
Novel Purine Chemotypes with Activity against Plasmodium falciparum and Trypanosoma cruzi

 , , , , , , , and
, , , , , , , and
Abstract
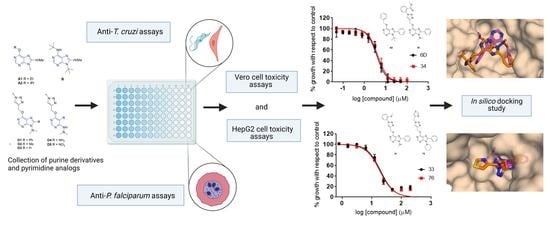
:
1. Introduction
2. Results
2.1. Primary Screening
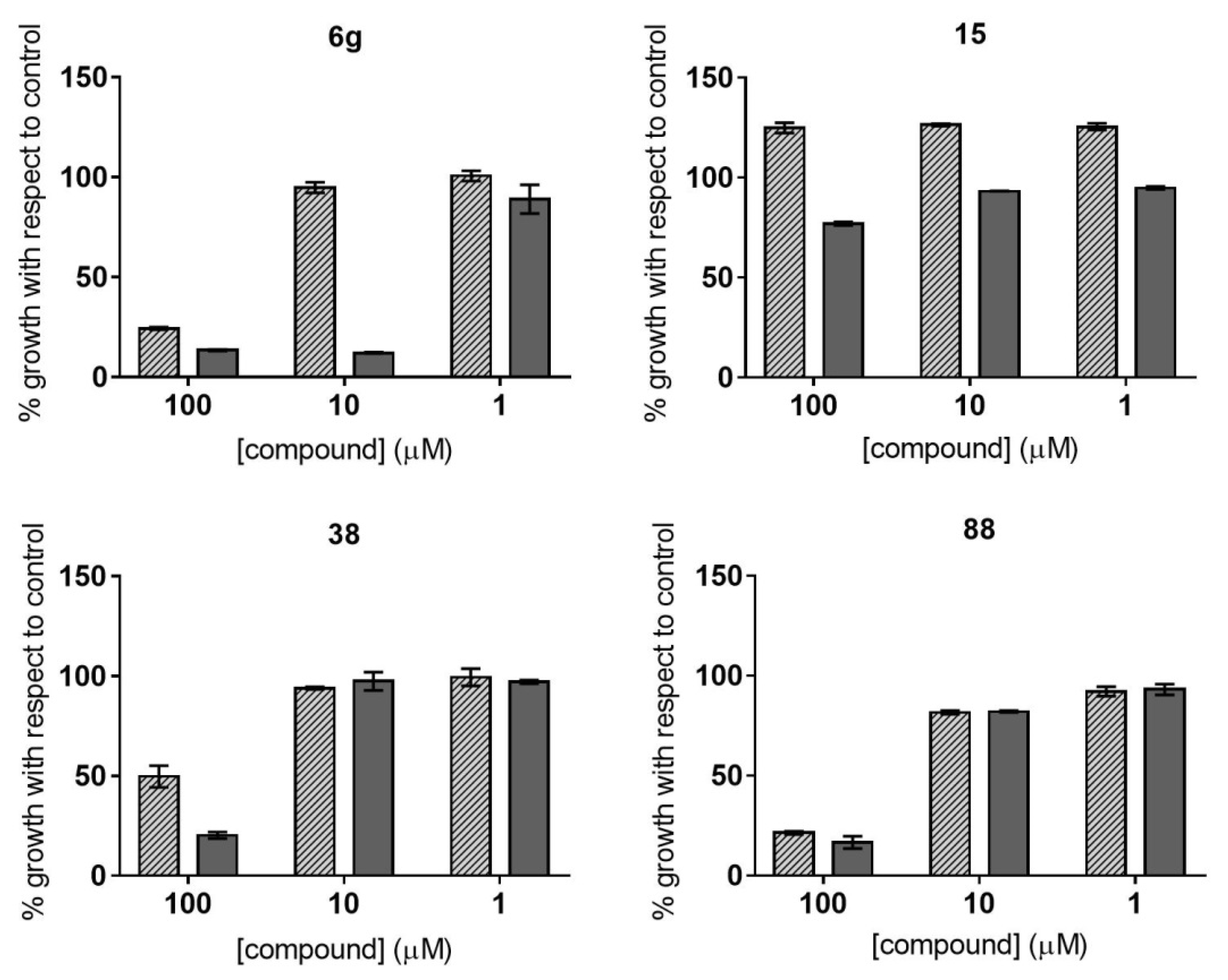
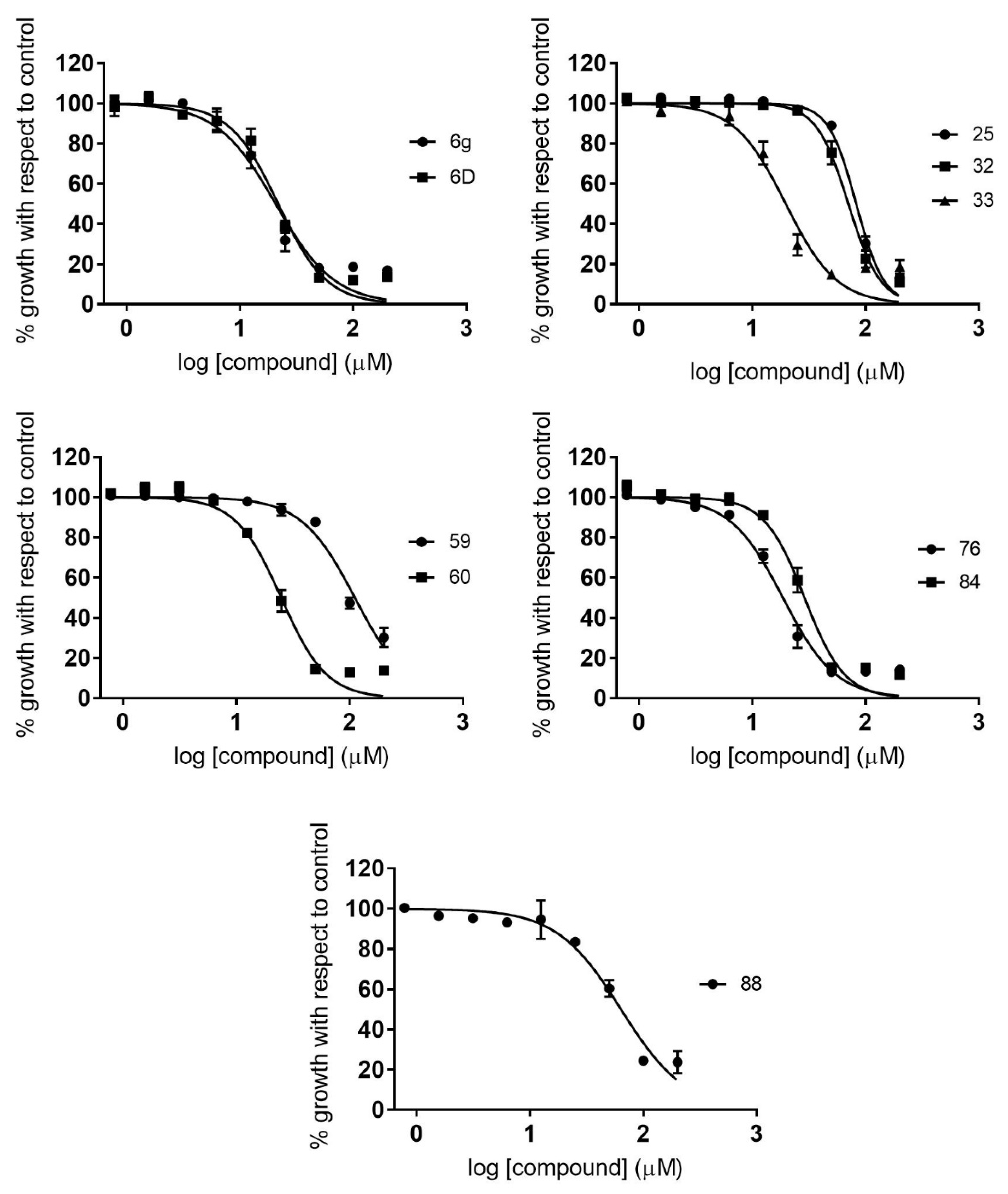
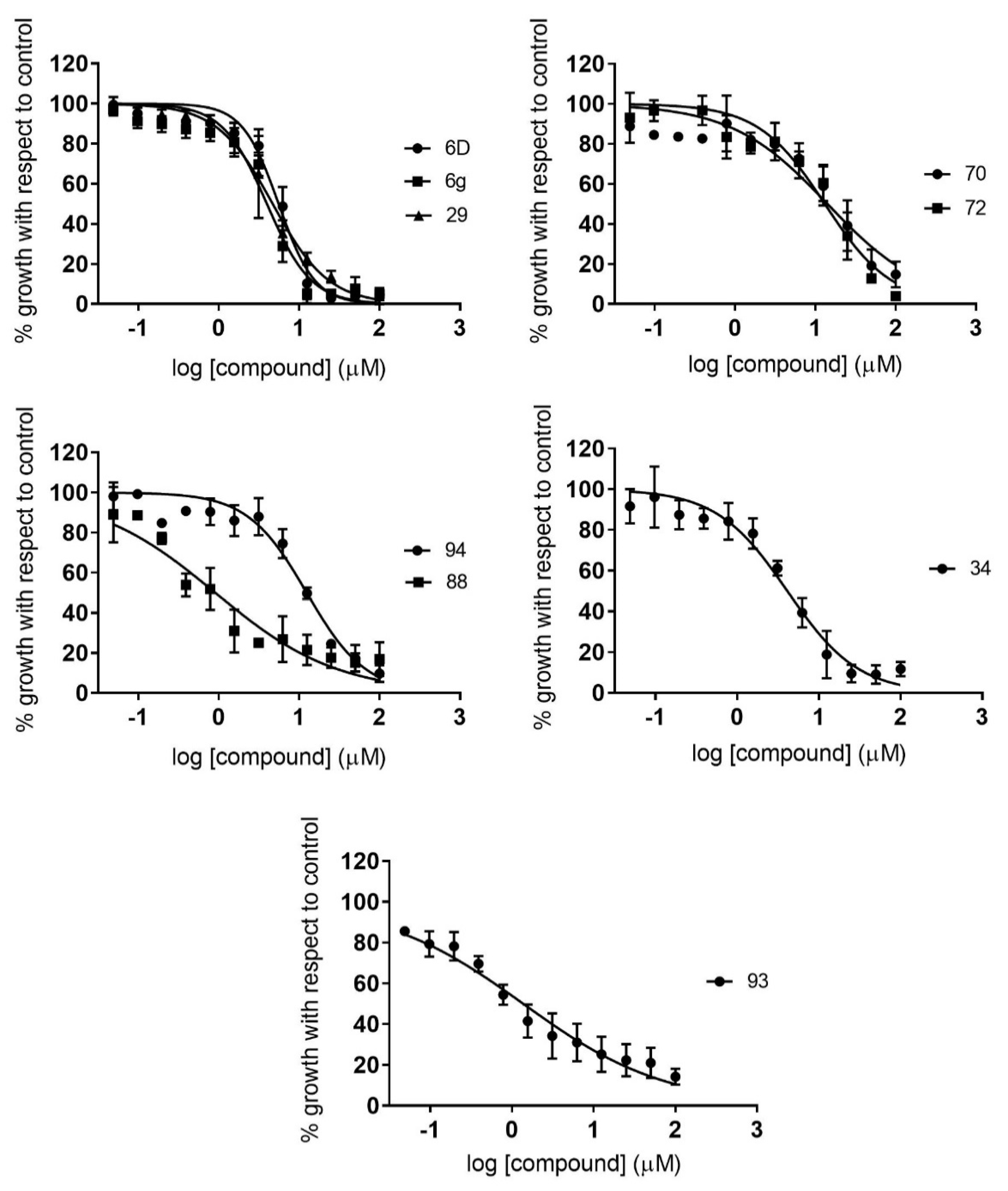
2.2. Dose–Response Growth Inhibition Assay
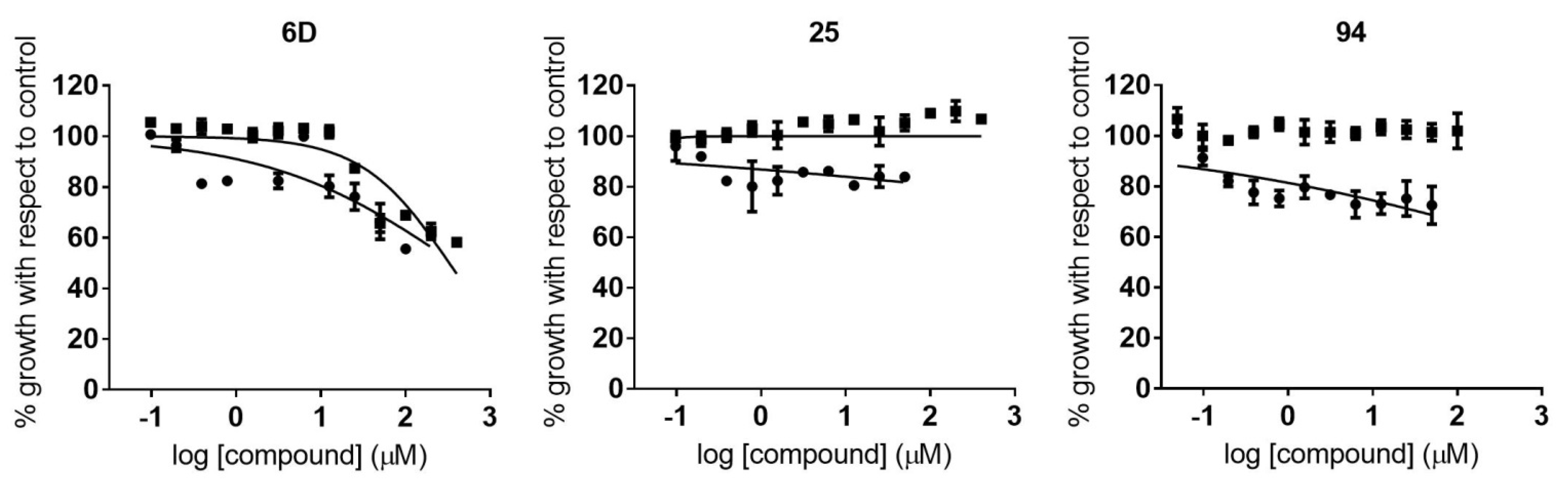
2.3. Identification of Compounds with Specific Antiparasitic Activity
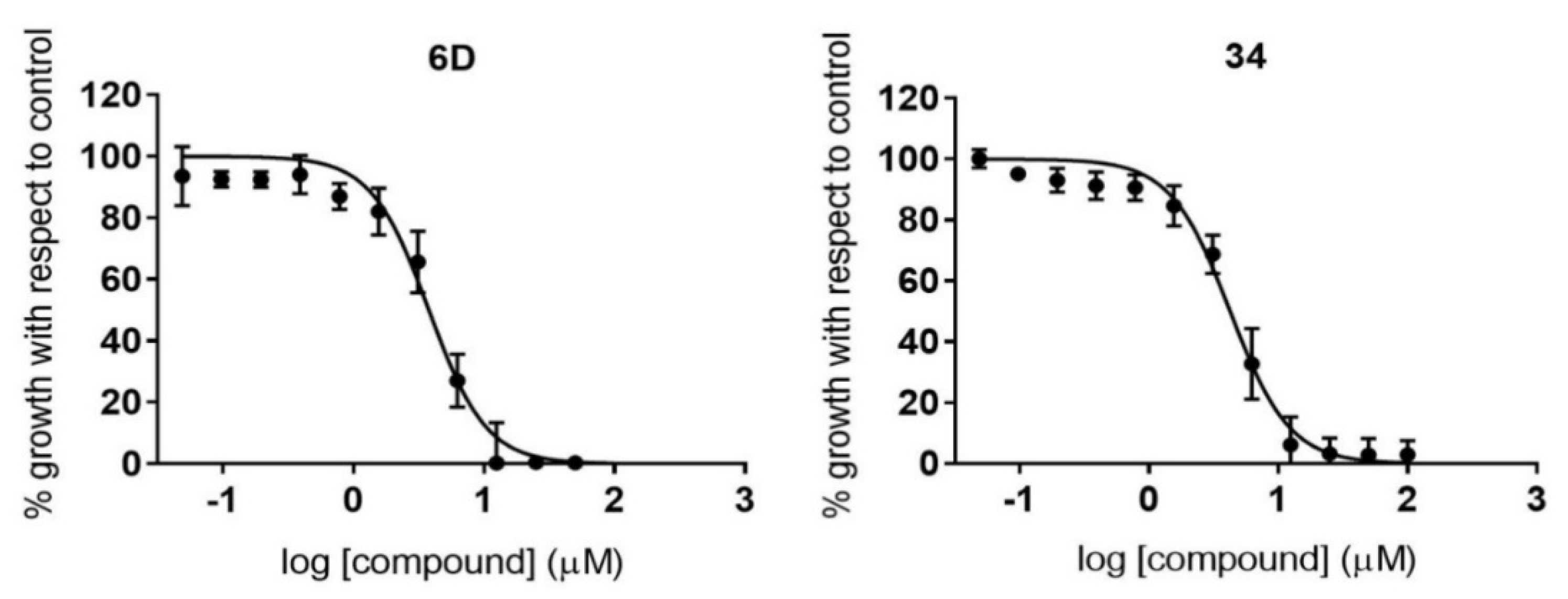
2.4. HepG2 Cell Toxicity Assay
2.5. Antiamastigote Growth Inhibition Assay
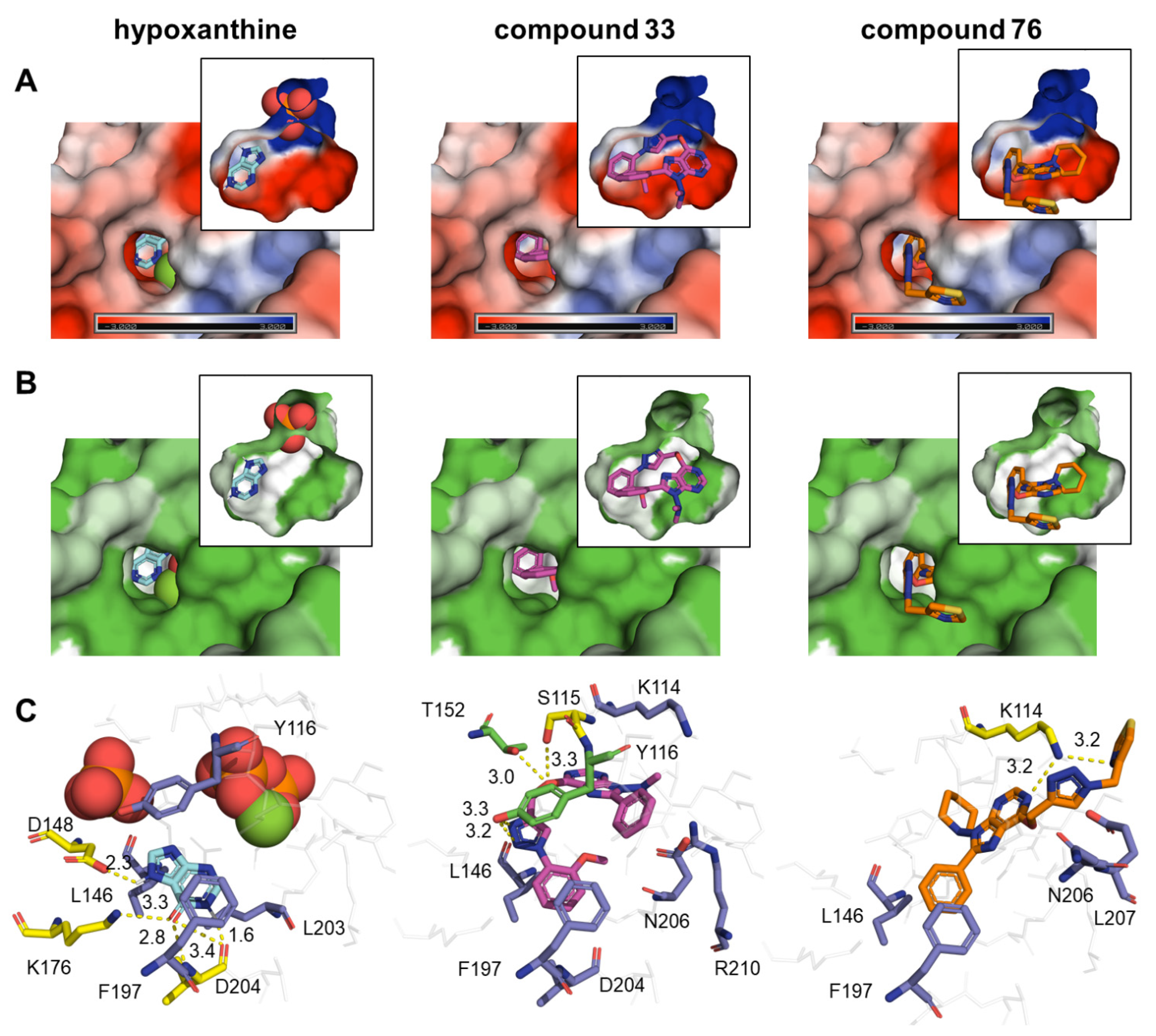
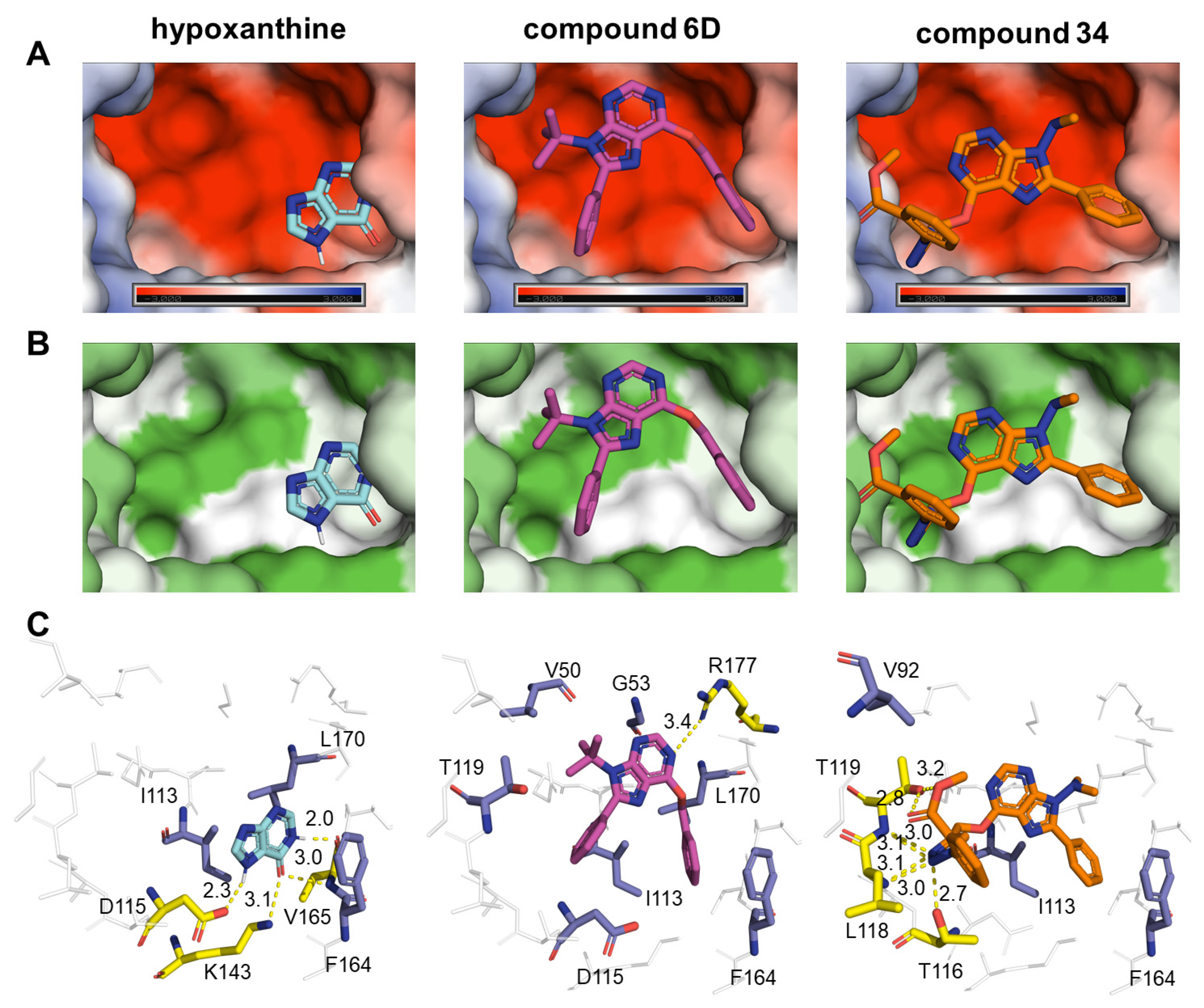
2.6. Computational Analysis
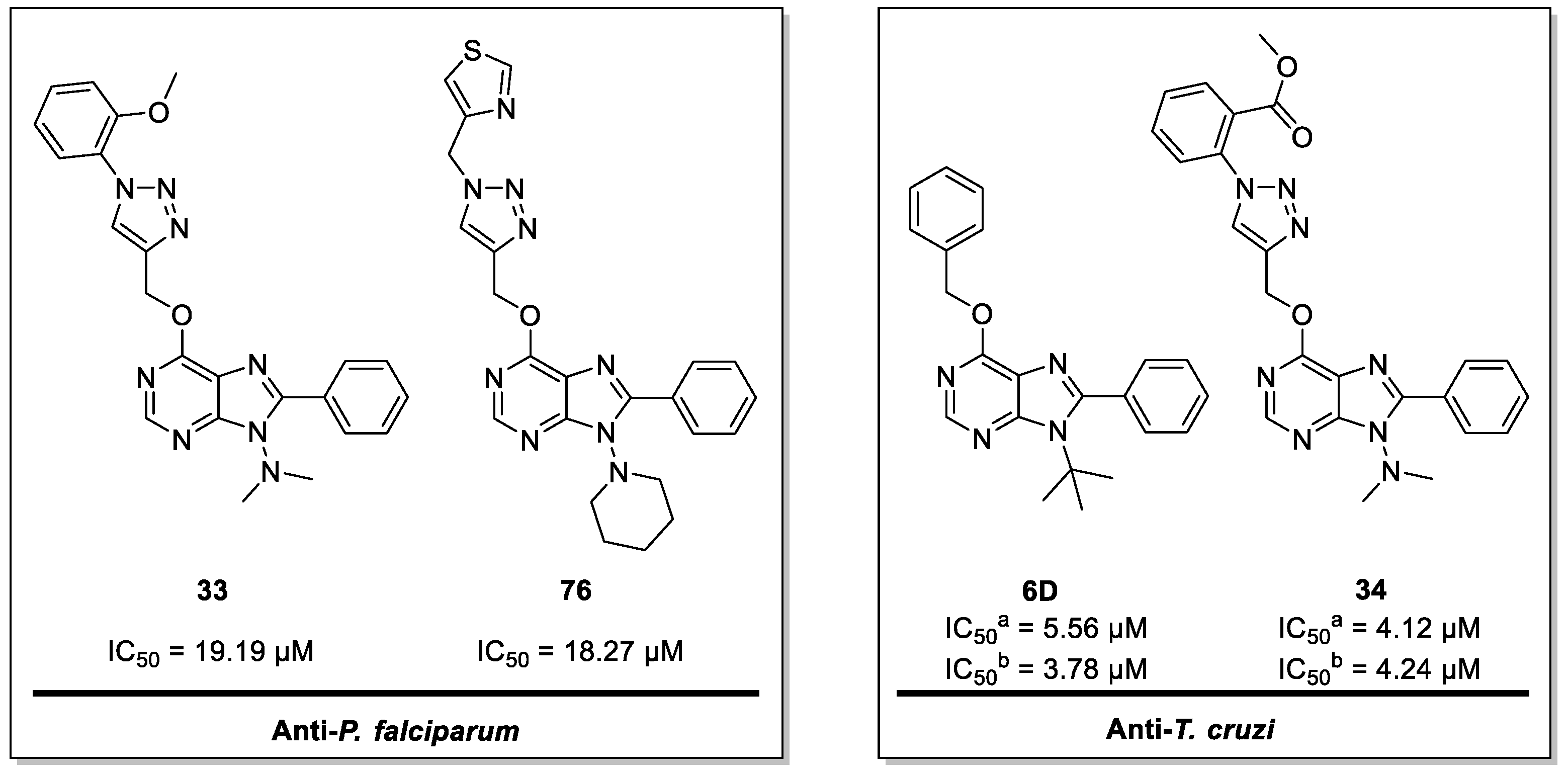
3. Discussion
4. Materials and Methods
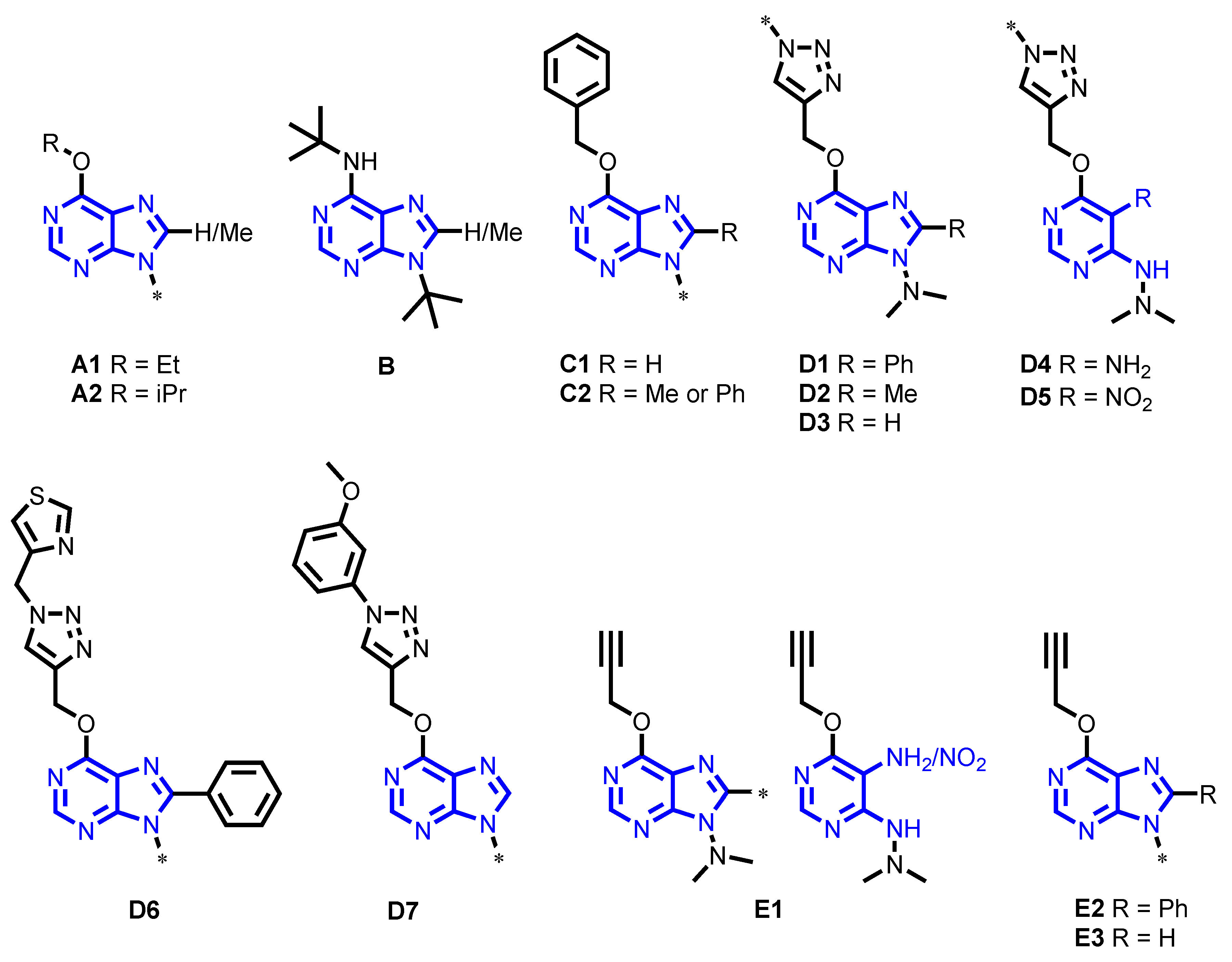
4.1. Chemical Collection
4.2. Host Cell Cultures
4.3. Culture of P. falciparum and T. cruzi Parasites
4.4. P. falciparum Primary Screening and Growth Inhibition Assay
4.5. T. cruzi Primary Screening and Growth Inhibition Assay
4.6. Vero and HepG2 Toxicity Assays
4.7. Antiamastigote Specific Assay
4.8. Computational Analysis
4.9. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization (WHO). World Malaria Report 2015. Available online: https://www.who.int/publications/i/item/9789241565721 (accessed on 10 March 2021).
- Howes, R.E.; Battle, K.E.; Mendis, K.N.; Smith, D.L.; Cibulskis, R.E.; Baird, J.K.; Hay, S.I. Global epidemiology of Plasmodium vivax. Am. J. Trop. Med. Hyg. 2016, 95, 15–34. [Google Scholar] [CrossRef] [Green Version]
- Guidelines for the Treatment of Malaria, 3rd ed.; World Health Organization: Geneva, Switzerland, 2015.
- Blasco, B.; Leroy, D.I.; Fidock, D.A. Antimalarial drug resistance: Linking Plasmodium falciparum parasite biology to the clinic. Nat. Med. 2017, 23, 917–928. [Google Scholar] [CrossRef] [PubMed]
- Dondorp, A.M.; Nosten, F.; Yi, P.; Debashish, D.; Phyo, A.P.; Tarning, J.; Lwin, K.M.; Ariey, F.; Hanpithakpong, W.; Lee, S.J.; et al. Artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med. 2009, 361, 455–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashley, E.A.; Dhorda, M.; Fairhurst, R.M.; Amaratunga, C.; Lim, P.; Suon, S.; Sreng, S.; Anderson, J.M.; Mao, S.; Sam, B.; et al. Spread of artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med. 2014, 371, 411–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization (WHO). Chagas Disease (American Trypanosomiasis). Available online: https://www.who.int/en/news-room/fact-sheets/detail/chagas-disease-(american-trypanosomiasis) (accessed on 10 March 2020).
- Gascon, J.; Bern, C.; Pinazo, M.J. Chagas disease in Spain, the United States and other non-endemic countries. Acta Trop. 2010, 115, 22–27. [Google Scholar] [CrossRef]
- Crespillo-Andujar, C.; Venanzi-Rullo, E.; López-Vélez, R.; Monge-Maillo, B.; Norman, F.; López-Polín, A.; Pérez-Molina, J. Safety profile of benznidazole in the treatment of chronic Chagas disease: Experience of a referral centre and systematic literature review with meta-analysis. Drug Saf. 2018, 41, 1035–1048. [Google Scholar] [CrossRef]
- Forsyth, C.J.; Hernandez, S.; Olmedo, W.; Abuhamidah, A.; Traina, M.I.; Sanchez, D.R.; Soverow, J.; Meymandi, S.K. Safety profile of nifurtimox for treatment of Chagas disease in the United States. Clin. Infect. Dis. 2016, 63, 1056–1062. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Peinado, N.; Cortes-Serra, N.; Losada-Galvan, I.; Alonso-Vega, C.; Urbina, J.A.; Rodríguez, A.; VandeBerg, J.L.; Pinazo, M.J.; Gascon, J.; Alonso-Padilla, J. Emerging agents for the treatment of Chagas disease: What is in the preclinical and clinical development pipeline? Expert Opin. Investig. Drugs 2020, 9, 947–959. [Google Scholar] [CrossRef]
- Berg, M.; Van der Veken, P.; Goeminne, A.; Haemers, A.; Augustyns, K. Inhibitors of the purine salvage pathway: A valuable approach for antiprotozoal chemotherapy? Curr. Med. Chem. 2010, 17, 2456–2481. [Google Scholar] [CrossRef]
- Chaudhary, K.; Darling, J.A.; Fohl, L.M.; Sullivan, W.J.; Donald, R.G.; Pfefferkorn, E.R.; Ullman, B.; Roos, D.S. Purine salvage pathways in the apicomplexan parasite Toxoplasma gondii. J. Biol. Chem. 2004, 279, 31221–31227. [Google Scholar] [CrossRef] [Green Version]
- Berens, R.L.; Marr, J.J.; LaFon, S.W.; Nelson, D.J. Purine metabolism in Trypanosoma cruzi. Mol. Biochem. Parasitol. 1981, 3, 187–196, 522. [Google Scholar] [CrossRef]
- El Kouni, M.H. Potential chemotherapeutic targets in the purine metabolism of parasites. Pharmacol. Ther. 2003, 99, 283–309, 524. [Google Scholar] [CrossRef]
- Downie, M.J.; Kirk, K.; Mamoun, C.B. Purine salvage pathways in the intraerythrocytic malaria parasite Plasmodium falciparum. Eukaryotic Cell 2008, 7, 1231–1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheviet, T.; Lefebvre-Tournier, I.; Wein, S.; Peyrottes, S. Plasmodium purine metabolism and its inhibition by nucleoside and nucleotide analogues. J. Med. Chem. 2019, 62, 8365–8391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freymann, D.M.; Wenck, M.A.; Engel, J.C.; Feng, J.; Focia, P.J.; Eakin, A.E.; Craig, S.P. Efficient identification of inhibitors targeting the closed active site conformation of the HPRT from Trypanosoma cruzi. Chem. Biol. 2000, 7, 957–968. [Google Scholar] [CrossRef] [Green Version]
- Fernández, D.; Wenck, M.A.; Craig, S.P.; Delfino, J.M. The purine transferase from Trypanosoma cruzi as a potential target for bisphosphonate-based chemotherapeutic compounds. Bioorg. Med. Chem. Lett. 2004, 14, 4501–4504. [Google Scholar] [CrossRef]
- Urbina, J.A.; Docampo, R. Specific chemotherapy of Chagas disease: Controversies and advances. Trends Parasitol. 2003, 19, 495–501. [Google Scholar] [CrossRef]
- Eakin, A.E.; Guerra, A.; Focia, P.J.; Torres-Martinez, J.; Craig, S.P. Hypoxanthine phosphoribosyltransferase from Trypanosoma cruzi as a target for structure-based inhibitor design: Crystallization and inhibition studies with purine analogs. Antimicrob. Agents Chemother. 1997, 41, 1686–1692. [Google Scholar] [CrossRef] [Green Version]
- Lawton, P. Purine analogues as antiparasitic agents. Expert Opin. Ther. Pat. 2005, 15, 987–994. [Google Scholar] [CrossRef]
- Singh, K.; Joshi, P.; Mahar, R.; Baranwal, P.; Shukla, S.K.; Tripathi, R.; Tripathi, R.P. Synthesis and antiplasmodial activity of purine-based C-nucleoside analogues. Med. Chem. Commun. 2018, 9, 1232–1238. [Google Scholar] [CrossRef]
- Hulpia, F.; Van Hecke, K.; França Da Silva, C.; da Gama Jaen Batista, D.; Maes, L.; Caljon, G.; de Nazaré, C.; Soeiro, M.; Van Calenbergh, S. Discovery of novel 7-aryl 7-deazapurine 3′-deoxy-ribofuranosyl nucleosides with potent activity against Trypanosoma cruzi. J. Med. Chem. 2018, 61, 9287–9300. [Google Scholar] [CrossRef] [PubMed]
- Cheviet, T.; Wein, S.; Bourchenin, G.; Lagacherie, M.; Périgaud, C.; Cerdan, R.; Peyrottes, S. β-Hydroxy- and β-aminophosphonate acyclonucleosides as potent inhibitors of Plasmodium falciparum growth. J. Med. Chem. 2020, 63, 8069–8087, 8543. [Google Scholar] [CrossRef] [PubMed]
- Tran, H.A.; Zheng, Z.; Wen, X.; Manivannan, S.; Pastor, A.; Kaiser, M.; Brun, R.; Snyder, F.F.; Back, T.G. Synthesis and activity of nucleoside-based antiprotozoan compounds. Bioorg. Med. Chem. 2017, 25, 2091–2104. [Google Scholar] [CrossRef] [PubMed]
- Rassi, A.; Luquetti Ostermayer, A.; Rassi, A., Jr.; Rassi, G.G.; Rasi, S.G.; DA Silva, I.G.; Rassi, A.G. Specific treatment for Trypanosoma cruzi: Lack of efficacy of allopurinol in the human chronic phase of Chagas disease. Am. J. Trop. Med. Hyg. 2007, 76, 58–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pineda de las Infantas y Villatoro, M.J.; Unciti-Broceta, J.D.; Contreras-Montoya, R.; Garcia-Salcedo, J.A.; Gallo Mezo, M.A.; Unciti-Broceta, A.; Diaz-Mochon, J.J. Amide-controlled, one-pot synthesis of tri-substituted purines generates structural diversity and analogues with trypanocidal activity. Sci. Rep. 2015, 5, 9139. [Google Scholar] [CrossRef] [Green Version]
- Lorente-Macías, Á.; Díaz-Mochón, J.J.; Pineda de las Infantas y Villatoro, M.J.; Unciti-Broceta, A. Design and synthesis of 9-dialkylamino-6-[(1H-1,2,3-triazol-4-yl)methoxy]-9H-purines. ChemRxiv 2021, preprint. [Google Scholar] [CrossRef]
- Lorente-Macías, Á.; Benítez-Quesada, M.; Molina, I.J.; Unciti-Broceta, A.; Díaz-Mochón, J.J.; Villatoro, M.J.P.d. 1 H and 13 C assignments of 6-, 8-, 9- substituted purines. Magn. Reson. Chem. 2018, 56, 852–859. [Google Scholar] [CrossRef]
- Martinez-Peinado, N.; Cortes-Serra, N.; Torras-Claveria, L.; Pinazo, M.-J.; Gascon, J.; Bastida, J.; Alonso-Padilla, J. Amaryllidaceae alkaloids with anti-Trypanosoma cruzi activity. Parasit. Vectors 2020, 13, 299. [Google Scholar] [CrossRef]
- Bettiol, E.; Samanovic, M.; Murkin, A.S.; Raper, J.; Buckner, F.; Rodriguez, A. Identification of three classes of heteroaromatic compounds with activity against intracellular Trypanosoma cruzi by chemical library screening. PLoS Negl. Trop. Dis. 2009, 3, e384. [Google Scholar] [CrossRef]
- Peña, I.; Pilar Manzano, M.; Cantizani, J.; Kessler, A.; Alonso-Padilla, J.; Bardera, A.I.; Alvarez, E.; Colmenarejo, G.; Cotillo, I.; Roquero, I.; et al. New compound sets identified from high throughput phenotypic screening against three kinetoplastid parasites: An open resource. Sci. Rep. 2015, 5, 8771. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.M.; Oh, S.J.; Lee, S.Y.; Im, J.H.; Oh, J.M.; Ryu, C.S.; Kwak, H.C.; Lee, J.Y.; Kang, K.W.; Kim, S.K. HepG2 cells as an in vitro model for evaluation of cytochrome P450 induction by xenobiotics. Arch Pharm Res 2015, 38, 691–704. [Google Scholar] [CrossRef]
- Crouch, S.P.M.; Kozlowski, R.; Slater, K.J.; Fletcher, J. The use of ATP bioluminescence as a measure of cell proliferation and cytotoxicity. J. Immunol. Methods 1993, 160, 81–88. [Google Scholar] [CrossRef]
- Lorente-Macías, Á. Design, Synthesis and Biological Evaluation of 6-Alkoxypurine Derivatives as Kinase Inhibitors. Ph.D. Thesis, University of Granada, Granada, Spain, 2019. [Google Scholar]
- Harmse, L.; Van Zyl, R.; Gray, N.; Schultz, P.; Leclerc, S.; Meijer, L.; Doerig, C.; Havlik, I. Structure-activity relationships and inhibitory effects of various purine derivatives on the in vitro growth of Plasmodium falciparum. Biochem. Pharmacol. 2001, 62, 341–348. [Google Scholar] [CrossRef]
- Chu, X.M.; Wang, C.; Wang, W.L.; Liang, L.L.; Liu, W.; Gong, K.K.; Sun, K.L. Triazole derivatives and their antiplasmodial and antimalarial activities. Eur. J. Med. Chem. 2019, 166, 206–223. [Google Scholar] [CrossRef] [PubMed]
- Leite, D.I.; Fontes, F.d.V.; Bastos, M.M.; Hoelz, L.V.B.; Bianco, M.D.C.A.D.; de Oliveira, A.P.; da Silva, P.B.; da Silva, C.F.; Batista, D.D.G.J.; da Gama, A.N.S.; et al. New 1,2,3-triazole-based analogues of benznidazole for use against Trypanosoma cruzi infection: In vitro and in vivo evaluations. Chem. Biol. Drug Des. 2018, 92, 1670–1682. [Google Scholar] [CrossRef] [PubMed]
- Faria, R.X.; Gonzaga, D.T.G.; Pacheco, P.A.F.; Souza, A.L.A.; Ferreira, V.F.; da Silva, F.C. Searching for new drugs for Chagas diseases: Triazole analogs display high in vitro activity against Trypanosoma cruzi and low toxicity toward mammalian cells. J. Bioenerg. Biomembr. 2018, 50, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Focia, P.J.; Craig, S.P.; Eakin, A.E. Approaching the transition state in the crystal structure of a phosphoribosyltransferase. Biochemistry 1998, 37, 17120–17127. [Google Scholar] [CrossRef] [PubMed]
- Ferreira de Freitas, R.; Schapira, M. A systematic analysis of atomic protein-ligand interactions in the PDB. Med. Chem. Commun. 2017, 8, 1970–1981. [Google Scholar] [CrossRef] [Green Version]
- Li, C.M.; Tyler, P.C.; Furneaux, R.H.; Kicska, G.; Xu, Y.; Grubmeyer, C.; Girvin, M.E.; Schramm, V.L. Transition-state analogs as inhibitors of human and malarial hypoxanthine-guanine phosphoribosyltransferases. Nat. Struct. Biol. 1999, 6, 582–587. [Google Scholar] [CrossRef]
- Evans, G.B.; Tyler, P.C.; Schramm, V.L. Immucillins in infectious diseases. AC Infect. Dis. 2018, 4, 107–117. [Google Scholar] [CrossRef]
- Cassera, M.B.; Hazleton, K.Z.; Merino, E.F.; Obaldia, N.; Ho, M.C.; Murkin, A.S.; DePinto, R.; Gutierrez, J.A.; Almo, S.C.; Evans, G.B.; et al. Plasmodium falciparum parasites are killed by a transition state analogue of purine nucleoside phosphorylase in a primate animal model. PLoS ONE 2011, 6, 26916. [Google Scholar] [CrossRef] [Green Version]
- Krečmerová, M.; Dračínský, M.; Hocková, D.; Holý, A.; Keough, D.T.; Guddat, L.W. Synthesis of purine N 9-[2-hydroxy-3-O-(phosphonomethoxy)propyl] derivatives and their side-chain modified analogs as potential antimalarial agents. Bioorg. Med. Chem. 2012, 20, 1222–1230. [Google Scholar] [CrossRef]
- Eads, J.C.; Scapin, G.; Xu, Y.; Grubmeyer, C.; Sacchettini, J.C. The crystal structure of human hypoxanthine-guanine phosphoribosyltransferase with bound GMP. Cell 1994, 78, 325–334. [Google Scholar] [CrossRef]
- De las Infantas, M.J.P.; Torres-Rusillo, S.; Unciti-Broceta, J.D.; Fernandez-Rubio, P.; Luque-Gonzalez, M.A.; Gallo, M.A.; Unciti-Broceta, A.; Molina, I.J.; Diaz-Mochon, J.J. Synthesis of 6,8,9 poly-substituted purine analogue libraries as pro-apoptotic inducers of human leukemic lymphocytes and dapk-1 inhibitors. Org. Biomol. Chem. 2015, 13, 5224–5234. [Google Scholar] [CrossRef] [Green Version]
- Lorente-Macías, Á.; Iañez, I.; Jiménez-López, M.C.; Benítez-Quesada, M.; Torres-Rusillo, S.; Díaz-Mochón, J.J.; Molina, I.J.; Pineda de las Infantas, M.J. Synthesis and screening of 6-alkoxy purine analogs as cell type-selective apoptotic inducers in Jurkat cells. Arch. Pharm. 2021, e2100095. [Google Scholar] [CrossRef]
- Trager, W.; Jensen, J.B. Human malaria parasites in continuous culture. Science 1976, 193, 673–675. [Google Scholar] [CrossRef]
- Lambros, C.; Vanderberg, J. Synchronization of Plasmodium falciparum erythrocytic stages in culture. J. Parasitol. 1979, 65, 418–420, 593. [Google Scholar] [CrossRef]
- Buckner, F.S.; Verlinde, C.L.M.J.; La Flamme, A.C.; Van Voorhis, W.C. Efficient technique for screening drugs for activity against Trypanosoma cruzi using parasites expressing β-galactosidase. Antimicrob. Agents Chemother. 1996, 40, 2592–2597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.H.; Chung, T.D.; Oldenburg, K.R. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 1999, 4, 67–73. [Google Scholar] [CrossRef]
- Martínez-Flórez, A.; Galizzi, M.; Izquierdo, L.; Bustamante, J.M.; Rodriguez, A.; Rodriguez, F.; Rodriguez-Cortes, A.; Alberola, J. Repurposing bioenergetic modulators against protozoan parasites responsible for tropical diseases. Int. J. Parasitol. Drugs Drug Resist. 2020, 14, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Peinado, N.; Martori, C.; Cortes-Serra, N.; Sherman, J.; Rodriguez, A.; Gascon, J.; Alberola, J.; Pinazo, M.J.; Rodriguez-Cortes, A.; Alonso-Padilla, J. Anti-Trypanosoma cruzi activity of metabolism modifier compounds. Int. J. Mol. Sci. 2021, 22, 688. [Google Scholar] [CrossRef]
- Warrenfeltz, S.; Basenko, E.Y.; Crouch, K.; Harb, O.S.; Kissinger, J.C.; Roos, D.S.; Shanmugasundram, A.; Silva-Franco, F. EuPathDB: The Eukaryotic Pathogen Genomics Database Resource. Methods Mol. Biol. 2018, 1757, 69–113. [Google Scholar] [CrossRef] [Green Version]
- Aurrecoechea, C.; Brestelli, J.; Brunk, B.P.; Dommer, J.; Fischer, S.; Gajria, B.; Gao, X.; Gingle, A.; Grant, G.; Harb, O.S.; et al. PlasmoDB: A functional genomic database for malaria parasites. Nucleic Acids Res. 2009, 37, D539–D543. [Google Scholar] [CrossRef] [Green Version]
- Aslett, M.; Aurrecoechea, C.; Berriman, M.; Brestelli, J.; Brunk, B.P.; Carrington, M.; Depledge, D.P.; Fischer, S.; Gajria, B.; Gao, X.; et al. TriTrypDB: A functional genomic resource for the Trypanosomatidae. Nucleic Acids Res. 2010, 38, D457–D462. [Google Scholar] [CrossRef] [Green Version]
- NCBI Resource Coordinators. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2018, 46, D8–D13. [Google Scholar] [CrossRef] [Green Version]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Šali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Fiser, A.; Šali, A. MODELLER: Generation and refinement of homology-based protein structure models. Methods Enzymol. 2003, 374, 461–491. [Google Scholar] [CrossRef]
- John, B.; Šali, A. Comparative protein structure modeling by iterative alignment, model building and model assessment. Nucleic Acids Res. 2003, 31, 3982–3992. [Google Scholar] [CrossRef]
- Shen, M.; Šali, A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006, 15, 2507–2524. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Wallace, A.C.; Laskowski, R.A.; Thornton, J.M. LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Prot. Eng. 1995, 8, 127–134. [Google Scholar] [CrossRef]
- DeLano, W.L. The PyMOL Molecular Graphics System; DeLano Scientific LLC: San Carlos, CA, USA, 2002; Available online: www.pymol.org/ (accessed on 1 June 2021).
- Kyte, J.; Doolittle, R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef] [Green Version]
- Jurrus, E.; Engel, D.; Star, K.; Monson, K.; Brandi, J.; Felberg, L.E.; Brookes, D.H.; Wilson, L.; Chen, J.; Liles, K.; et al. Improvements to the APBS biomolecular solvation software suite. Protein Sci. 2018, 27, 112–128. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parasite Growth Rate Relative to the Assay Negative Control | ||||||
|---|---|---|---|---|---|---|
| Compound Concentration (µM) | P. falciparum | T. cruzi | ||||
| <30% | 30–50% | >50% | <30% | 30–50% | >50% | |
| 100 | 10 (12.3) * | 10 (12.3) | 61 (75.3) | 28 (33.3) | 8 (9.8) | 45 (56.8) |
| 10 | 1 (1.2) | 0 (0) | 80 (98.8) | 2 (2.4) | 2 (2.4) | 77 (95.0) |
| 1 | 0 (0) | 0 (0) | 81 (100) | 0 (0) | 0 (0) | 81 (100) |
| P. falciparum | T. cruzi | Vero Cells | HepG2 Cells | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound | Subgroup | IC50 | IC50 a | IC50 b | TC50 * | SI c | SI a | SI b | TC50 * |
| BNZ | - | NT | 1.72 | 1.82 | 209.5 | - | 121.80 | 115.11 | 255.3 |
| 29 | C1 | NT | 4.86 | 157.3 | >1000 | - | >205.76 | >6.36 | >1000 |
| 6D | C2 | 18.9 | 5.56 | 3.78 | >150 | >7.94 | >26.98 | >39.68 | >200 |
| 6g | C2 | 19.08 | 3.97 | 19.64 | >150 | >7.86 | >37.78 | >7.63 | >300 |
| 25 | D1 | 81.54 | NT | NT | >6000 | >73.58 | - | - | NA |
| 32 | D1 | 69.66 | NT | NT | >1000 | >14.36 | - | - | NA |
| 33 | D1 | 19.19 | NT | NT | >300 | >15.63 | - | - | NA |
| 34 | D1 | NT | 4.12 | 4.24 | >6000 | - | >1456.31 | >1415.10 | NA |
| 59 | D5 | 110.1 | NT | NT | >300 | >2.73 | - | - | NT |
| 60 | D5 | 23.3 | NT | NT | >300 | >12.87 | - | - | NA |
| 70 | D6 | NT | 13.72 | 29.08 | >50 | - | >3.64 | >1.72 | NT |
| 76 | D6 | 18.27 | NT | NT | >500 | >27.37 | - | - | NA |
| 84 | D6 | 28.01 | NT | NT | >70 | >2.50 | - | - | NT |
| 88 | D7 | 65.35 | 12.72 | 35.43 | >180 | >2.75 | >14.15 | >5.08 | NA |
| 94 | D7 | NT | 0.92 | 75.11 | >60 | - | >63.16 | >0.79 | NA |
| 93 | E3 | NT | 1.42 | 218.2 | >2000 | - | >1408.45 | >9.17 | NA |
| Enzyme | Natural Ligand | Energy Binding with Natural Ligand (Kcal/mol) | Energy Binding with Compound 33 (Kcal/mol) | Energy Binding with Compound 76 (Kcal/mol) | Energy Difference with 33 (Kcal/mol) | Energy Difference with 76 (Kcal/mol) |
|---|---|---|---|---|---|---|
| PfHGXPRT (XP_001347406.1) | Xanthine | −6.80 (0.00) | −9.91 (0.08) | −10.30 (0.02) | −3.11 | −3.50 |
| Hypoxanthine | −5.73 (0.26) | −4.18 | −4.58 | |||
| Guanine | −6.65 (0.05) | −3.26 | −3.65 | |||
| PfADA (XP_001347573.1) | Adenosine | −7.10 (0.00) | −9.05 (0.05) | −9.22 (0.09) | −2.12 | −1.95 |
| PfGMPS (XP_001347408.1) | XMP | −6.63 (0.12) | −8.19 (0.19) | −8.51 (0.06) | −1.56 | −1.88 |
| PfPNP (XP_001351690.1) | Inosine | −7.13 (0.04) | −8.51 (0.14) | −8.83 (0.46) | −1.38 | −1.71 |
| PfADSL (XP_001349577.1) | SAICAR | −8.10 (0.13) | −9.27 (0.11) | −9.15 (0.06) | −1.17 | −1.05 |
| Adenylosuccinate | −8.41 (0.12) | −0.86 | −0.74 | |||
| PfADSS (XP_001350257.1) | IMP | −9.49 (0.03) | −9.62 (1.02) | −8.51 (0.31) | −0.13 | 0.98 |
| Enzyme | Natural Ligand | Energy Binding with Natural Ligand (Kcal/mol) | Energy Binding with Compound 6D (Kcal/mol) | Energy Binding with Compound 34 (Kcal/mol) | Energy Difference with 6D (Kcal/mol) | Energy Difference with 34 (Kcal/mol) |
|---|---|---|---|---|---|---|
| TcHGPRT (XP_813396.1) | Hypoxanthine | −4.80 0.00) | −7.92 (0.04) | −8.64 (0.13) | −3.12 | −3.84 |
| Guanine | −5.49 (0.03) | −2.43 | −3.15 | |||
| TcAPRT (XP_818435.1) | Adenine | −5.40 (0.02) | −7.64 (0.34) | −8.37 (0.36) | −2.23 | −2.97 |
| TcAK (XP_820251.1) | Adenosine | −7.24 (0.10) | −9.20 (0.00) | −9.72 (0.09) | −1.96 | −2.48 |
| TcIGNH (XP_818171.1) | Inosine | −7.62 (0.09) | −9.10 (0.02) | −9.25 (0.11) | −1.48 | −1.63 |
| Guanosine | −8.22 (0.20) | −0.88 | −1.03 | |||
| TcIMPDH (XP_805772.1) | IMP | −7.99 (0.03) | −8.19 (0.04) | −9.12 (0.24) | −0.20 | −1.13 |
| TcIAGNH (XP_804829.1) | Inosine | −9.40 (0.00) | −8.92 (0.13) | −10.09 (0.17) | 0.48 | −0.69 |
| Guanosine | −9.33 (0.27) | 0.41 | −0.76 | |||
| Adenosine | −8.85 (0.05) | −0.07 | −1.21 | |||
| TcADSL (XP_811726.1) | SAICAR | −9.26 (0.08) | −8.50 (0.00) | −10.08 (0.32) | 0.76 | −0.82 |
| Adenylosuccinate | −9.45 (0.06) | 0.95 | −0.63 |
| Group | Scaffold | Sub-Group | Chemical Name | Number of Compounds |
|---|---|---|---|---|
| A | 6-Alkoxy-purines | A1 | 6-Ethoxy-8-(H or methyl)-9-substituted-9H-purines | 4 |
| A2 | 8-(H or Methyl)-6-isopropoxy-9-isopropyl-9H-purines | 2 | ||
| B | 6-Tert-butyl amino purines | - | 9-tert-Butyl-6-(tert-butylamino)-8-substituted-9H-purines | 3 |
| C | 6-Benzoxy-purines | C1 | 6-Benzoxy-9-substituted-9H-purines | 4 |
| C2 | 6-Benzoxy-9-substituted-8-(methyl or phenyl)-9H-purines | 10 | ||
| D | 6-{[1-(substituted)-1H-1,2,3-triazol-4-yl]methoxy}-purines and pyrimidine analogs | D1 | 6-{[1-(substituted)-1H-1,2,3-triazol-4-yl]methoxy}-9-(dimethylamino)-8-phenyl-9H-purines | 7 |
| D2 | 8-Methyl-9-(dimethylamino)-6-{[1-(substituted)-1H-1,2,3-triazol-4-yl]methoxy}-9H-purines. | 7 | ||
| D3 | 6-{[1-(substituted)-1H-1,2,3-triazol-4-yl]methoxy}-9-(dimethylamino)-9H-purines | 7 | ||
| D4 | 6-{[1-(substituted)-1H-1,2,3-triazol-4-yl]methoxy}-4-(2,2-dimethylhydrazinyl)-5-nitropyrimidines | 7 | ||
| D5 | 5-Amino-4-(2,2-dimethylhydrazinyl)-6-{[1-(substituted)-1H-1,2,3-triazol-4-yl]methoxy}pyrimidines | 7 | ||
| D6 | 9-(Alkyl or Dialkylamino)-8-phenyl-6-{[1-(thiazol-4-ylmethyl)-1H-1,2,3-triazol-4-yl]methoxy}-9H-purines | 5 | ||
| D7 | 9-(Alkyl or Dialkylamino)-6-{[1-(3-methoxyphenyl)-1H-1,2,3-triazol-4-yl]methoxy}-9H-purines | 5 | ||
| E | 6-Propargyloxy-purines and pyrimidine analogs | E1 | 9-(N,N-dimethyl)-6-propargyloxy-8-substituted-9H-purines and pyrimidine analogs | 5 |
| E2 | 9-(Alkyl or Dialkylamino)-8-phenyl-6-propargyloxy-9H-purines | 4 | ||
| E3 | 9-(Alkyl or Dialkylamino)-6-propargyloxy-9H-purines | 4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martinez-Peinado, N.; Lorente-Macías, Á.; García-Salguero, A.; Cortes-Serra, N.; Fenollar-Collado, Á.; Ros-Lucas, A.; Gascon, J.; Pinazo, M.-J.; Molina, I.J.; Unciti-Broceta, A.; et al. Novel Purine Chemotypes with Activity against Plasmodium falciparum and Trypanosoma cruzi. Pharmaceuticals 2021, 14, 638. https://doi.org/10.3390/ph14070638
Martinez-Peinado N, Lorente-Macías Á, García-Salguero A, Cortes-Serra N, Fenollar-Collado Á, Ros-Lucas A, Gascon J, Pinazo M-J, Molina IJ, Unciti-Broceta A, et al. Novel Purine Chemotypes with Activity against Plasmodium falciparum and Trypanosoma cruzi. Pharmaceuticals. 2021; 14(7):638. https://doi.org/10.3390/ph14070638
Chicago/Turabian StyleMartinez-Peinado, Nieves, Álvaro Lorente-Macías, Alejandro García-Salguero, Nuria Cortes-Serra, Ángel Fenollar-Collado, Albert Ros-Lucas, Joaquim Gascon, Maria-Jesus Pinazo, Ignacio J. Molina, Asier Unciti-Broceta, and et al. 2021. "Novel Purine Chemotypes with Activity against Plasmodium falciparum and Trypanosoma cruzi" Pharmaceuticals 14, no. 7: 638. https://doi.org/10.3390/ph14070638