
Design, Rational Repurposing, Synthesis, In Vitro Evaluation, Homology Modeling and In Silico Study of Sulfuretin Analogs as Potential Antileishmanial Hit Compounds

 ,
, 
Abstract
:1. Introduction
2. Results
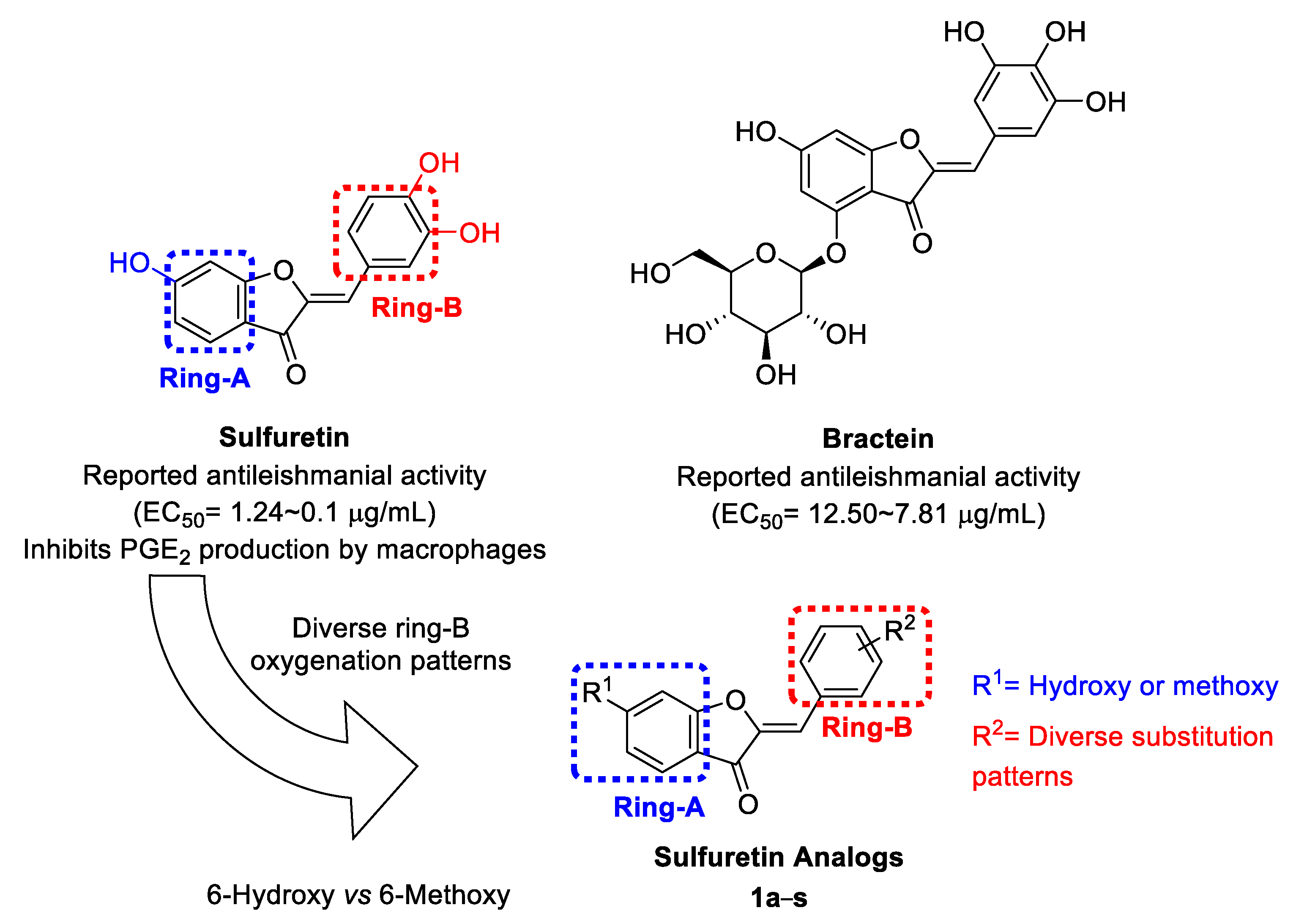
2.1. Design and Repurposing Rational
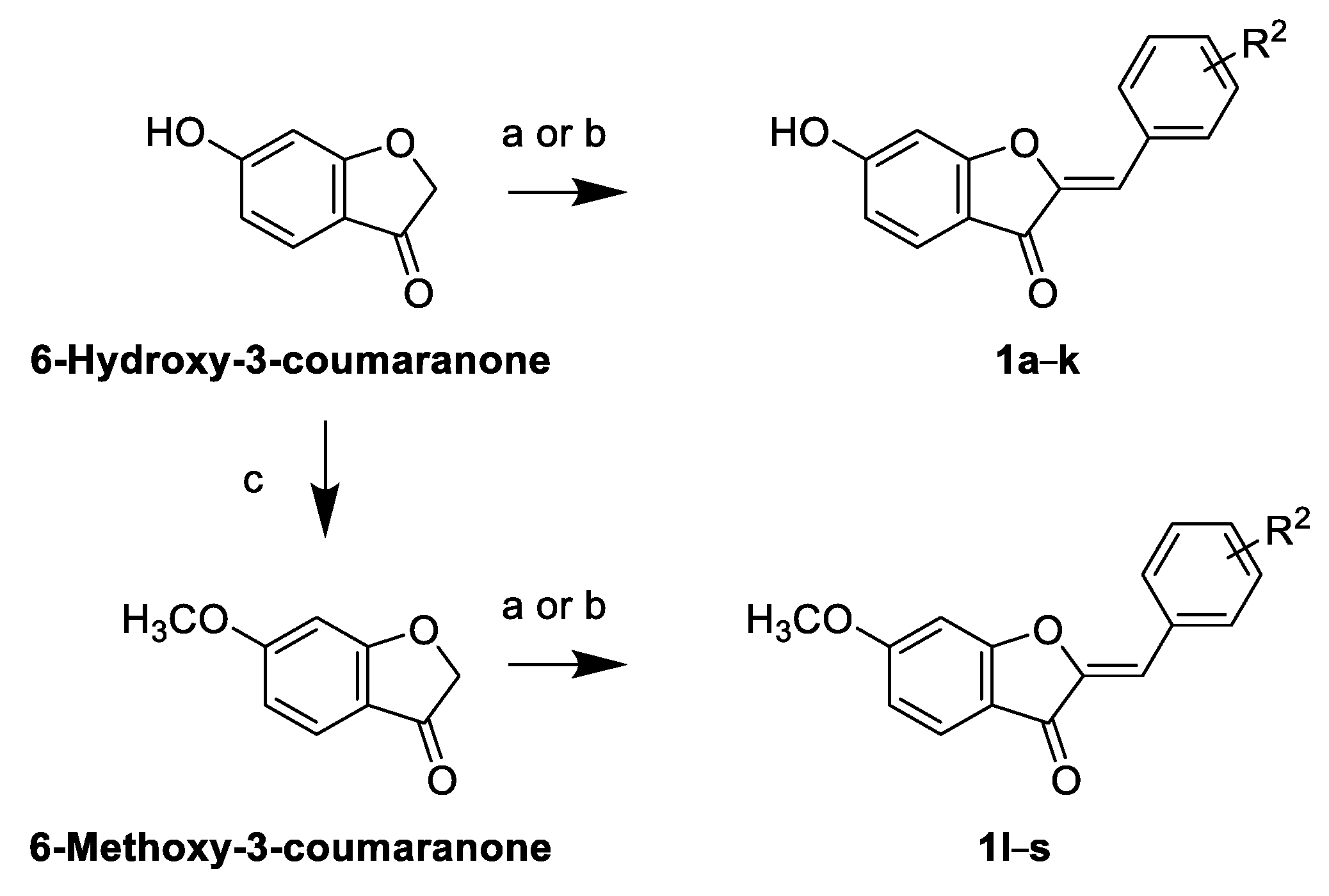
2.2. Synthesis of Sulfuretin Analogs
2.3. In Vitro Evaluation at 50 and 25 µM Concentrations
2.4. Cell Viability and Cytotoxicity Evaluation
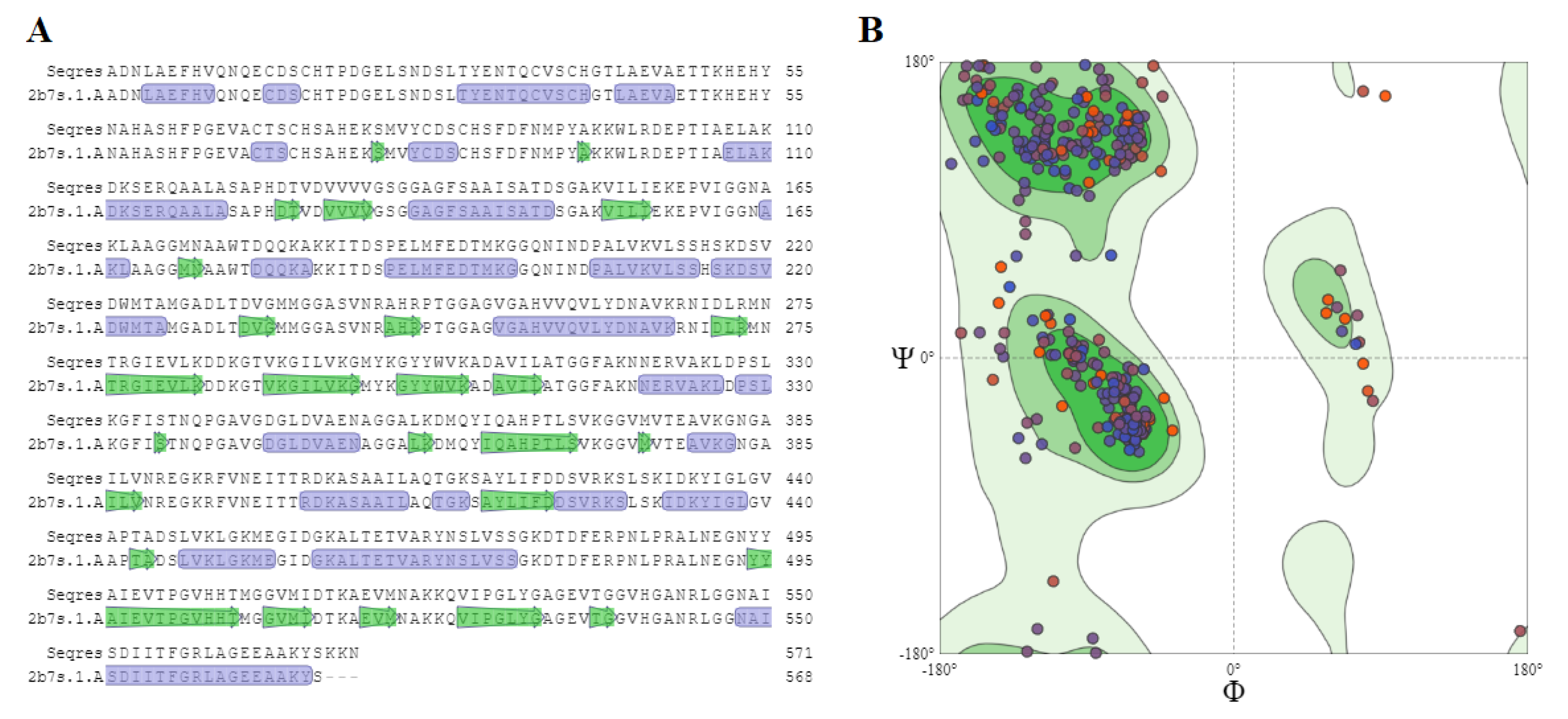
2.5. Homology Modeling
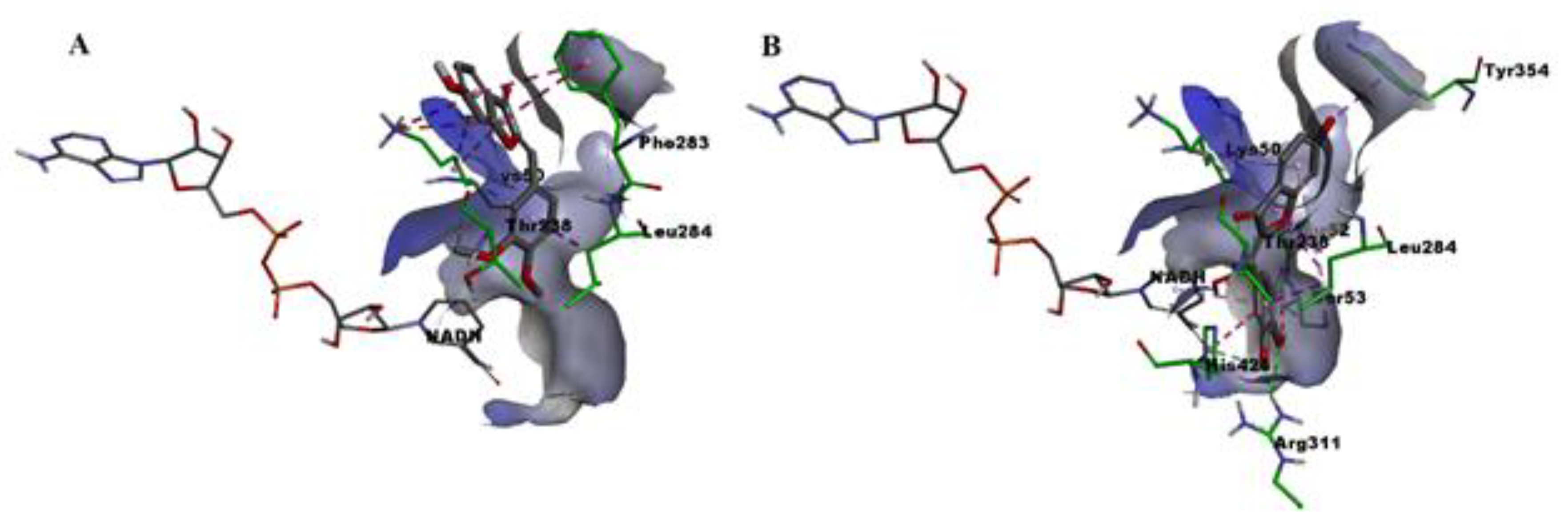
2.6. Molecular Docking
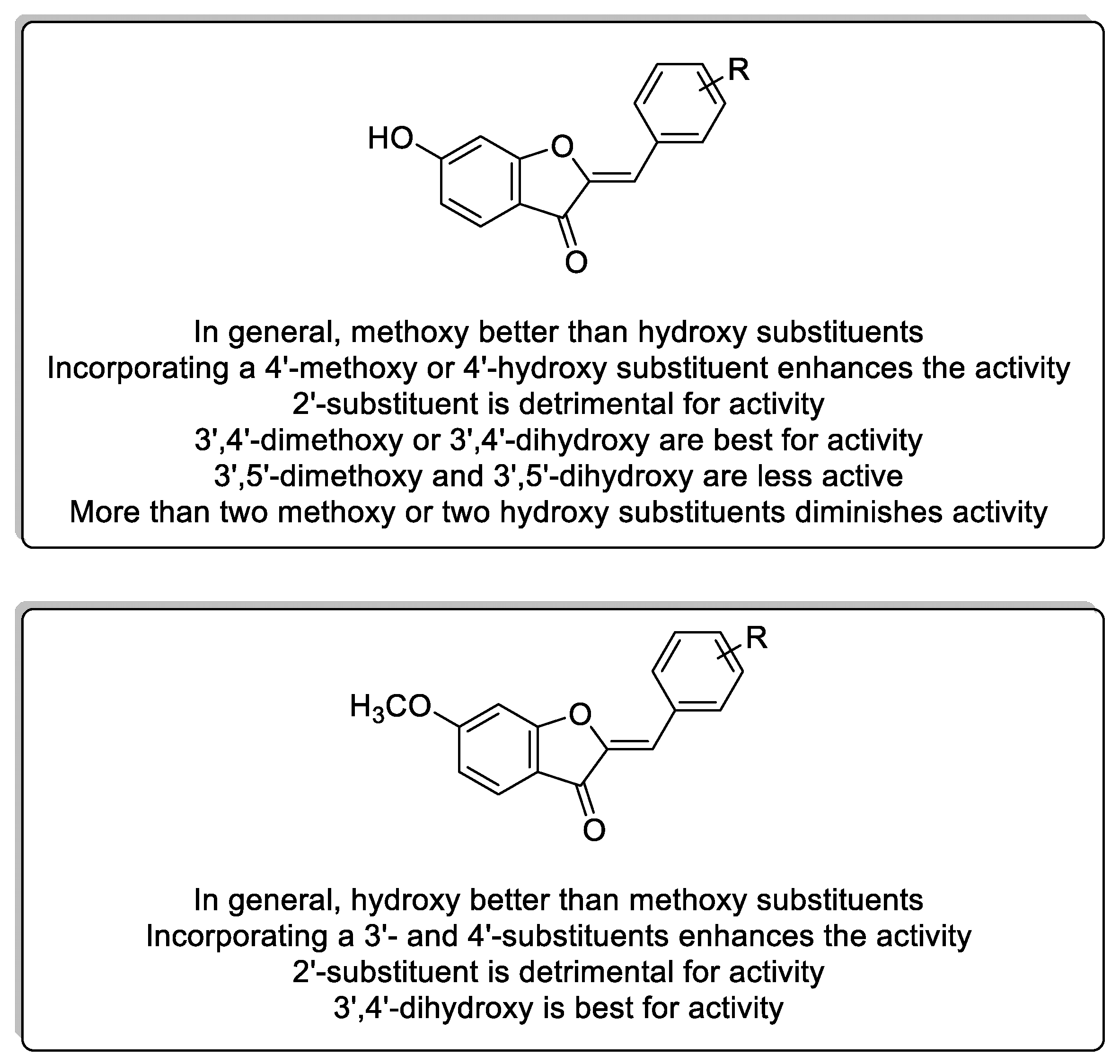
3. Discussion
4. Materials and Methods
4.1. Chemistry
General Method for Acid-Catalyzed Cross-Aldol Condensation to Prepare Sulfuretin Analogs
4.2. In Vitro Evaluation against L. donovani Promastigotes
4.3. In Vitro Host Cells Viability and Cytotoxicity
4.4. Statistical Analysis
4.5. In Silico Simulation
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Steverding, D. The history of leishmaniasis. Parasit. Vectors 2017, 10, 82. [Google Scholar] [CrossRef] [PubMed]
- Hassan, A.H.E.; Phan, T.-N.; Yoon, S.; Lee, C.J.; Jeon, H.R.; Kim, S.-H.; No, J.H.; Lee, Y.S. Pyrrolidine-based 3-deoxysphingosylphosphorylcholine analogs as possible candidates against neglected tropical diseases (NTDs): Identification of hit compounds towards development of potential treatment of Leishmania donovani. J. Enzyme Inhib. Med. Chem. 2021, 36, 1922–1930. [Google Scholar] [CrossRef] [PubMed]
- Matte, C.; Maion, G.; Mourad, W.; Olivier, M. Leishmania donovani-induced macrophages cyclooxygenase-2 and prostaglandin E2 synthesis. Parasite Immunol. 2001, 23, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Saha, A.; Biswas, A.; Srivastav, S.; Mukherjee, M.; Das, P.K.; Ukil, A. Prostaglandin E2 Negatively Regulates the Production of Inflammatory Cytokines/Chemokines and IL-17 in Visceral Leishmaniasis. J. Immunol. 2014, 193, 1400399. [Google Scholar] [CrossRef]
- Bhattacharjee, A.; Majumder, S.; Das, S.; Ghosh, S.; Biswas, S.; Majumdar, S.; Appleton, J.A. Leishmania donovani-Induced Prostaglandin E2 Generation Is Critically Dependent on Host Toll-Like Receptor 2–Cytosolic Phospholipase A2 Signaling. Infect. Immun. 2016, 84, 2963–2973. [Google Scholar] [CrossRef]
- Sakai, C.; Tomitsuka, E.; Esumi, H.; Harada, S.; Kita, K. Mitochondrial fumarate reductase as a target of chemotherapy: From parasites to cancer cells. Biochim. Biophys. Acta 2012, 1820, 643–651. [Google Scholar] [CrossRef]
- Chen, M.; Bennedsen, M.; Zhai, L.; Kharazmi, A. Purification and enzymatic activity of an NADH-fumarate reductase and other mitochondrial activities of Leishmania parasites. Apmis 2001, 109, 801–808. [Google Scholar] [CrossRef]
- Santhamma, K.R.; Bhaduri, A. Characterization of the respiratory chain of Leishmania donovani promastigotes. Mol. Biochem. Parasitol. 1995, 75, 43–53. [Google Scholar] [CrossRef]
- Chen, M.; Zhai, L.; Christensen Søren, B.; Theander Thor, G.; Kharazmi, A. Inhibition of Fumarate Reductase in Leishmania major and L. donovani by Chalcones. Antimicrob. Agents Chemother. 2001, 45, 2023–2029. [Google Scholar] [CrossRef]
- Alam, M.M.; Hassan, A.H.E.; Lee, K.W.; Cho, M.C.; Yang, J.S.; Song, J.; Min, K.H.; Hong, J.; Kim, D.-H.; Lee, Y.S. Design, synthesis and cytotoxicity of chimeric erlotinib-alkylphospholipid hybrids. Bioorg. Chem. 2019, 84, 51–62. [Google Scholar] [CrossRef]
- Farag, A.K.; Hassan, A.H.E.; Jeong, H.; Kwon, Y.; Choi, J.G.; Oh, M.S.; Park, K.D.; Kim, Y.K.; Roh, E.J. First-in-class DAPK1/CSF1R dual inhibitors: Discovery of 3,5-dimethoxy-N-(4-(4-methoxyphenoxy)-2-((6-morpholinopyridin-3-yl)amino)pyrimidin-5-yl)benzamide as a potential anti-tauopathies agent. Eur. J. Med. Chem. 2019, 162, 161–175. [Google Scholar] [CrossRef]
- Bansal, Y.; Silakari, O. Multifunctional compounds: Smart molecules for multifactorial diseases. Eur. J. Med. Chem. 2014, 76, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Shen, B. A New Golden Age of Natural Products Drug Discovery. Cell 2015, 163, 1297–1300. [Google Scholar] [CrossRef] [PubMed]
- Harvey, A.L.; Edrada-Ebel, R.; Quinn, R.J. The re-emergence of natural products for drug discovery in the genomics era. Nat. Rev. Drug Discov. 2015, 14, 111–129. [Google Scholar] [CrossRef]
- Kayser, O.; Kiderlen, A.F.; Folkens, U.; Kolodziej, H. In vitro leishmanicidal activity of aurones. Planta Med. 1999, 65, 316–319. [Google Scholar] [CrossRef]
- Kayser, O.; Chen, M.; Kharazmi, A.; Kiderlen, A.F. Aurones interfere with Leishmania major mitochondrial fumarate reductase. Z. Naturforsch. C 2002, 57, 717–720. [Google Scholar] [CrossRef]
- Roussaki, M.; Costa Lima, S.; Kypreou, A.M.; Kefalas, P.; Cordeiro da Silva, A.; Detsi, A. Aurones: A promising heterocyclic scaffold for the development of potent antileishmanial agents. Int. J. Med. Chem. 2012, 2012, 196921. [Google Scholar] [CrossRef]
- Shin, S.Y.; Shin, M.C.; Shin, J.-S.; Lee, K.-T.; Lee, Y.S. Synthesis of aurones and their inhibitory effects on nitric oxide and PGE2 productions in LPS-induced RAW 264.7 cells. Bioorg. Med. Chem. Lett. 2011, 21, 4520–4523. [Google Scholar] [CrossRef]
- Gupta, S.; Nishi. Visceral leishmaniasis: Experimental models for drug discovery. Indian J. Med. Res. 2011, 133, 27–39. [Google Scholar]
- Kulshrestha, A.; Bhandari, V.; Mukhopadhyay, R.; Ramesh, V.; Sundar, S.; Maes, L.; Dujardin, J.C.; Roy, S.; Salotra, P. Validation of a simple resazurin-based promastigote assay for the routine monitoring of miltefosine susceptibility in clinical isolates of Leishmania donovani. Parasitol. Res. 2013, 112, 825–828. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertoni, M.; Kiefer, F.; Biasini, M.; Bordoli, L.; Schwede, T. Modeling protein quaternary structure of homo- and hetero-oligomers beyond binary interactions by homology. Sci. Rep. 2017, 7, 10480. [Google Scholar] [CrossRef] [PubMed]
- Studer, G.; Tauriello, G.; Bienert, S.; Biasini, M.; Johner, N.; Schwede, T. ProMod3—A versatile homology modelling toolbox. PLoS Comput. Biol. 2021, 17, e1008667. [Google Scholar] [CrossRef] [PubMed]
- El Hajj, R.; El Hajj, H.; Khalifeh, I. Fatal Visceral Leishmaniasis Caused by Leishmania infantum, Lebanon. Emerg. Infect. Dis. 2018, 24, 906–907. [Google Scholar] [CrossRef]
- Gaich, T.; Baran, P.S. Aiming for the Ideal Synthesis. J. Org. Chem. 2010, 75, 4657–4673. [Google Scholar] [CrossRef]
- Hassan, A.H.E.; Kim, H.J.; Gee, M.S.; Park, J.-H.; Jeon, H.R.; Lee, C.J.; Choi, Y.; Moon, S.; Lee, D.; Lee, J.K.; et al. Positional scanning of natural product hispidol’s ring-B: Discovery of highly selective human monoamine oxidase-B inhibitor analogues downregulating neuroinflammation for management of neurodegenerative diseases. J. Enzyme Inhib. Med. Chem. 2022, 37, 768–780. [Google Scholar] [CrossRef]
- Łączkowski, K.Z.; Pakulski, M.M.; Krzemiński, M.P.; Jaisankar, P.; Zaidlewicz, M. Asymmetric synthesis of N-substituted N-hydroxyureas. Tetrahedron Asymmetry 2008, 19, 788–795. [Google Scholar] [CrossRef]
- Stuhr-Hansen, N.; Sørensen, J.K.; Moth-Poulsen, K.; Christensen, J.B.; Bjørnholm, T.; Nielsen, M.B. Synthetic protocols and building blocks for molecular electronics. Tetrahedron 2005, 61, 12288–12295. [Google Scholar] [CrossRef]
- Lee, Y.H.; Shin, M.C.; Yun, Y.D.; Shin, S.Y.; Kim, J.M.; Seo, J.M.; Kim, N.-J.; Ryu, J.H.; Lee, Y.S. Synthesis of aminoalkyl-substituted aurone derivatives as acetylcholinesterase inhibitors. Bioorg. Med. Chem. 2015, 23, 231–240. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Comp. | R1 | R2 | PGE2 IC50 (µM) 1 | Comp. | R1 | R2 | PGE2 IC50 (µM) 1 |
|---|---|---|---|---|---|---|---|
| 1a | 6-Hydroxy | 3′,5′-Dihydroxy | ND 2 | 1k | 6-Hydroxy | 2′,3′,4′-Trimethoxy | ND 2 |
| 1b | 6-Hydroxy | 3′,4′,5′-Trihydroxy | 37.62 | 1l | 6-Methoxy | 2′-Hydroxy | 2.22 |
| 1c | 6-Hydroxy | 2′-Methoxy | 18.10 | 1m | 6-Methoxy | 3′-Hydroxy | 13.15 |
| 1d | 6-Hydroxy | 3′-Methoxy | 3.79 | 1n | 6-Methoxy | 4′-Hydroxy | 35.82 |
| 1e | 6-Hydroxy | 4′-Methoxy | 2.00 | 1o | 6-Methoxy | 2′,3′-Dihydroxy | ND 2 |
| 1f | 6-Hydroxy | 4′-Methoxymethoxy | ND 2 | 1p | 6-Methoxy | 2′,4′-Dihydroxy | 8.80 |
| 1g | 6-Hydroxy | 2′,3′-Dimethoxy | ND 2 | 1q | 6-Methoxy | 3′,4′-Dihydroxy | 4.90 |
| 1h | 6-Hydroxy | 2′,5′-Dimethoxy | ND 2 | 1r | 6-Methoxy | 2′-Methoxy | 59.50 |
| 1i | 6-Hydroxy | 3′,4′-Dimethoxy | 2.90 | 1s | 6-Methoxy | 3′-Methoxy | 2.50 |
| 1j | 6-Hydroxy | 3′,5′-Dimethoxy | 1.67 | Sulfuretin | 6-Hydroxy | 3′,4′-Dihydroxy | 5.90 |

| Comp. | R1 | R2 | % Inhibition ± SD at 50 µM 1 | % Inhibition ± SD at 25 µM 1 |
|---|---|---|---|---|
| 1a | 6-Hydroxy | 3′,5′-Dihydroxy | 18.16 ± 2.32 | 12.14 ± 2.22 |
| 1b | 6-Hydroxy | 3′,4′,5′-Trihydroxy | 36.74 ± 6.24 | NI 2 |
| 1c | 6-Hydroxy | 2′-Methoxy | 31.55 ± 3.76 | 1.06 ± 10.98 |
| 1d | 6-Hydroxy | 3′-Methoxy | 29.19 ± 10.02 | 12.13 ± 0.60 |
| 1e | 6-Hydroxy | 4′-Methoxy | 54.80 ± 1.21 | 43.34 ± 1.10 |
| 1f | 6-Hydroxy | 4′-Methoxymethoxy | 29.34 ± 0.93 | 20.61 ± 3.96 |
| 1g | 6-Hydroxy | 2′,3′-Dimethoxy | 25.37 ± 5.24 | 8.74 ± 1.05 |
| 1h | 6-Hydroxy | 2′,5′-Dimethoxy | 19.05 ± 3.74 | 3.28 ± 1.31 |
| 1i | 6-Hydroxy | 3′,4′-Dimethoxy | 109.83 ± 0.37 | 106.54 ± 0.29 |
| 1j | 6-Hydroxy | 3′,5′-Dimethoxy | 70.92 ± 1.19 | 78.10 ± 1.91 |
| 1k | 6-Hydroxy | 2′,3′,4′-Trimethoxy | 34.10 ± 0.37 | 13.58 ± 3.39 |
| 1l | 6-Methoxy | 2′-Hydroxy | 27.02 ± 6.19 | NI 2 |
| 1m | 6-Methoxy | 3′-Hydroxy | 72.10 ± 1.49 | 35.47 ± 4.38 |
| 1n | 6-Methoxy | 4′-Hydroxy | 63.95 ± 1.88 | 55.78 ± 1.48 |
| 1o | 6-Methoxy | 2′,3′-Dihydroxy | 83.55 ± 1.65 | 80.05 ± 1.32 |
| 1p | 6-Methoxy | 2′,4′-Dihydroxy | NI 2 | NI 2 |
| 1q | 6-Methoxy | 3′,4′-Dihydroxy | 108.11 ± 0.75 | 105.35 ± 0.13 |
| 1r | 6-Methoxy | 2′-Methoxy | 38.68 ± 0.67 | 20.45 ± 2.98 |
| 1s | 6-Methoxy | 3′-Methoxy | 70.44 ± 0.56 | 59.23 ± 0.34 |
| Sulfuretin | 6-Hydroxy | 3′,4′-Dihydroxy | 93.71 ± 1.25 | 53.72 ± 0.57 |
| Erufosine | 107.63 ± 0.27 | 106.37 ± 0.97 | ||

| Comp. | R1 | R2 | CC50 (µM) 1 | |
|---|---|---|---|---|
| THP-1 Cell Line | HEK293T Cell Line | |||
| 1i | 6-Hydroxy | 3′,4′-Dimethoxy | 96.86 ± 2.12 | >200 |
| 1q | 6-Methoxy | 3′,4′-Dihydroxy | 15.08 ± 0.52 | 29.13 ± 0.98 |
| Erufosine | 1.91 ± 0.51 | 28.24 ± 3.87 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hassan, A.H.E.; Phan, T.-N.; Choi, Y.; Moon, S.; No, J.H.; Lee, Y.S. Design, Rational Repurposing, Synthesis, In Vitro Evaluation, Homology Modeling and In Silico Study of Sulfuretin Analogs as Potential Antileishmanial Hit Compounds. Pharmaceuticals 2022, 15, 1058. https://doi.org/10.3390/ph15091058
Hassan AHE, Phan T-N, Choi Y, Moon S, No JH, Lee YS. Design, Rational Repurposing, Synthesis, In Vitro Evaluation, Homology Modeling and In Silico Study of Sulfuretin Analogs as Potential Antileishmanial Hit Compounds. Pharmaceuticals. 2022; 15(9):1058. https://doi.org/10.3390/ph15091058
Chicago/Turabian StyleHassan, Ahmed H.E., Trong-Nhat Phan, Yeonwoo Choi, Suyeon Moon, Joo Hwan No, and Yong Sup Lee. 2022. "Design, Rational Repurposing, Synthesis, In Vitro Evaluation, Homology Modeling and In Silico Study of Sulfuretin Analogs as Potential Antileishmanial Hit Compounds" Pharmaceuticals 15, no. 9: 1058. https://doi.org/10.3390/ph15091058