



Genes 2023, 14(9), 1745; https://doi.org/10.3390/genes14091745 - 31 Aug 2023
Viewed by 2856
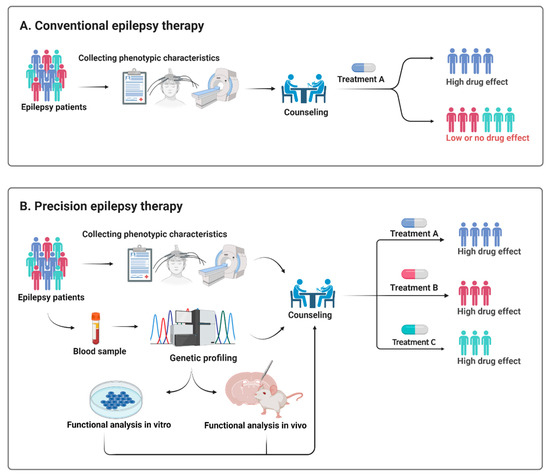
Abstract
►
Show Figures
In 1990, Gorlin et al. described four types of craniofacial duplications: (1) single mouth with duplication of the maxillary arch; (2) supernumerary mouth laterally placed with rudimentary segments; (3) single mouth with replication of the mandibular segments; and (4) true facial duplication, namely
[...] Read more.
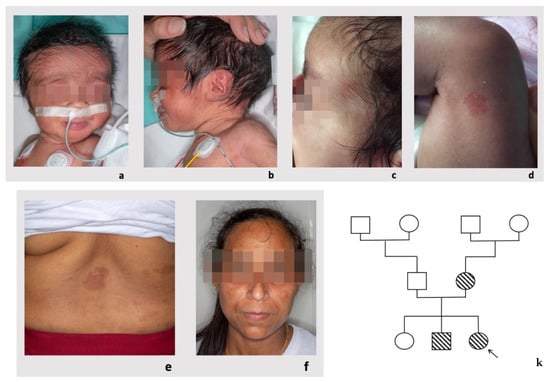
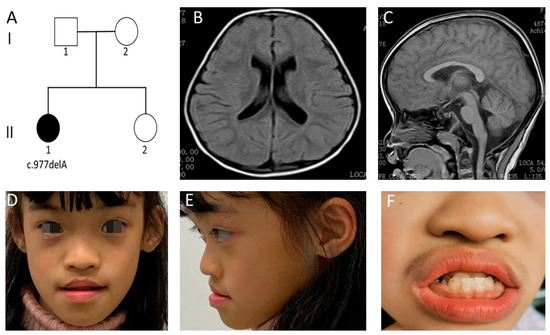
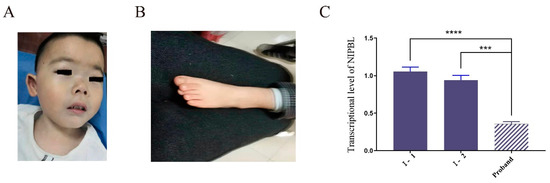
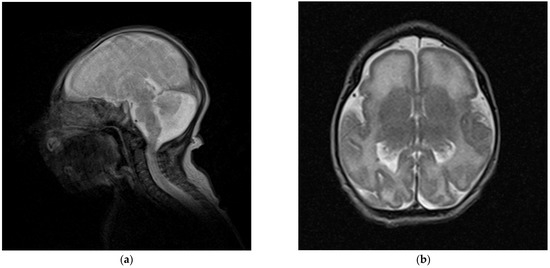
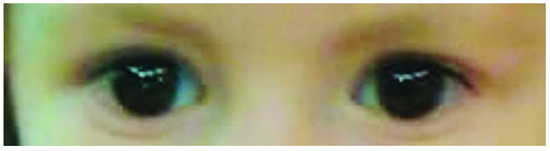
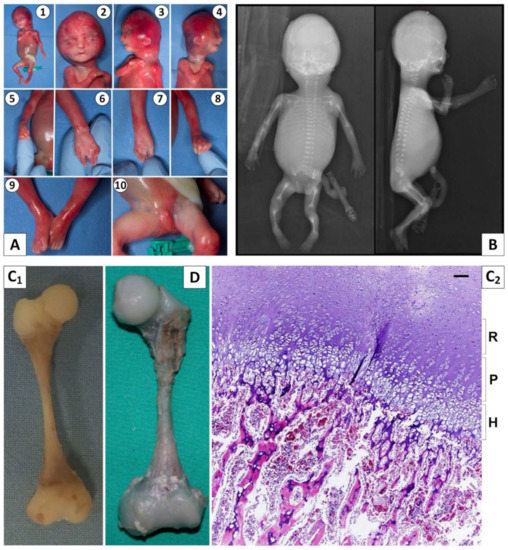
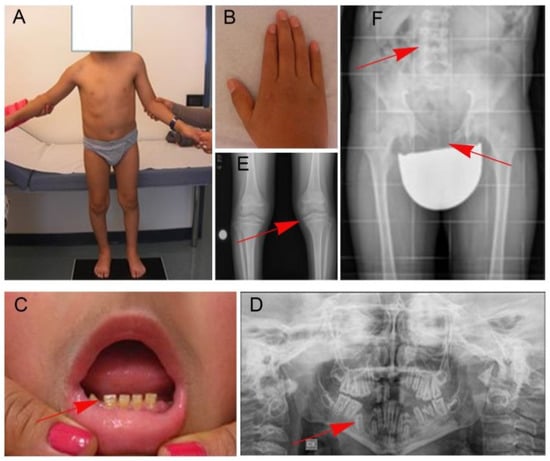
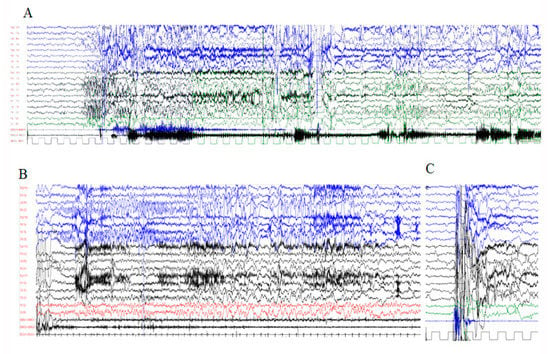
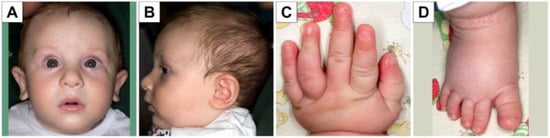
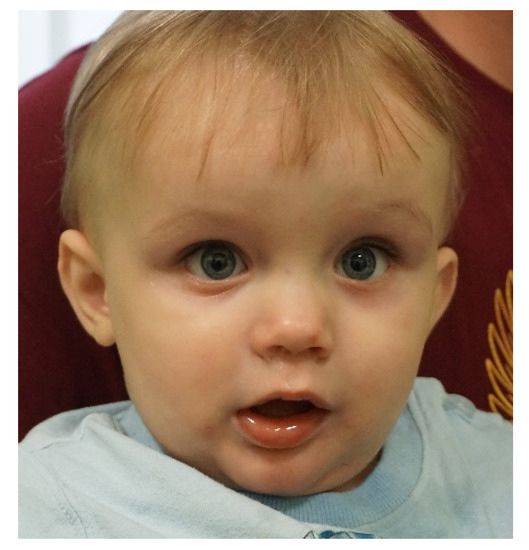
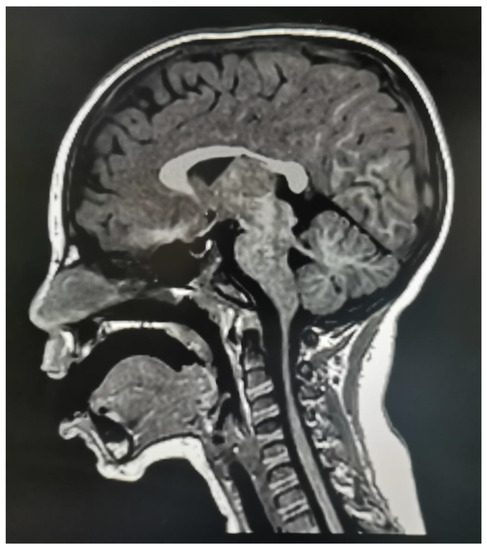
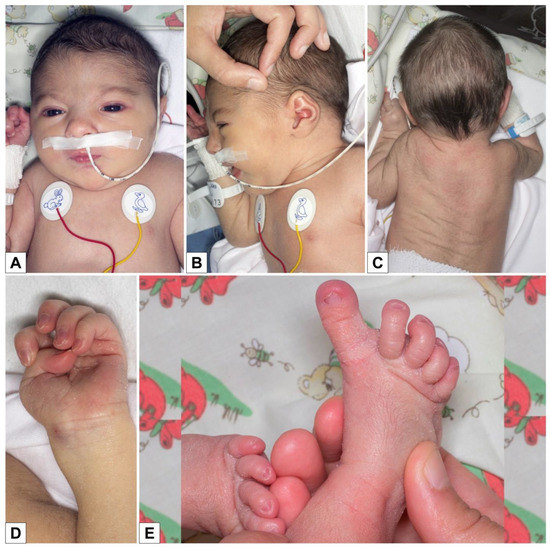
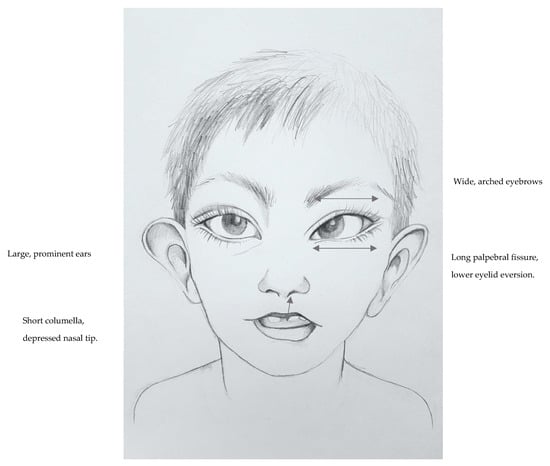
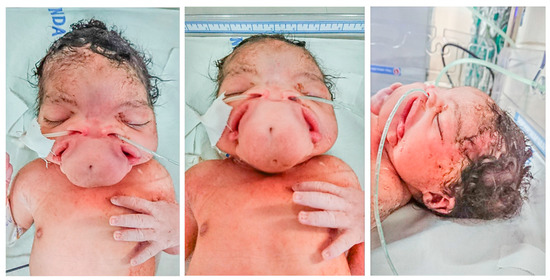
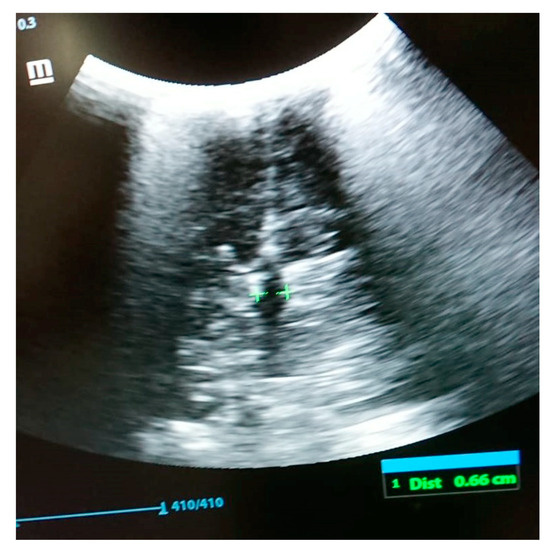
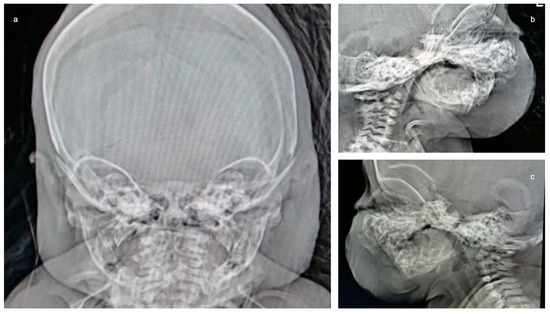
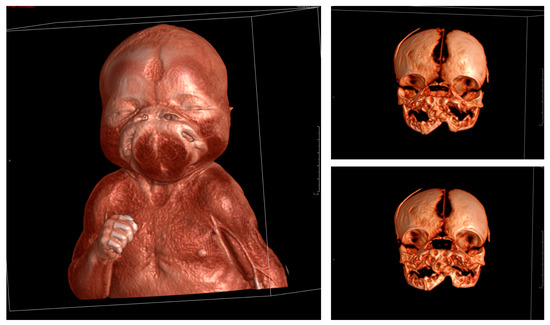
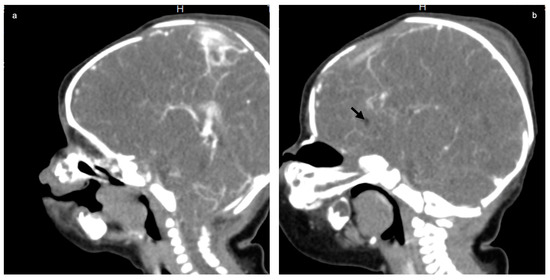
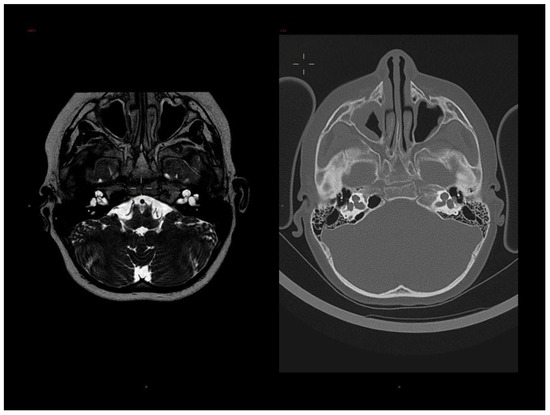
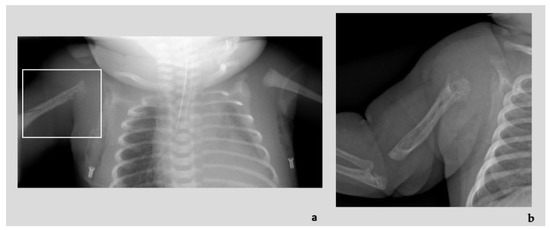
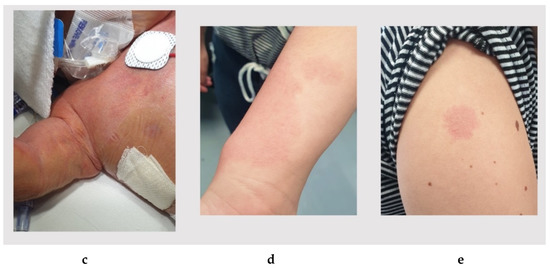
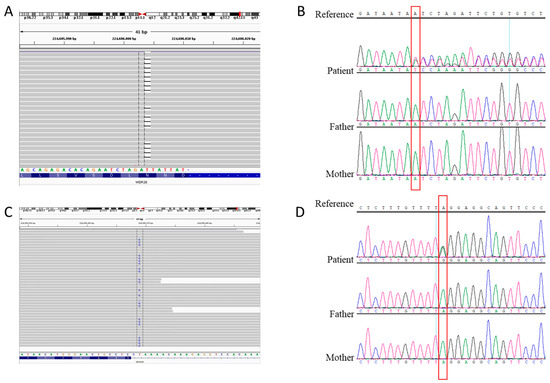
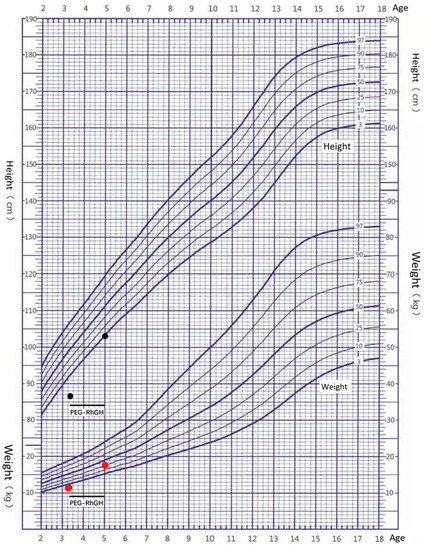
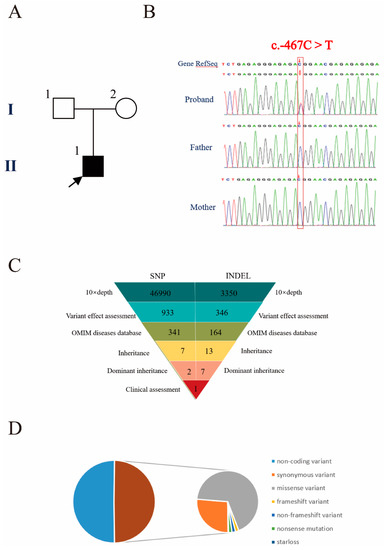
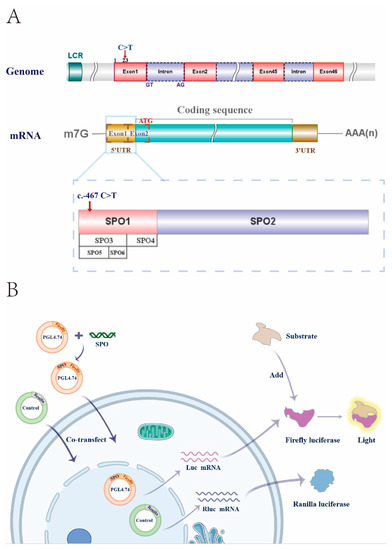
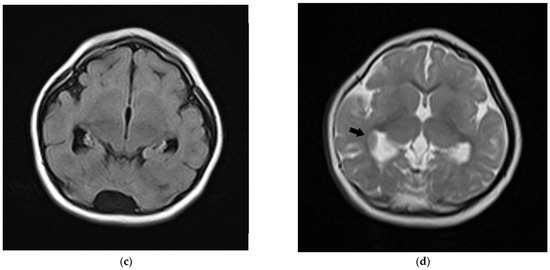
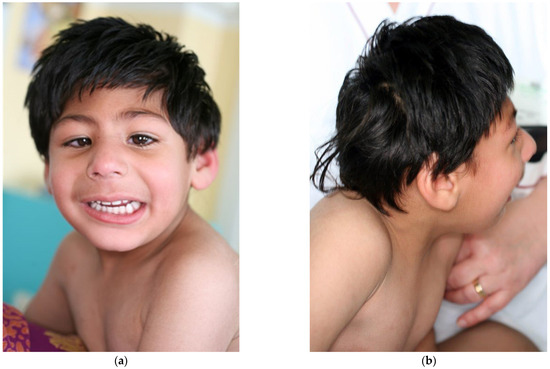
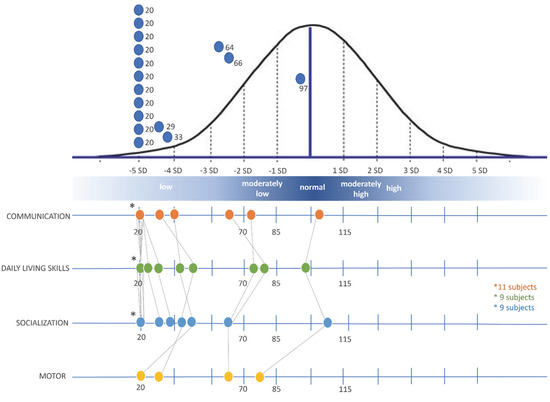
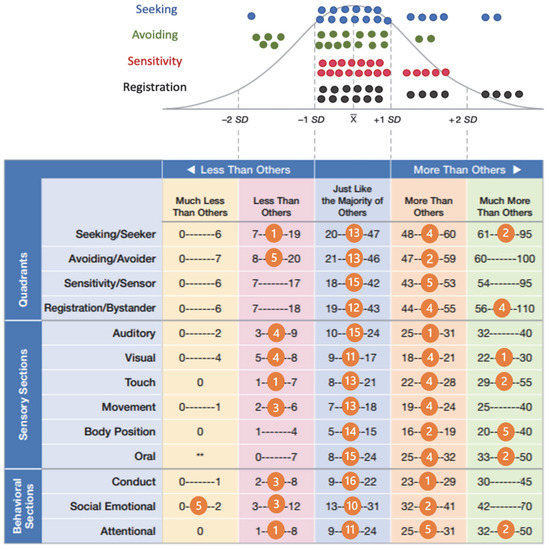
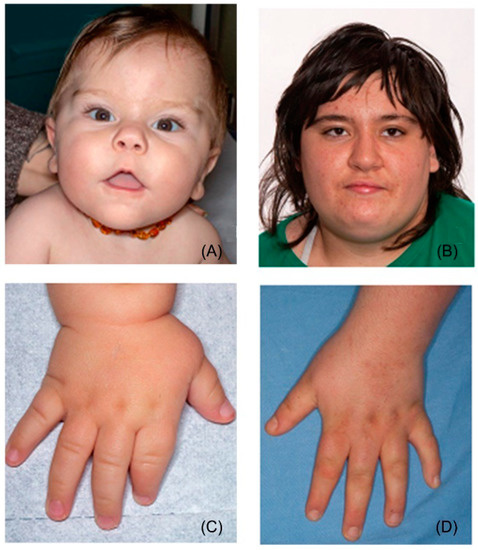
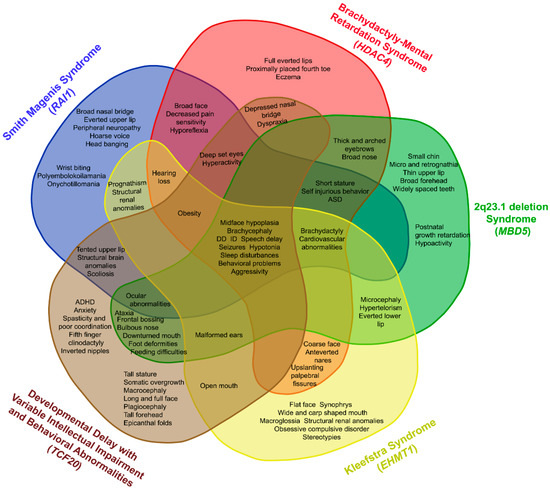
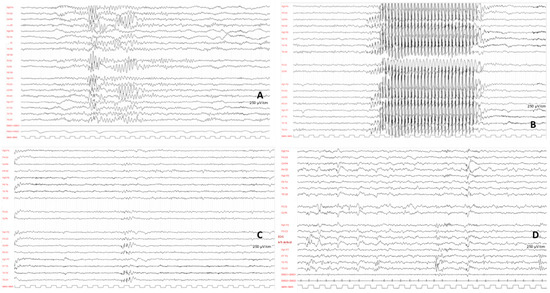
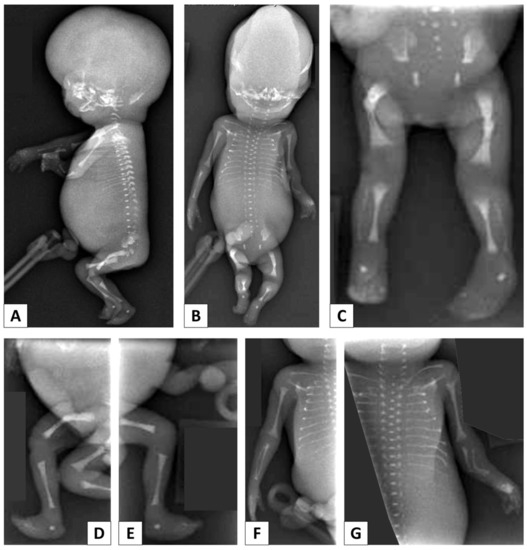
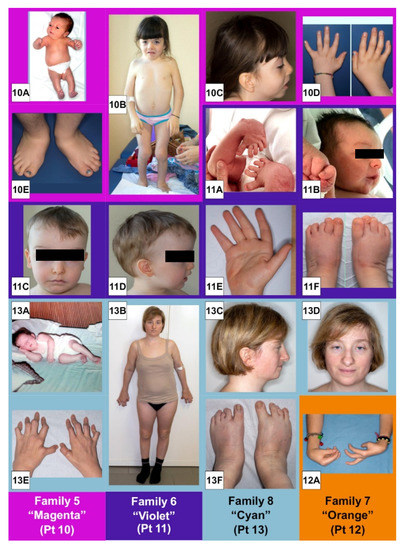
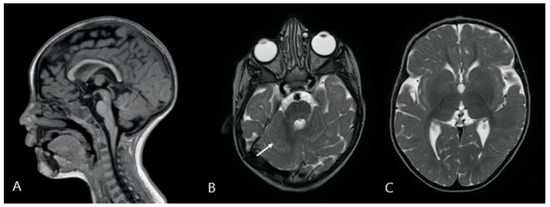
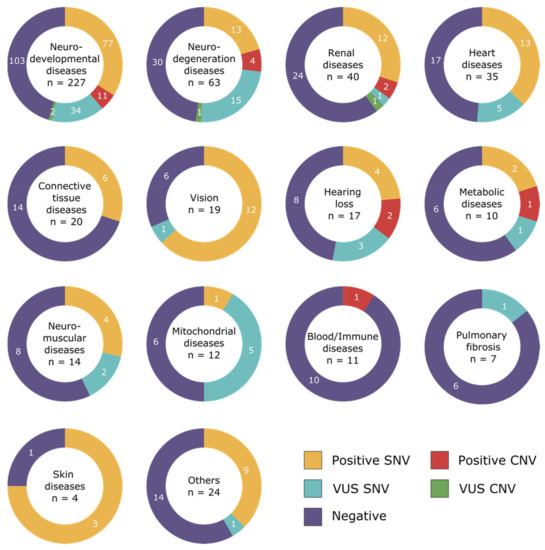
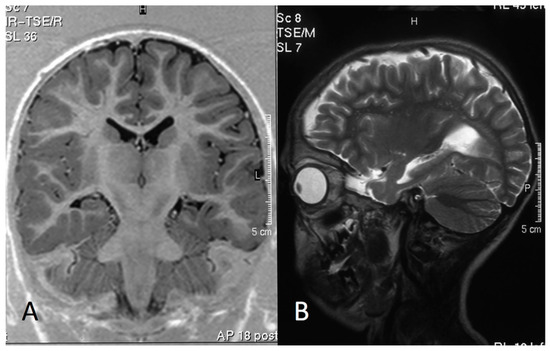
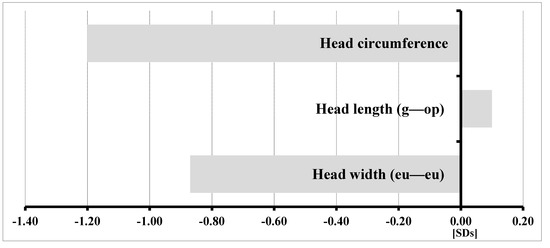
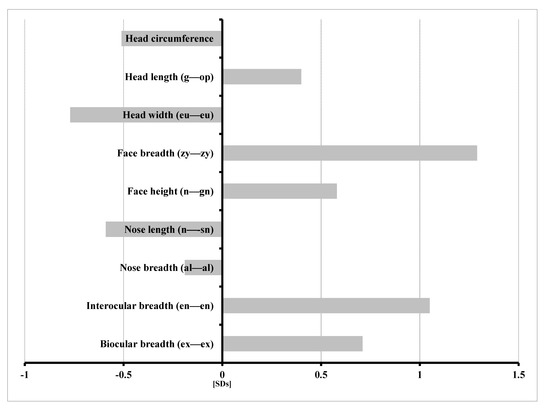
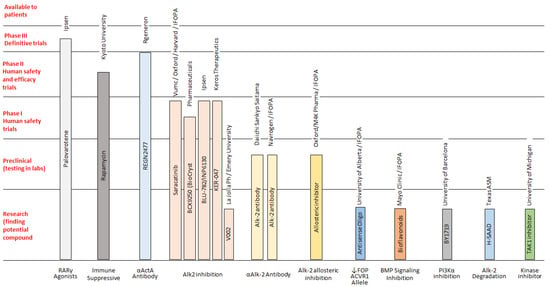
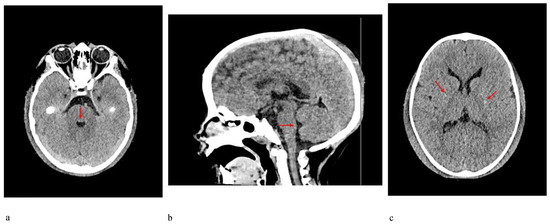
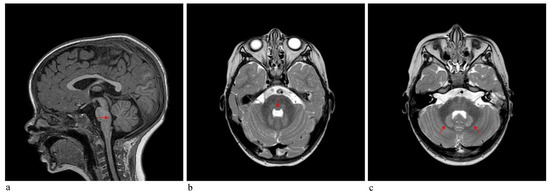
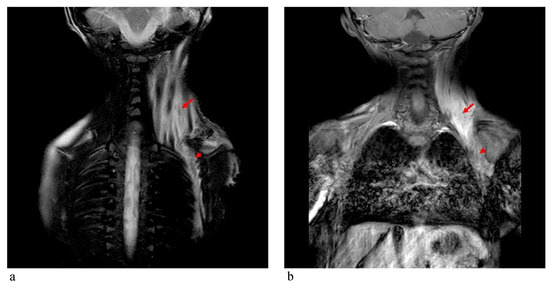
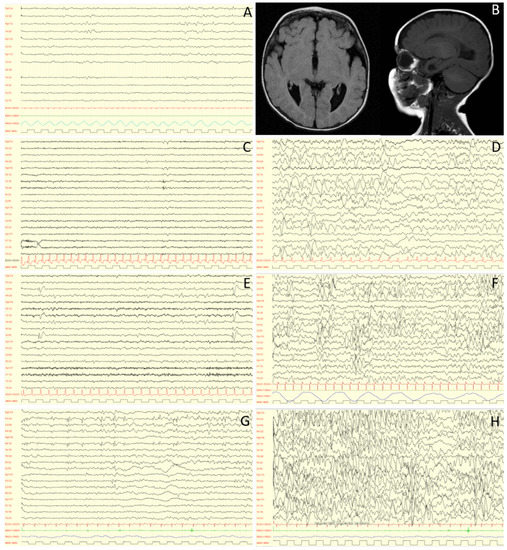
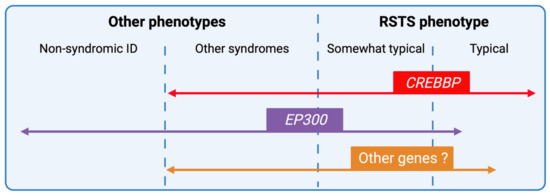
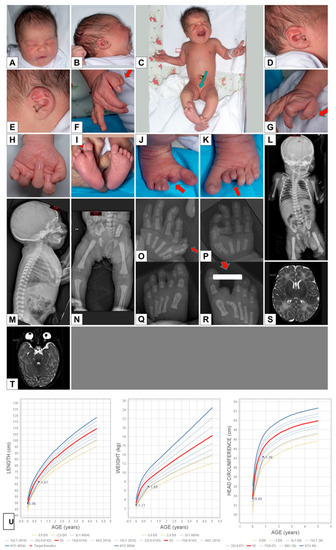
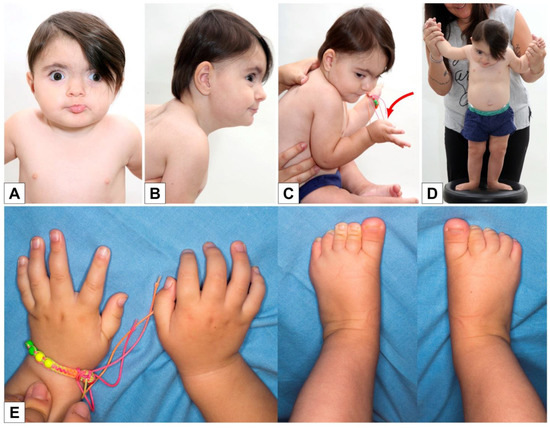
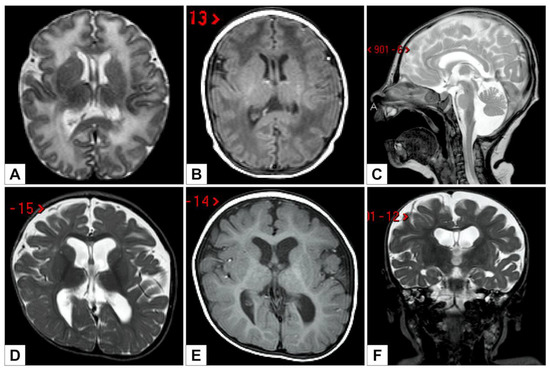
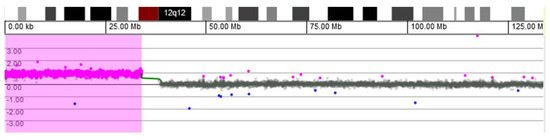
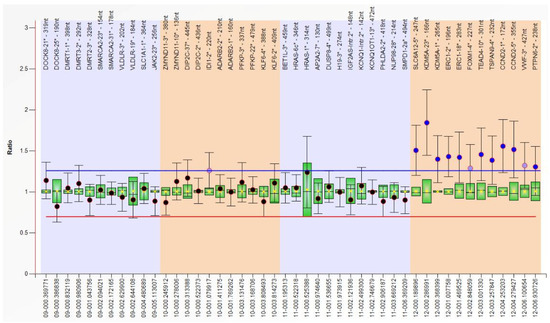
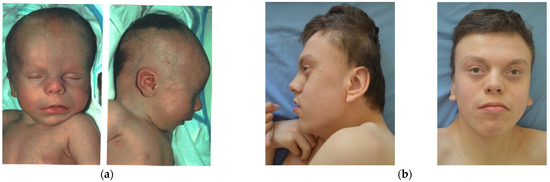
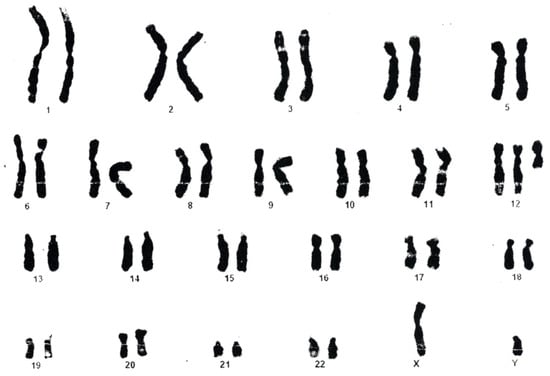
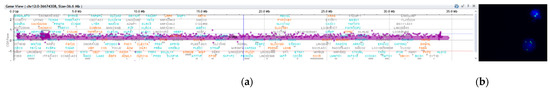
In 1990, Gorlin et al. described four types of craniofacial duplications: (1) single mouth with duplication of the maxillary arch; (2) supernumerary mouth laterally placed with rudimentary segments; (3) single mouth with replication of the mandibular segments; and (4) true facial duplication, namely diprosopus. We describe a newborn born with wide-spaced eyes, a very broad nose, and two separate mouths. Workup revealed the absence of the corpus callosum and the presence of a brain midline lipoma, wide sutures, and a Chiari I malformation with cerebellar herniation. We conducted a systematic review of the literature and compared all the cases described as diprosopus. In 96% of these, the central nervous system is affected, with anencephaly being the most commonly associated abnormality. Other associated anomalies include cardiac malformations (86%), cleft palate (63%), diaphragmatic hernia (13%), and disorder of sex development (DSD) (13%). Although the facial features are those that first strike the eye, the almost obligate presence of cerebral malformations suggests a disruptive event in the cephalic pole of the forming embryo. No major monogenic contribution has been recognized today for this type of malformation.
Full article
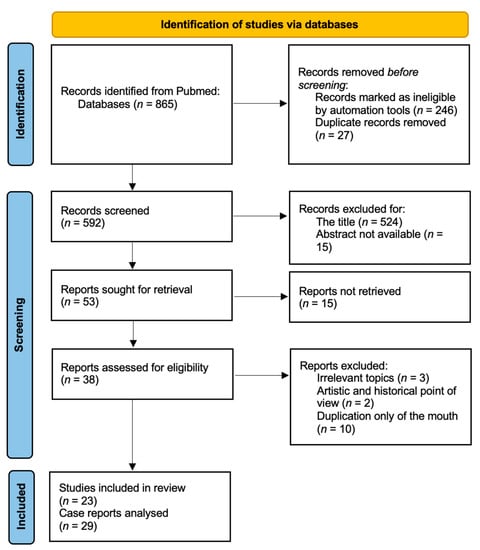
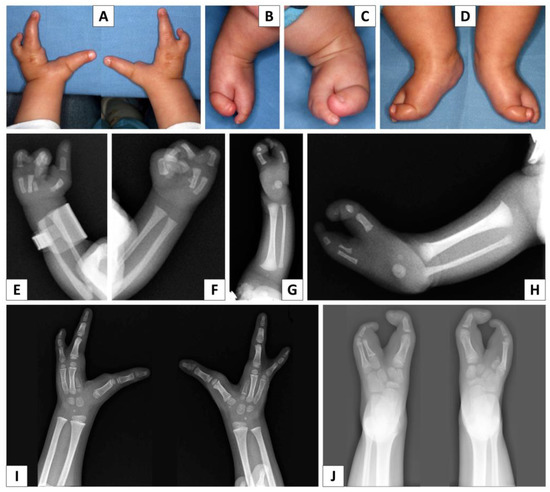
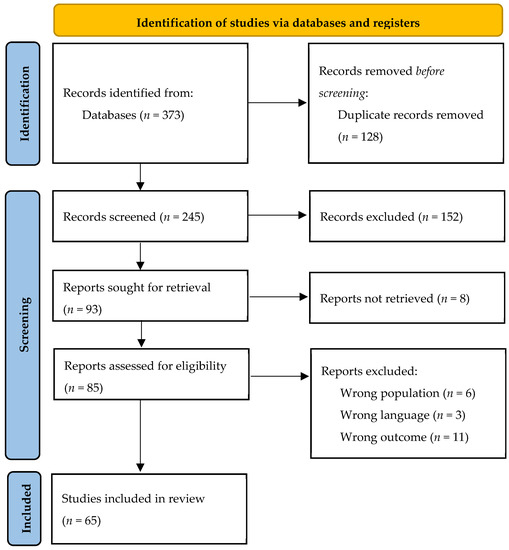
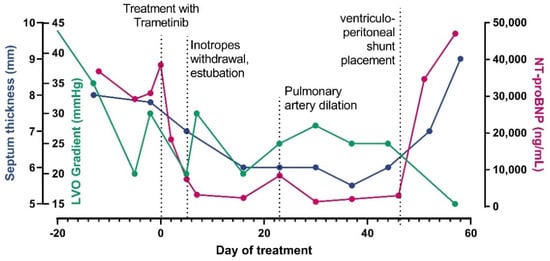
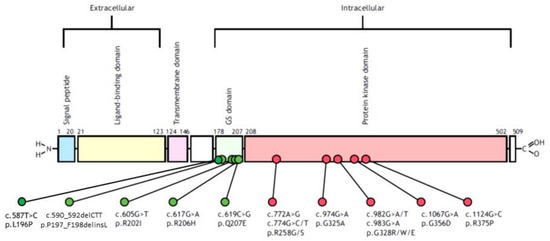
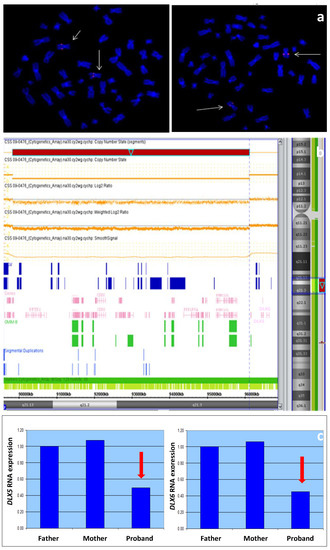
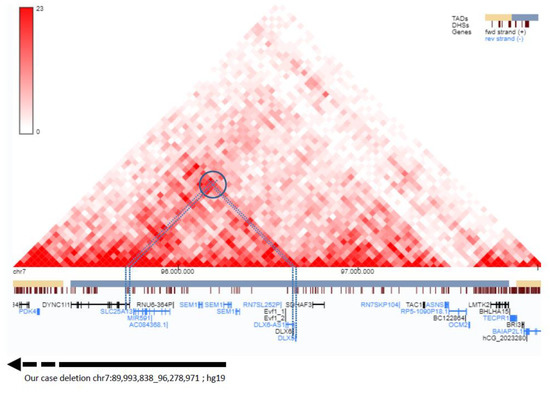
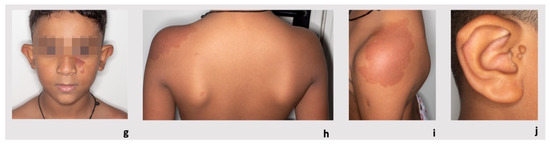
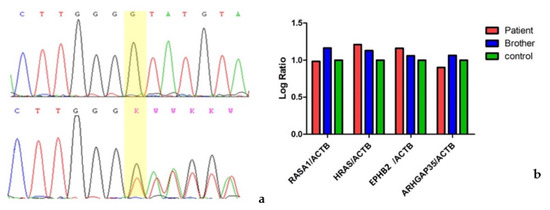
Figure 1



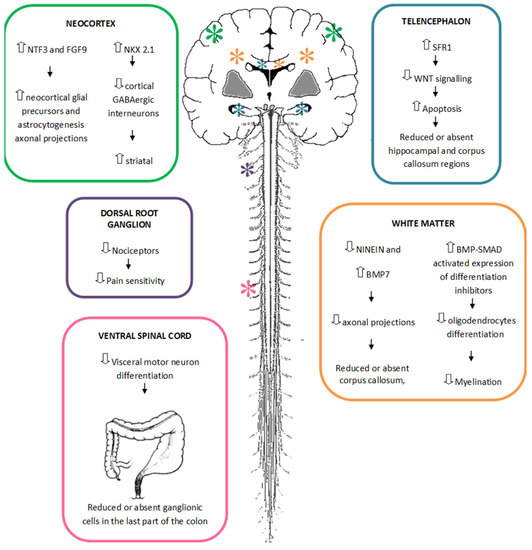

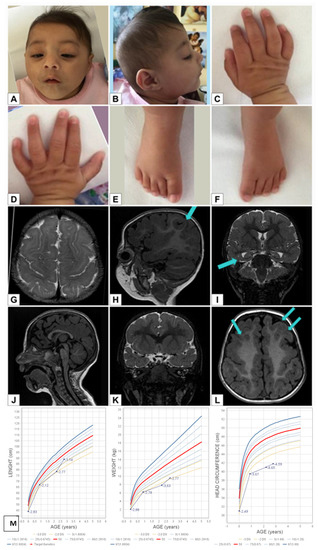
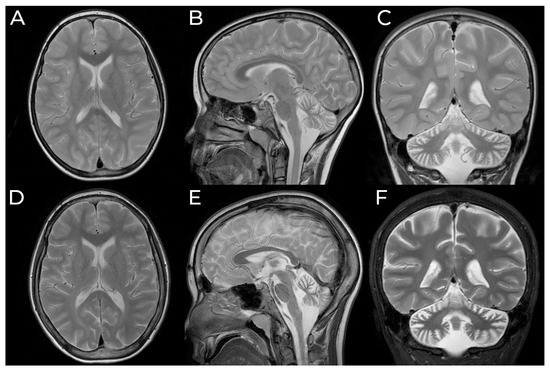
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}