vale, N. Combination of Gemcitabine with Cell-Penetrating Peptides: A Pharmacokinetic Approach Using in Silico Tools. Biomolecules 2019, 9, 693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, A.; Martins, H.; Oliveira, J.C.; Lapa, R.; Vale, N. In silico pharmacokinetic study of vancomycin using PBPK modeling and therapeutic drug monitoring. Curr. Drug Metabol. 2021. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Human Plasma | PBS | ||
|---|---|---|---|
| Compound | t1/2 (h) a | t1/2 (h) b | t1/2 (h) c |
| PQ | 3.46 | 3.70 | ND |
| 8 | 3.86 | ND | >72 |
| Compound | T. cruzi | T. brucei | P. falciparum | L. infantum | S. aureus | MRC-5 |
|---|---|---|---|---|---|---|
| 1 | 1.7 ± 0.1 | 8.2 ± 0.4 | 2.2 ± 0.2 | 4.0 ± 0.5 | >64.0 | 34.8 ± 1.8 |
| 8 | 1.4 ± 0.05 | 8.4 ± 0.6 | 10.2 ± 0.5 | 20.3 ± 1.1 | >64.0 | 30.1 ± 1.6 |
| Benznidazole | 1.6 ± 0.03 | - | - | - | - | - |
| Suramin | - | 0.04 ± 0.02 | - | - | - | - |
| Chloroquine | - | - | 0.2 ± 0.02 | - | - | - |
| Amphotericin B | - | - | - | 1.2 ± 0.1 | - | - |
| Erythromycin | - | - | - | - | 10.4 ± 0.7 | - |
| Tamoxifen | - | - | - | - | - | 10.8 ± 0.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pereira, M.; Caljon, G.; Gouveia, M.J.; Maes, L.; Vale, N. Synthesis, Biological Activity and In Silico Pharmacokinetic Prediction of a New 2-Thioxo-Imidazoldidin-4-One of Primaquine. Pharmaceuticals 2021, 14, 196. https://doi.org/10.3390/ph14030196
Pereira M, Caljon G, Gouveia MJ, Maes L, Vale N. Synthesis, Biological Activity and In Silico Pharmacokinetic Prediction of a New 2-Thioxo-Imidazoldidin-4-One of Primaquine. Pharmaceuticals. 2021; 14(3):196. https://doi.org/10.3390/ph14030196
Chicago/Turabian StylePereira, Mariana, Guy Caljon, Maria João Gouveia, Louis Maes, and Nuno Vale. 2021. "Synthesis, Biological Activity and In Silico Pharmacokinetic Prediction of a New 2-Thioxo-Imidazoldidin-4-One of Primaquine" Pharmaceuticals 14, no. 3: 196. https://doi.org/10.3390/ph14030196