


Genes 2024, 15(5), 558; https://doi.org/10.3390/genes15050558 (registering DOI) - 27 Apr 2024
Abstract
Uterine leiomyomas (ULs) are the most common benign tumor of the uterus. They can be associated with symptoms including abnormal uterine bleeding, pelvic pain, urinary frequency, and pregnancy complications. Despite the high prevalence of UL, its underlying pathophysiology mechanisms have historically been poorly
[...] Read more.
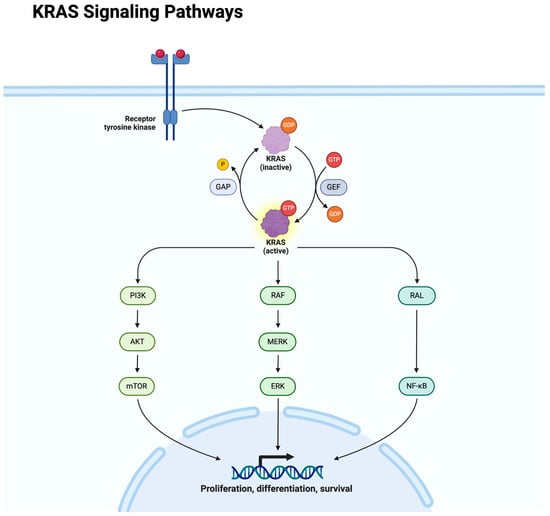
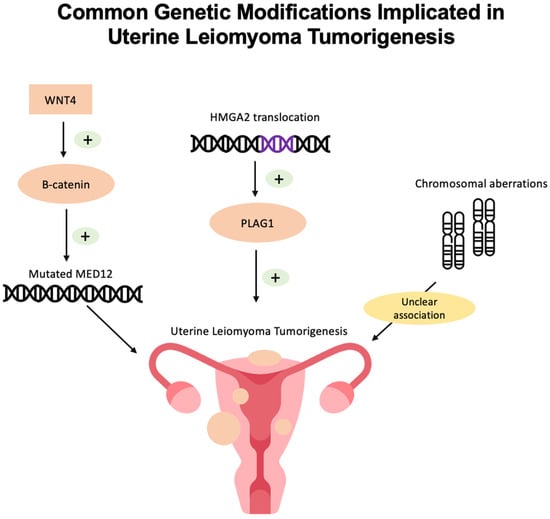
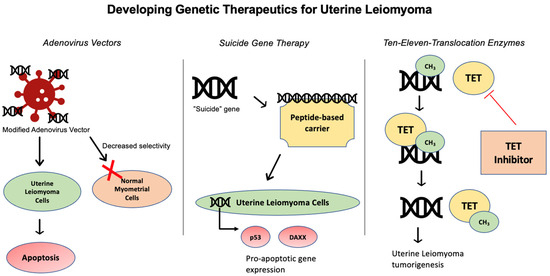
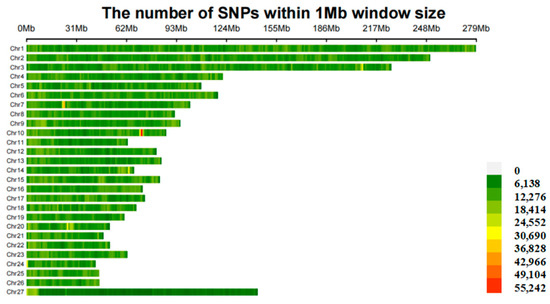
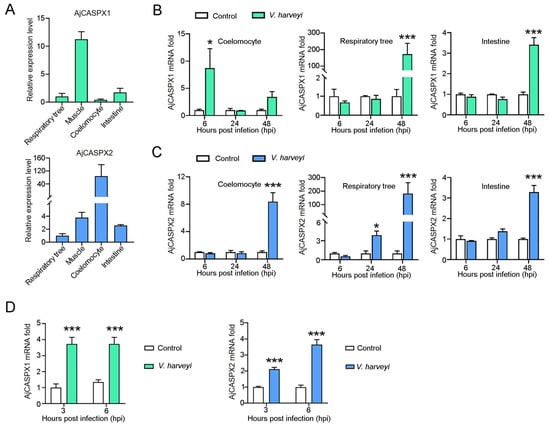
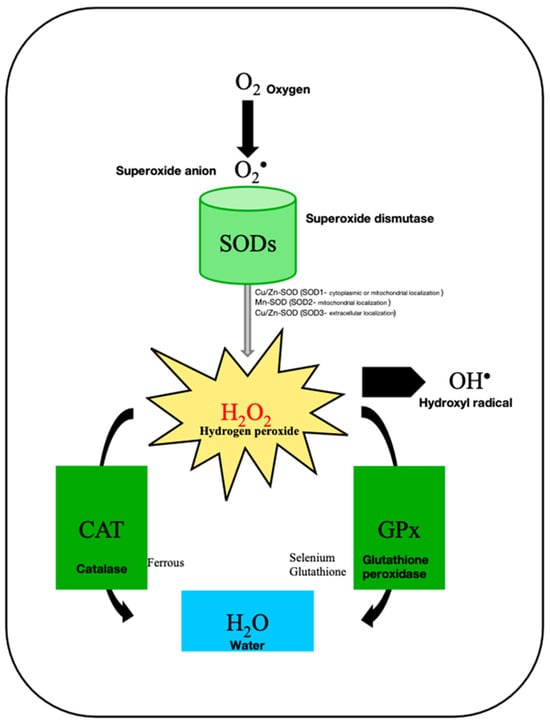
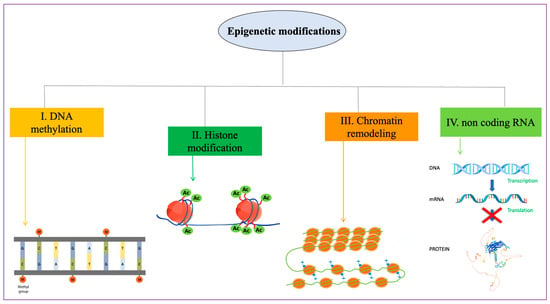
Uterine leiomyomas (ULs) are the most common benign tumor of the uterus. They can be associated with symptoms including abnormal uterine bleeding, pelvic pain, urinary frequency, and pregnancy complications. Despite the high prevalence of UL, its underlying pathophysiology mechanisms have historically been poorly understood. Several mechanisms of pathogenesis have been suggested, implicating various genes, growth factors, cytokines, chemokines, and microRNA aberrations. The purpose of this study is to summarize the current research on the relationship of genetics with UL. Specifically, we performed a literature review of published studies to identify how genetic aberrations drive pathophysiology, epidemiology, and therapeutic approaches of UL. With regards to pathophysiology, research has identified MED12 mutations, HMGA2 overexpression, fumarate hydratase deficiency, and cytogenetic abnormalities as contributors to the development of UL. Additionally, epigenetic modifications, such as histone acetylation and DNA methylation, have been identified as contributing to UL tumorigenesis. Specifically, UL stem cells have been found to contain a unique DNA methylation pattern compared to more differentiated UL cells, suggesting that DNA methylation has a role in tumorigenesis. On a population level, genome-wide association studies (GWASs) and epidemiologic analyses have identified 23 genetic loci associated with younger age at menarche and UL growth. Additionally, various GWASs have investigated genetic loci as potential drivers of racial disparities in UL incidence. For example, decreased expression of Cytohesin 4 in African Americans has been associated with increased UL risk. Recent studies have investigated various therapeutic options, including ten-eleven translocation proteins mediating DNA methylation, adenovirus vectors for drug delivery, and “suicide gene therapy” to induce apoptosis. Overall, improved understanding of the genetic and epigenetic drivers of UL on an individual and population level can propel the discovery of novel therapeutic options.
Full article
(This article belongs to the Special Issue Genetics and Genomics of Female Reproduction)
►
Show Figures

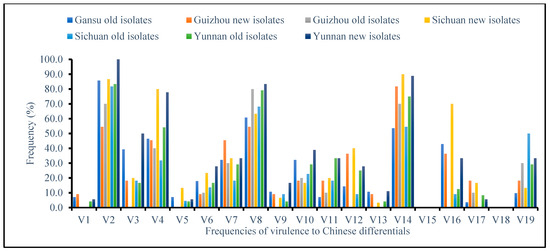
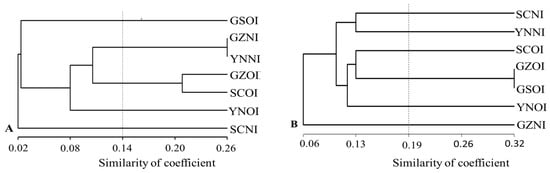
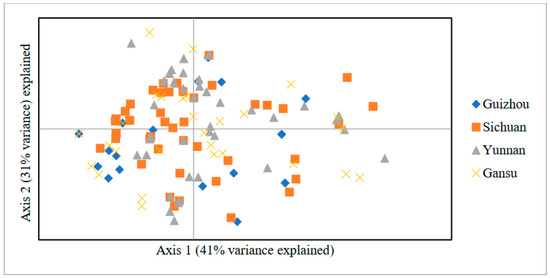
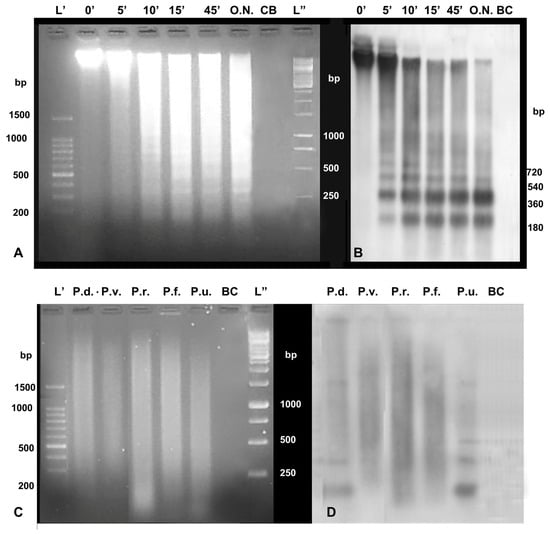
Figure 1







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}