
Synthesis, Molecular Docking, and Antimalarial Activity of Hybrid 4-Aminoquinoline-pyrano[2,3-c]pyrazole Derivatives

 , , , , , and
, , , , , and 
Abstract
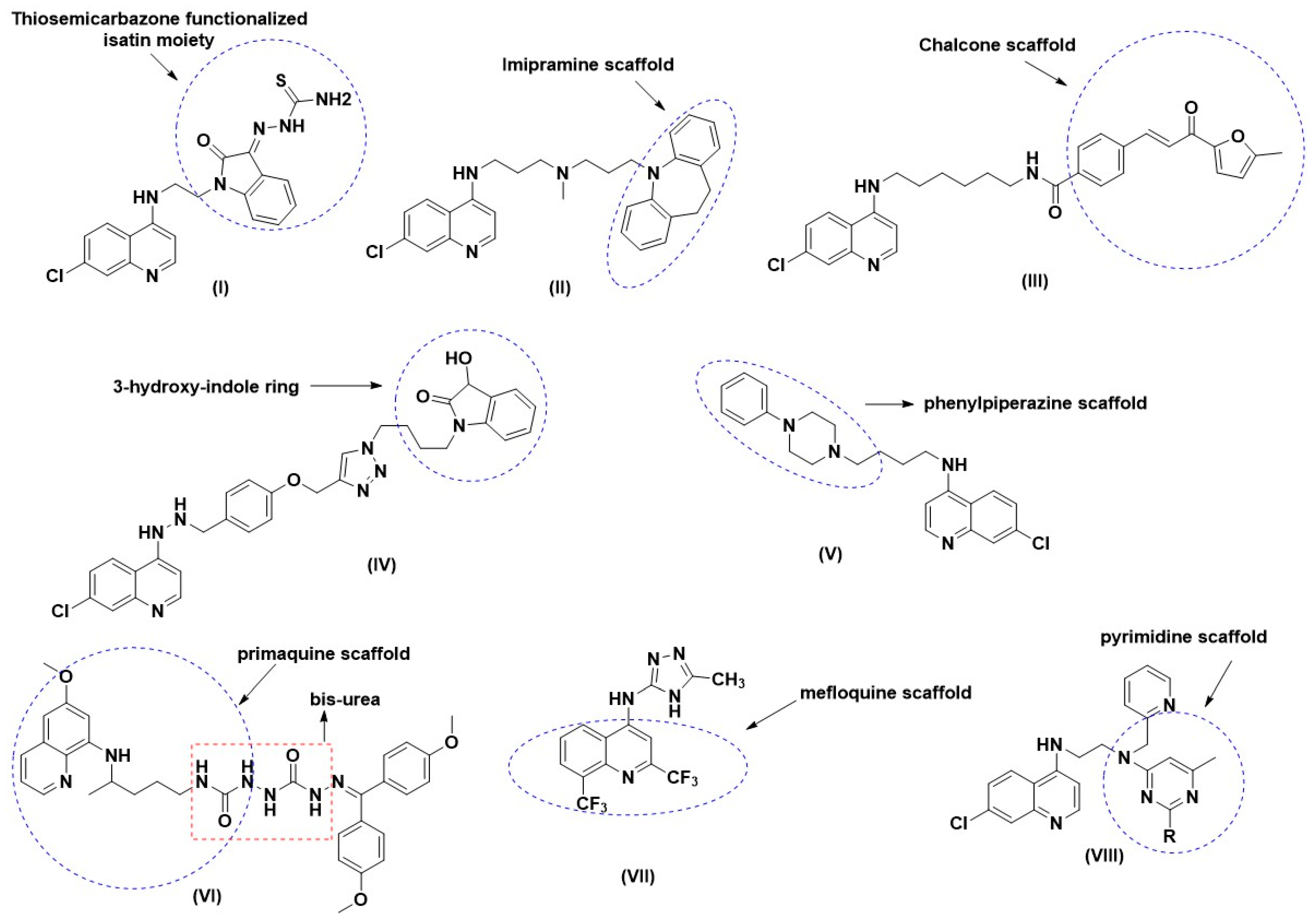
:1. Introduction
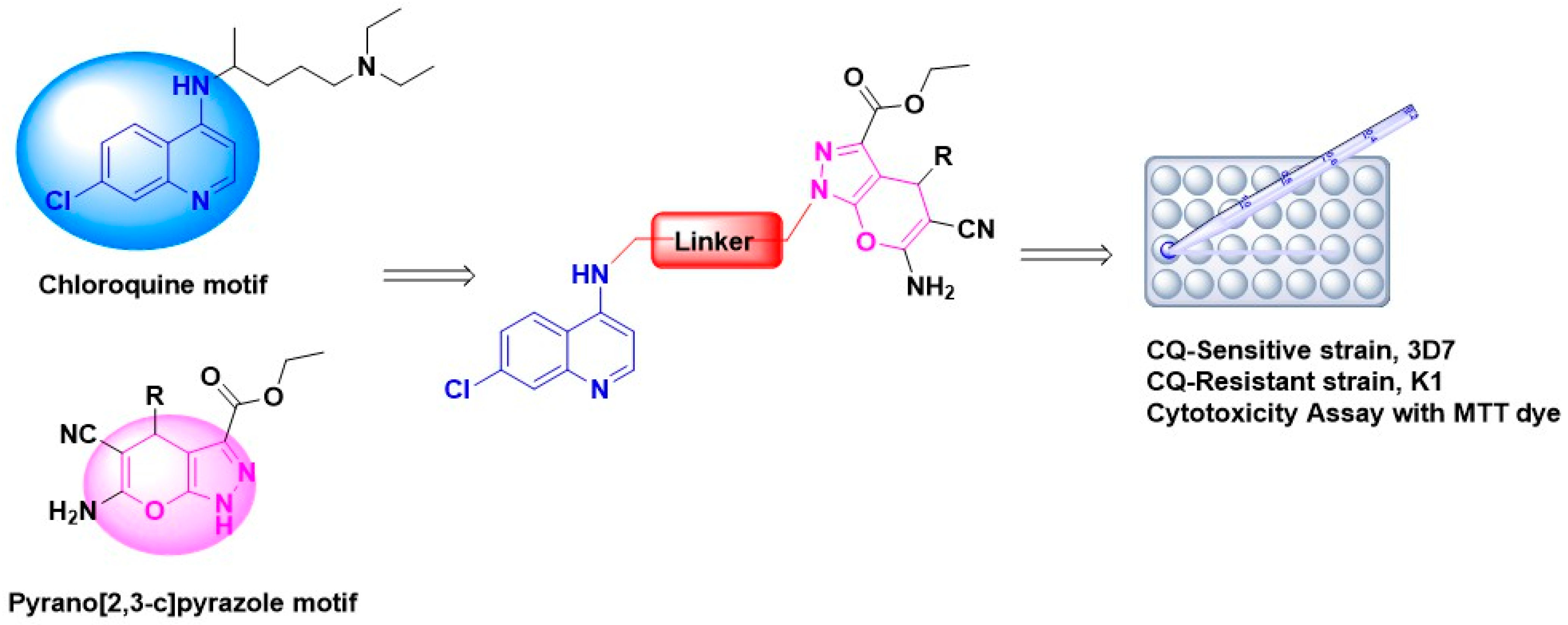
2. Results and Discussion
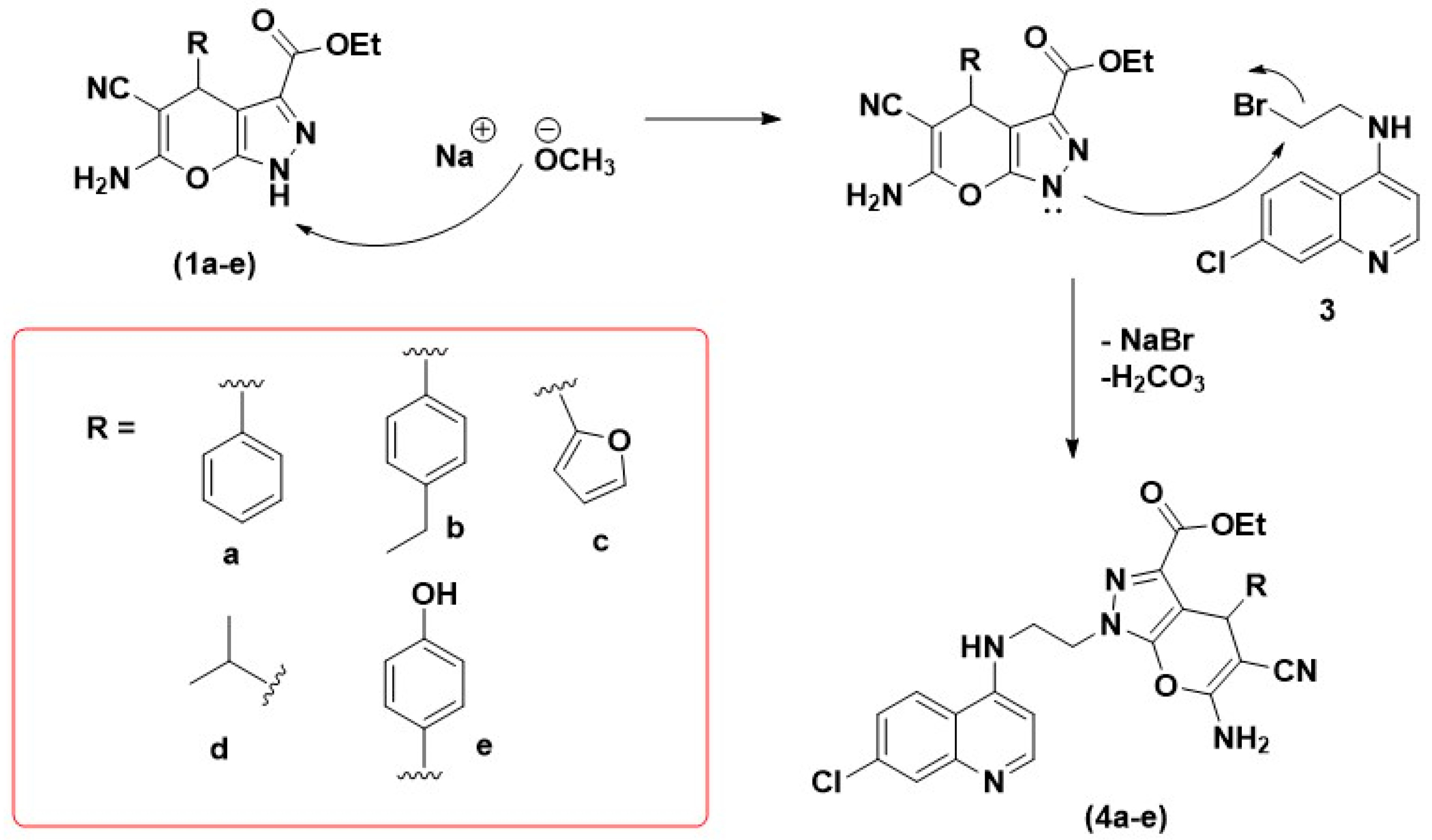
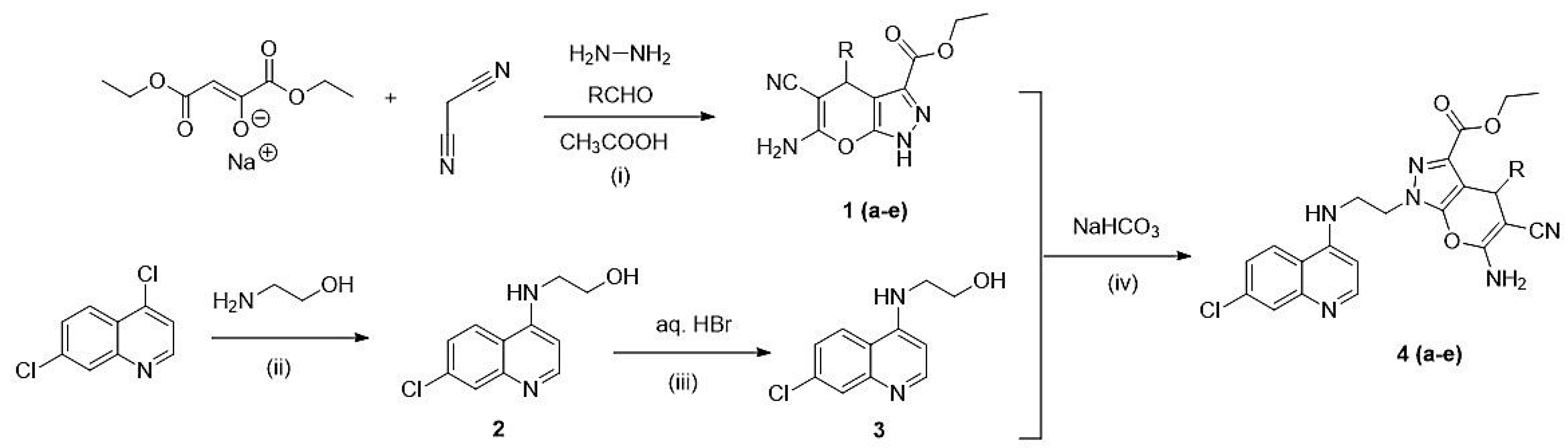
2.1. Chemistry: Reaction and Mechanism
2.2. Antimalarial (P. falciparum) and Cytotoxic (Vero Cells) Activities
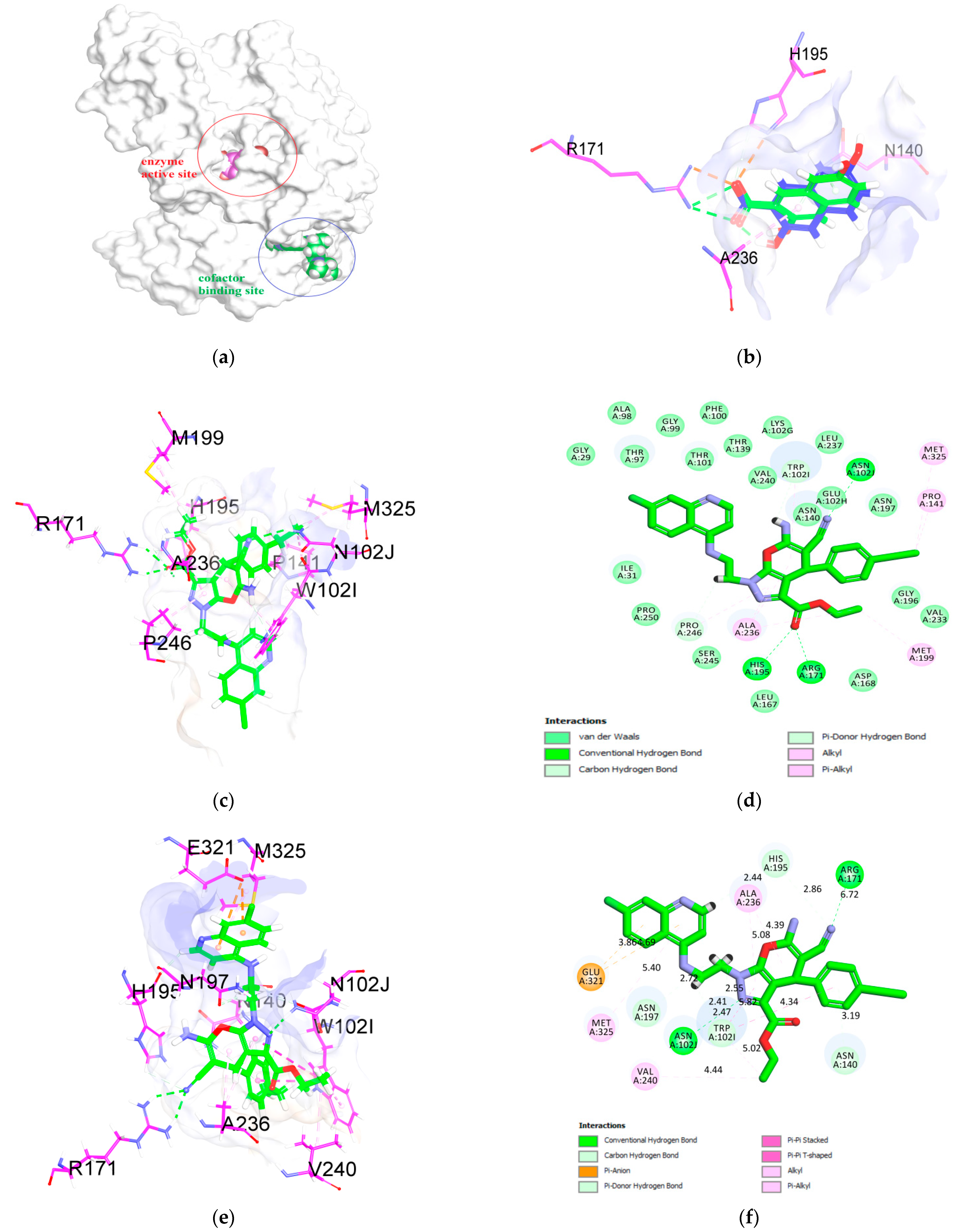
2.3. Molecular Docking Analysis
2.4. ADMET Profile of Compounds 4a–e
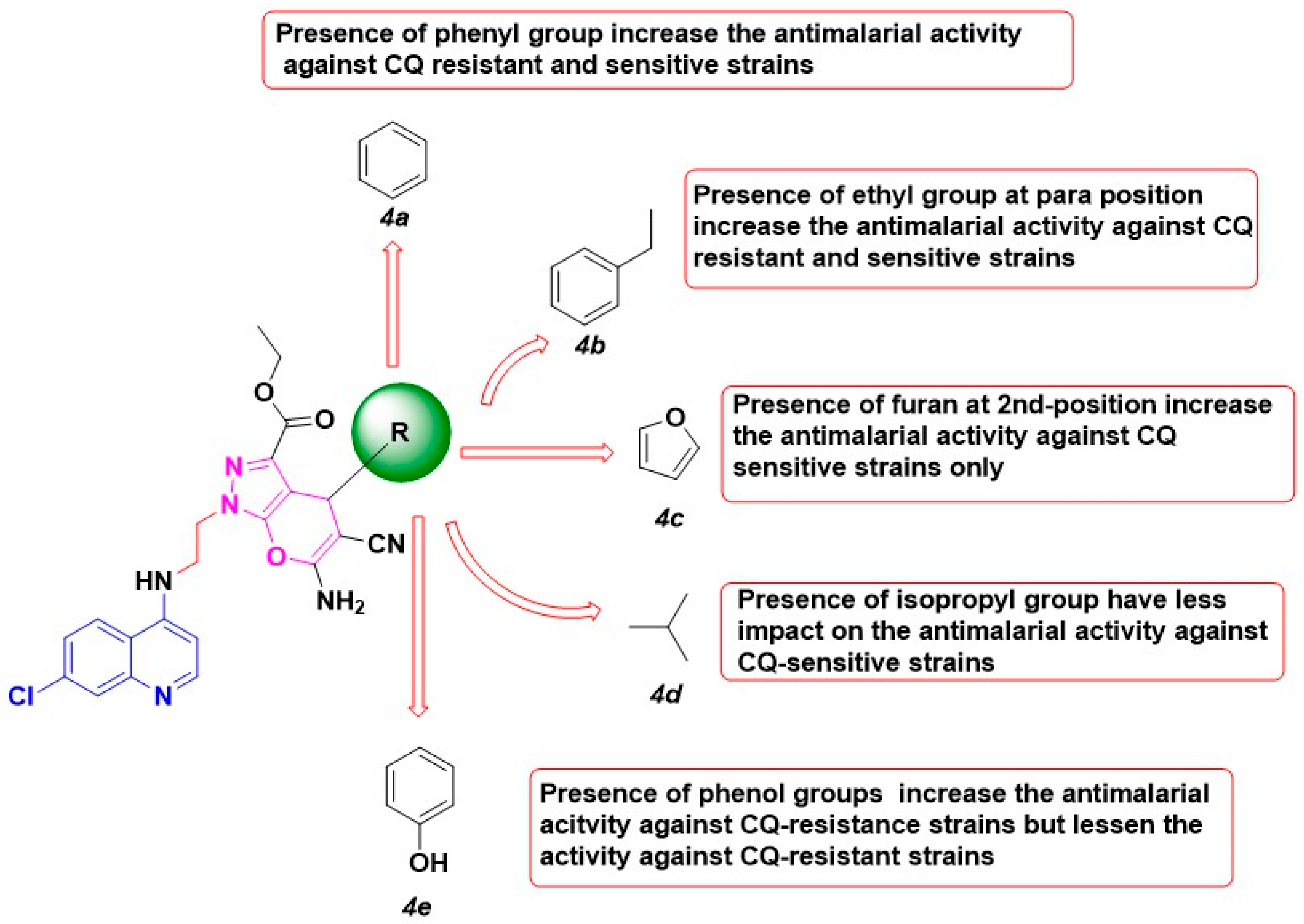
2.5. Structure-Activity Relationship
3. Materials and Methods
3.1. General Methods
3.2. General Procedure for the Synthesis of Pyrano[2,3-c]pyrazole
3.2.1. Synthesis of Pyrano[2,3-c]pyrazole-3-carboxylate (1a–e)
3.2.2. Synthesis of 4-(Ethanolamino)-7-chloroquine (2)
3.2.3. Synthesis of 4-(Bromoethylamino)-7-chloroquinoline (3)
3.3. General Procedure for the Synthesis of Hybrid (4a–e)
3.3.1. Ethyl 6-Amino-1-(2-((7-chloroquinolin-4-yl)amino)ethyl)-5-cyano-4-phenyl-1,4-dihydropyrano[2,3-c]pyrazole-3-carboxylate (4a)
3.3.2. Ethyl 6-Amino-1-(2-((7-chloroquinolin-4-yl)amino)ethyl)-5-cyano-4-(4-ethylphenyl)-1,4-dihydropyrano[2,3-c]pyrazole-3-carboxylate (4b)
3.3.3. Ethyl 6-Amino-1-(2-((7-chloroquinolyn-4-yl)amino)ethyl)-5-cyano-4-(furan-2-yl)-1,4-dihydropyrano[2,3-c]pyrazole-3-carboxylate (4c)
3.3.4. Ethyl 6-Amino-1-(2-((7-chloroquinolyn-4-yl)amino)ethyl)-5-cyano-4-isopropyl-1,4-dihydropyrano[2,3-c]pyrazole-3-carboxylate (4d)
3.3.5. Ethyl 6-Amino-1-(2-((7-chloroquinolyn-4-yl)amino)ethyl)-5-cyano-4-(4-hydroxyphenyl)-1,4-dihydropyrano[2,3-c]pyrazole-3-carboxylate (4e)
3.4. Antimalarial Assessments of Hybrid Molecules (4a–e) Using In Vitro P. falciparum 3D7 and K1 Cultures
3.4.1. Antimalarial Assay
3.4.2. Cytotoxicity Assay
3.4.3. Selectivity and Resistance Indexes Calculation
3.5. Computational Studies
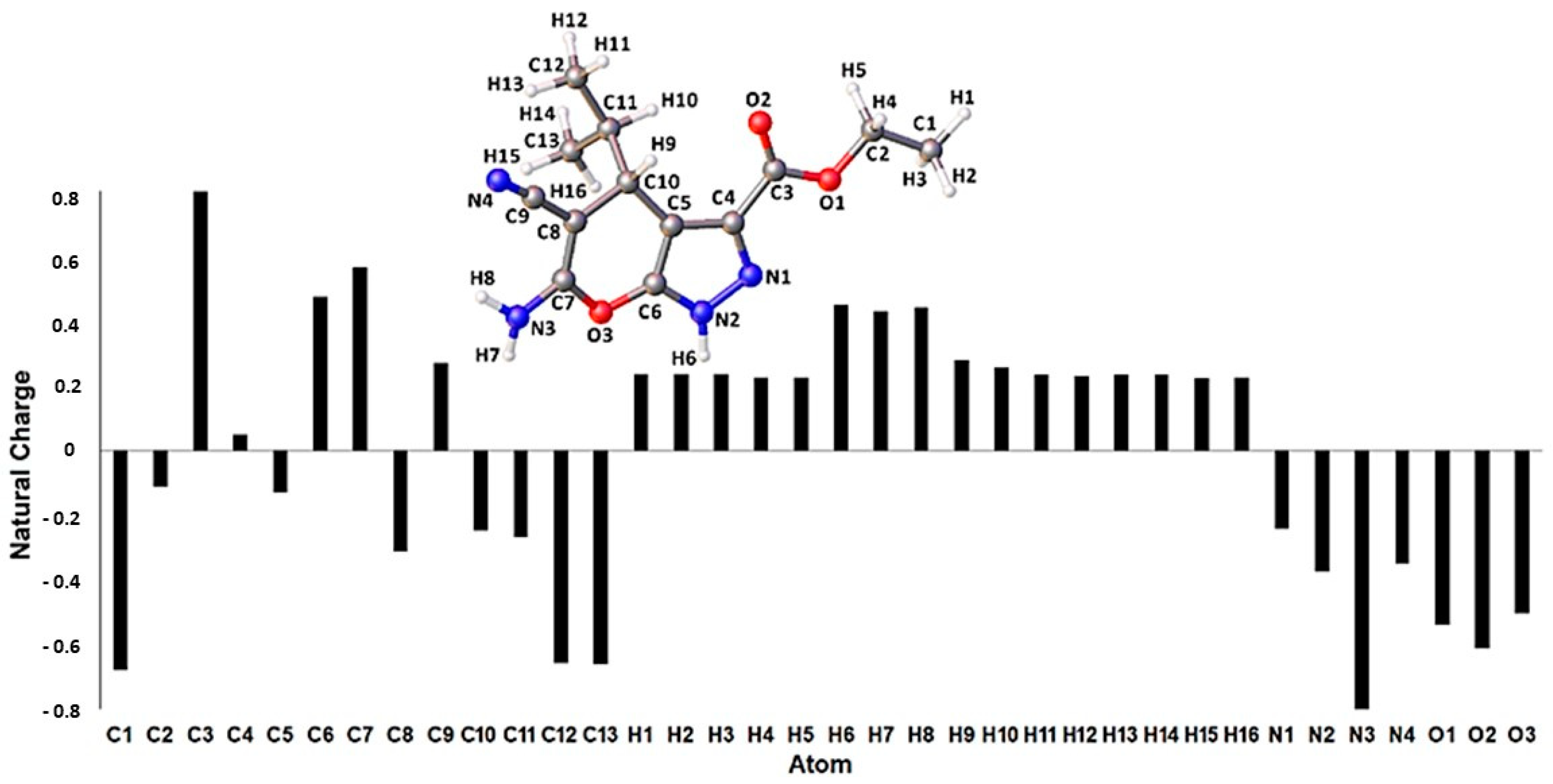
3.5.1. DFT Calculations
3.5.2. Preparation and Optimization of Ligands
3.5.3. Preparation and Optimization of Protein Receptors and Molecular Docking
3.5.4. Pharmacological Properties of the Hybrid Compounds
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alonso, P.L.; Tanner, M. Public health challenges and prospects for malaria control and elimination. Nat. Med. 2013, 19, 150–155. [Google Scholar] [CrossRef]
- Tibon, N.S.; Ng, C.H.; Cheong, S.L. Current progress in antimalarial pharmacotherapy and multi-target drug discovery. Eur. J. Med. Chem. 2020, 188, 111983. [Google Scholar] [CrossRef]
- Morphy, R.; Rankovic, Z. Designed multiple ligands. An emerging drug discovery paradigm. J. Med. Chem. 2005, 48, 6523–6543. [Google Scholar] [CrossRef]
- Meunier, B. Hybrid molecules with a dual mode of action: Dream or reality? Acc. Chem. Res. 2008, 41, 69–77. [Google Scholar] [CrossRef]
- Gasco, A.; Boschi, D.; Chegaev, K.; Cena, C.; Di Stilo, A.; Fruttero, R.; Lazzarato, L.; Rolando, B.; Tosco, P. Multitarget drugs: Focus on the NO-donor hybrid drugs. Pure Appl. Chem. 2008, 80, 1693–1701. [Google Scholar] [CrossRef] [Green Version]
- Pretorius, S.I.; Breytenbach, W.J.; de Kock, C.; Smith, P.J.; N’Da, D.D. Synthesis, characterization and antimalarial activity of quinoline-pyrimidine hybrid with potent antimalarial activity. Bioorg. Med. Chem. 2013, 21, 269–277. [Google Scholar] [CrossRef]
- Joubert, J.P.; Smit, F.J.; du Plessis, L.; Smith, P.J.; N’Da, D.D. Synthesis and in vitro biological evaluation of aminoacridines and artemisinin-acridine hybrids. Eur. J. Pharm. Sci. 2014, 56, 16–27. [Google Scholar] [CrossRef]
- Agarwal, D.; Gupta, R.D.; Awasthi, S.K. Are antimalarial hybrid molecules a close reality or a distant dream? Antimicrob. Agents Chemother. 2017, 61, e0024917. [Google Scholar] [CrossRef] [Green Version]
- Kouznetsov, V.V.; Gomez-Barrio, A. Recent developments in the design and Synthesis of hybrid molecules based on aminoquinoline ring and their antiplasmodial evaluation. Eur. J. Med. Chem. 2009, 44, 3091–3113. [Google Scholar] [CrossRef]
- Manohar, S.; Tripathi, M.; Rawat, D.S. 4-aminoquinoline based molecular hybrids as anti-malarials: An overview. Curr. Top. Med. Chem. 2014, 14, 1706–1733. [Google Scholar] [CrossRef]
- Pandey, A.V.; Bisht, H.; Babbarwal, V.K.; Srivastava, J.; Pandey, K.C.; Chauhan, V.S. Mechanism of malarial haem detoxification inhibition by chloroquine. Biochem. J. 2001, 355, 333–338. [Google Scholar] [CrossRef]
- De, D.; Krogstad, F.M.; Byers, L.D.; Krogstad, D.J. Structure-activity relationships for antiplasmodial activity among 7- substituted 4-aminoquinolines. J. Med. Chem. 1998, 41, 4918–4926. [Google Scholar] [CrossRef]
- Chiyanzu, I.; Clarkson, C.; Smith, P.J.; Lehman, J.; Gut, J.; Rosenthal, P.J.; Chibale, K. Design, Synthesis and anti-plasmodial evaluation in vitro of new 4-aminoquinoline isatin derivatives. Bioorg. Med. Chem. 2005, 13, 3249–3261. [Google Scholar] [CrossRef]
- Burgess, S.J.; Selzer, A.; Kelly, J.X.; Smilkstein, M.J.; Riscoe, M.K.; Peyton, D.H. A chloroquine-like molecule designed to reverse resistance in Plasmodium falciparum. J. Med. Chem. 2006, 49, 5623–5625. [Google Scholar] [CrossRef] [Green Version]
- Smit, F.J.; N’Da, D.D. Synthesis, in vitro antimalarial activity and cytotoxicity of novel 4-aminoquinolinyl-chalcone amides. Bioorg. Med. Chem. 2014, 22, 1128–1138. [Google Scholar] [CrossRef]
- Kondaparla, S.; Soni, A.; Manhas, A.; Srivasta, K.; Puri, S.K.; Katti, S.B. Antimalarial activity of novel 4-aminoquinolines active against drug-resistant strains. Bioorg. Chem. 2017, 70, 74–85. [Google Scholar] [CrossRef]
- Pavic, K.; Perkovic, I.; Pospisilova, S.; Machado, M.; Fontinha, D.; Prudencio, M.; Jampilek, J.; Coffey, A.; Endersen, L.; Rimac, H.; et al. Primaquine hybrids as promising antimycobacterial and antimalarial agents. Eur. J. Med. Chem. 2018, 143, 769–779. [Google Scholar] [CrossRef]
- Maurya, S.S.; Bahuguna, A.; Khan, S.I.; Kumar, D.; Kholiya, R.; Rawat, D.S. N-substituted aminoquinoline-pyrimidine hybrids: Synthesis, in vitro antimalarial activity evaluation and docking studies. Eur. J. Med. Chem. 2019, 162, 277–289. [Google Scholar] [CrossRef]
- Da Silva, R.M.; Gandi, M.O.; Mendonça, J.S.; Carvalho, A.S.; Coutinho, J.P.; Aguiar, A.C.; Krettli, A.U.; Boechat, N. New hybrid trifluoromethylquinolines as antiplasmodium agents. Bioorg. Med. Chem. 2019, 27, 1002–1008. [Google Scholar] [CrossRef]
- Satasia, S.P.; Kalaria, P.N.; Raval, D.K. Catalytic regioselective Synthesis of pyrazole based pyrido[2,3-d]pyrimidine-diones and their biological evaluation. Org. Biomol. Chem. 2014, 12, 1751–1758. [Google Scholar] [CrossRef]
- Insuasty, B.; Ramirez, J.; Beceraa, D.; Echeverry, C.; Quiroga, J.; Abonia, R.; Robledo, S.M.; Vélez, I.D.; Upegui, Y.; Muñoz, J.A.; et al. An efficient synthesis of new caffeine-based chalcones, pyrazolines and pyrazolo[3,4-b][1,4]diazepines as potential antimalarial, antitrypanosomal and antileishmanial agents. Eur. J. Med. Chem. 2015, 93, 401–413. [Google Scholar] [CrossRef]
- Bekhit, A.A.; Hassan, A.M.; El Razik, H.A.A.; El-Miligy, M.M.; El-Agroudy, E.J.; Bekhit, A.-D. New heterocyclic hybrids of pyrazile and its bioisosteres: Design, Synthesis, and biological evaluation as dual-acting antimalarial-antileishmanial agents. Eur. J. Med. Chem. 2015, 94, 30–44. [Google Scholar] [CrossRef]
- Karad, S.C.; Purohit, V.B.; Avalani, J.R.; Sapariya, N.H.; Raval, D.K. Design, Synthesis, and characterization of a fluoro substituted novel pyrazole nucleus clubbed with 1, 3, 4-oxadiazole scaffolds and their biological applications. RSC Adv. 2016, 6, 41532–41541. [Google Scholar] [CrossRef]
- Mark-Lee, W.F.; Chong, Y.Y.; Lahuri, A.H.; Kassim, M.B. Synthesis and solid-state structural elucidation of rhenium (i) complex with 1-cinnamoyl-3-(pyridin-2-yl) pyrazole. Malays. J. Anal. Sci. 2020, 24, 11–20. [Google Scholar]
- Dolabela, M.F.N.; Oliveira, S.G.; Nascimento, J.M.; Peres, J.M.; Wagner, H.; Póvoa, M.M.; de Oliveira, A.B. In vitro antiplasmodial activity of extract and constituents from Esenbeckia febrifuga, a plant traditionally used to treat malaria in the Brazilian Amazon. Phytomedicine 2008, 15, 367–372. [Google Scholar] [CrossRef]
- Katsuno, K.; Burrows, J.N.; Duncan, K.; Van Huijsduijnen, R.H.; Kaneko, T.; Kita, K.; Mowbray, C.E.; Schmatz, D.; Warner, P.; Slingsby, B.T. Hit and lead criteria in drug discovery for infectious diseases of the developing world. Nat. Rev. Drug Discov. 2015, 14, 751–758. [Google Scholar] [CrossRef]
- Burger, A.M.; Fiebig, H.H. Preclinical Screening for New Anticancer Agents. In Handbook of Anticancer Pharmacokinetics and Pharmacodynamics; Springer: Berlin, Germany, 2014; pp. 23–38. [Google Scholar]
- Wiji Prasetyaningrum, P.; Bahtiar, A.; Hayun, H. Synthesis and cytotoxicity evaluation of novel asymmetrical mono-carbonyl analogs of curcumin (AMACs) against Vero, HeLa, and MCF7 cell lines. Sci. Pharm. 2018, 86, 25. [Google Scholar] [CrossRef] [Green Version]
- Singh, K.; Kaur, H.; Chibale, K.; Balzarini, J. Synthesis of 4-aminoquinoline–pyrimidine hybrids as potent anti-malarials and their mode of action studies. Eur. J. Med. Chem. 2013, 66, 314–323. [Google Scholar] [CrossRef]
- Mohammat, M.F.; Maarop, M.S.; Shaameri, Z.; Wibowo, A.; Johari, S.A.; Hamzah, A.S. Practical synthesis and electronic study of non-spiro and spiropyrano [2, 3-c] pyrazole-3-carboxylate derivatives via uncatalyzed domino one-pot, four-component reactions. Org. Commun. 2018, 11, 149–162. [Google Scholar] [CrossRef]
- Trager, W.; Jensen, J.B. Continuous culture of Plasmodium falciparum: Its impact on malaria research. Int. J. Parasitol. 1997, 27, 989–1006. [Google Scholar] [CrossRef]
- Makler, M.T.; Hinrichs, D.J. Measurement of the lactate dehydrogenase activity of Plasmodium falciparum as an assessment of parasitemia. Am. J. Trop. Med. Hyg. 1993, 48, 205–210. [Google Scholar] [CrossRef]
- Nkhoma, S.; Molyneux, M.; Ward, S. In vitro antimalarial susceptibility profile and prcrt/pfmdr-1 genotypes of Plasmodium falciparum field isolates from Malawi. Am. J. Trop. Med. Hyg. 2007, 76, 1107–1112. [Google Scholar] [CrossRef] [Green Version]
- Mosmann, T. Rapid colourimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Sarr, S.O.; Perrotey, S.; Fall, I.; Ennahar, S.; Zhao, M.; Diop, Y.M.; Candolfi, E.; Marchioni, E. Icacina senegalensis (Icacinaceae), traditionally used for the treatment of malaria, inhibits in vitro Plasmodium falciparum growth without host cell toxicity. Malar. J. 2011, 10, 85–95. [Google Scholar] [CrossRef] [Green Version]
- Iwaniuk, D.P.; Whetmore, E.D.; Rosa, N.; Ekoue-Kovi, K.; Alumasa, J.; de Dios, A.C.; Roepe, P.D.; Wolf, C. Synthesis and antimalarial activity of new chloroquine analogues carrying a multifunctional linear side chain. Bioorg. Med. Chem. 2009, 17, 6560–6566. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.E.A.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision d. 01; Gaussian. Inc.: Wallingford, CT, USA, 2015; Volume 2009, p. 201. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Test Compounds | P. falciparum 3D7 (CQ-Sensitive) EC50 (µM) ± S.D. | P. falciparum K1 (CQ-Resistant) EC50 (µM) ± S.D. | Vero Cell (Normal Cell) CC50 (µM) ± S.D. | Selectivity Index (SI), | Resistance Index (RI), | |
|---|---|---|---|---|---|---|
| CQ-Sensitive | CQ-Resistant | |||||
| 4a | 0.19 ± 0.07 | 0.25 ± 0.03 | 102.54 ± 22.15 | 539.7 | 410.16 | 1.32 |
| 4b | 0.0130 ± 0.0002 | 0.02 ± 0.01 | 17.60 ± 1.50 | 1353.9 | 880 | 1.53 |
| 4c | 0.113 ± 0.002 | 1.61 ± 0.15 | 84.52 ± 6.45 | 747.9 | 52.49 | 14.24 |
| 4d | 3.39 ± 1.89 | 0.30 ± 0.01 | 86.14 ± 7.33 | 25.4 | 287 | 0.09 |
| 4e | 0.026 ± 0.009 | 7.12 ± 3.72 | 9.24 ± 1.13 | 355.3 | 1.3 | 273.84 |
| 4-aminoquinoline | 9.29 ± 0.12 | 140.43 ± 27.61 | 113.85 ± 1.63 | 12.25 | 0.81 | 15.11 |
| CQ | 0.002 | 0.33 | 138.40 ± 8.77 | >2000 | >2000 | 16.5 |
| ART | 0.0001 | 0.00017 | 434.60 ± 64.21 | >2000 | >2000 | 1.7 |
| Compounds | Cdocker Energy (kcal/mol) | Cdocker Interaction Energy (kcal/mol) | Residues of the Active Site Interacting with the Ligands |
|---|---|---|---|
| 4a | −13.7 | −53.9 | Thr97, Thr101, Trp102, Ser245, Ser245, Pro246, Val240, Asn102, Leu237, Glu102, Pro141, Asn234, Asn197, Gly196, Val233, Met199, Asp168, Arg171, His195, Asn140, Leu167, Tyr247, Pro250, Ile31, Val138, Thr139, Thr97 |
| 4b | −20.7 (* −16.4) | −60.5 (* −54.3) | Refer to Figure 5d * (Figure 5f) |
| 4c | −19.4 | −56.9 | Ile31, Thr97, Val138, Thr139, Pro250, Arg171, Leu167, His195, Pro141, Val142, Asn140, Gly196, Asn197, Asb234, Val233, Met199, Val240, Asn102, Leu237, Ala236, Pro246, Trp102, Ser245, Thr101 |
| 4d | −19.0 | −53.7 | Gly29, Met30, Thr97, Thr101, Gly99, Phe100, Thr139, Lys102, Glu102, Asn140, Pro141, Val142, His195, His196, Val233, Asn197, Asn234, Met199, Leu237, Ala236, Asn102, Val240, Trp102, Pro246, Tyr247, Ile31 |
| 4e | −17.4 | −56.9 | Met30, Ile31, Thr97, Thr101, Thr139, Val138, Leu163, Pro246, Leu167, Ala236, Asn140, Leu167, His195, Asn197, Met199, Arg171, Gly196, Val233, Asp168, Glu102, Asn102, Leu237, Trp102, Val240, Ser245, Tyr247 |
| 3,5-dihydroxy-2-naphthoic acid | −32.7 | −44.5 | Gly196, Asn102, Asn197, Trp102, Ala236, Val240, Pro246, Pro250, Leu167, Asn140, Arg171, His195 |
| Compound | ADMET Parameter | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Human Intestinal Absorption | Aqueous Solubility | Blood-Brain Barrier (BBB) Penetration | Plasma Protein Binding (PPB) | Cytochrome P450 2D6 (CYP2D6) | Hepatotoxicity | |||||
| PSA a | ALogP98 b | Level c | Log(Sw) d | Level e | LogBB f | Level g | Prediction h | Prediction i | Prediction j | |
| 4a | 125.316 | 4.577 | 2 | −6.68 | 1 | - | 4 | 0 | 0 | 1 |
| 4b | 125.316 | 5.519 | 2 | −7.285 | 1 | - | 4 | 0 | 0 | 1 |
| 4c | 146.132 | 4.335 | 2 | −6.601 | 1 | - | 4 | 0 | 0 | 1 |
| 4d | 137.870 | 3.761 | 2 | −6.278 | 1 | - | 4 | 0 | 0 | 1 |
| 4e | 125.316 | 4.252 | 2 | −6.402 | 1 | - | 4 | 0 | 0 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shamsuddin, M.A.; Ali, A.H.; Zakaria, N.H.; Mohammat, M.F.; Hamzah, A.S.; Shaameri, Z.; Lam, K.W.; Mark-Lee, W.F.; Agustar, H.K.; Mohd Abd Razak, M.R.; et al. Synthesis, Molecular Docking, and Antimalarial Activity of Hybrid 4-Aminoquinoline-pyrano[2,3-c]pyrazole Derivatives. Pharmaceuticals 2021, 14, 1174. https://doi.org/10.3390/ph14111174
Shamsuddin MA, Ali AH, Zakaria NH, Mohammat MF, Hamzah AS, Shaameri Z, Lam KW, Mark-Lee WF, Agustar HK, Mohd Abd Razak MR, et al. Synthesis, Molecular Docking, and Antimalarial Activity of Hybrid 4-Aminoquinoline-pyrano[2,3-c]pyrazole Derivatives. Pharmaceuticals. 2021; 14(11):1174. https://doi.org/10.3390/ph14111174
Chicago/Turabian StyleShamsuddin, Mohd Asyraf, Amatul Hamizah Ali, Nur Hanis Zakaria, Mohd Fazli Mohammat, Ahmad Sazali Hamzah, Zurina Shaameri, Kok Wai Lam, Wun Fui Mark-Lee, Hani Kartini Agustar, Mohd Ridzuan Mohd Abd Razak, and et al. 2021. "Synthesis, Molecular Docking, and Antimalarial Activity of Hybrid 4-Aminoquinoline-pyrano[2,3-c]pyrazole Derivatives" Pharmaceuticals 14, no. 11: 1174. https://doi.org/10.3390/ph14111174