
Complete Chloroplast Genome of Alternanthera sessilis and Comparative Analysis with Its Congeneric Invasive Weed Alternanthera philoxeroides
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plants Collection
2.2. DNA Extraction and Sequencing
2.3. Assembly and Annotation of A. sessilis Chloroplast Genome
2.4. Analysis Data Collection
2.5. Chloroplast Genome Structure
2.6. Codon Usage Bias Analysis of A. sessilis and A. philoxeroides cp Genomes
2.7. Repeat Sequence Analysis
2.8. Analysis of Hotspots and ka/ks and Identification of Highly Divergent Regions
2.9. Contraction and Expansion Analysis of IRs Boundaries
2.10. Genome Analysis and Comparison with Other Amaranthaceae Species cp Genomes
2.11. Phylogenetic Analysis and Divergence Time Estimate
3. Results
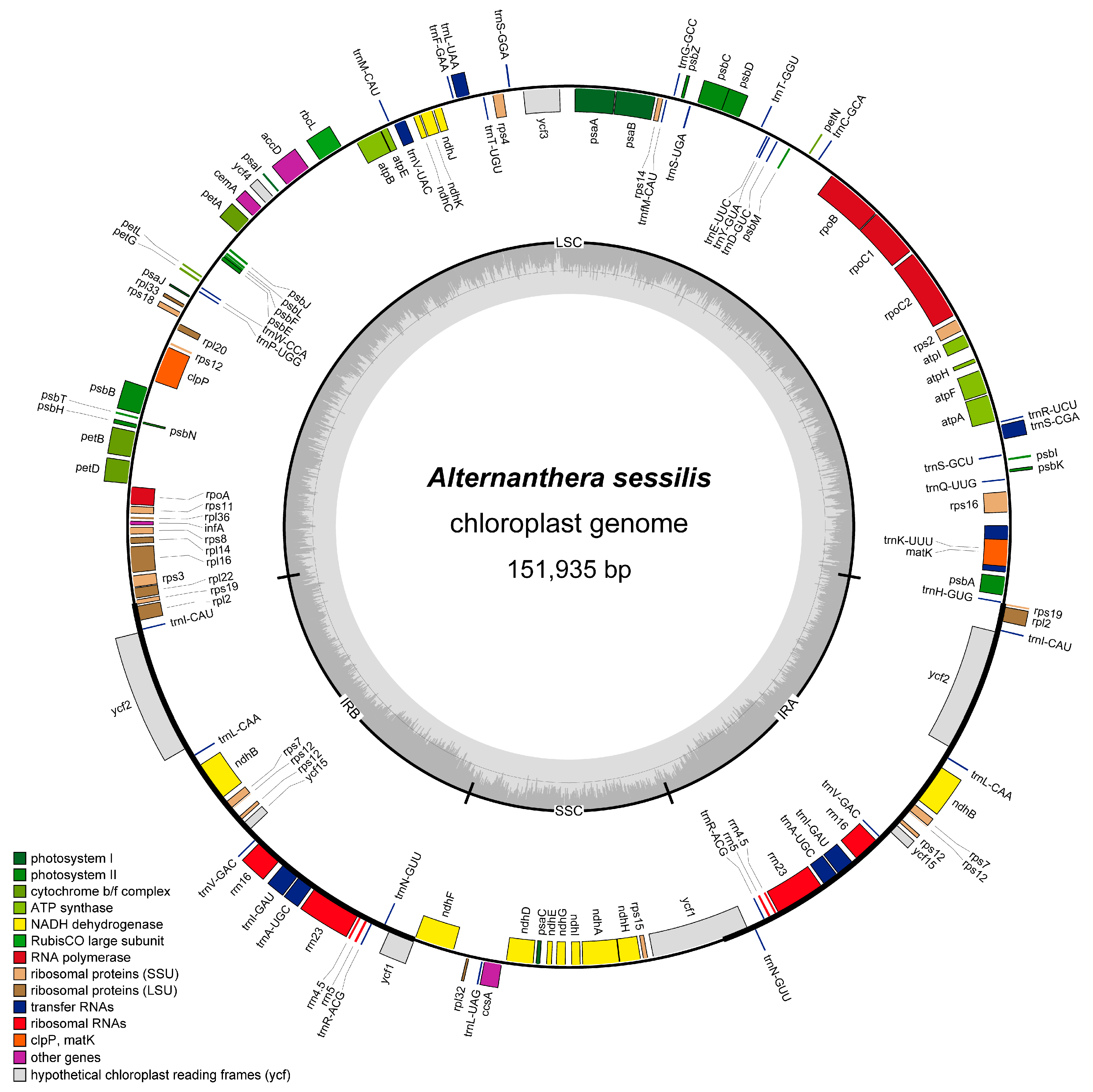
3.1. Sequencing, Assembly, and Annotation of A. sessilis cp Genome
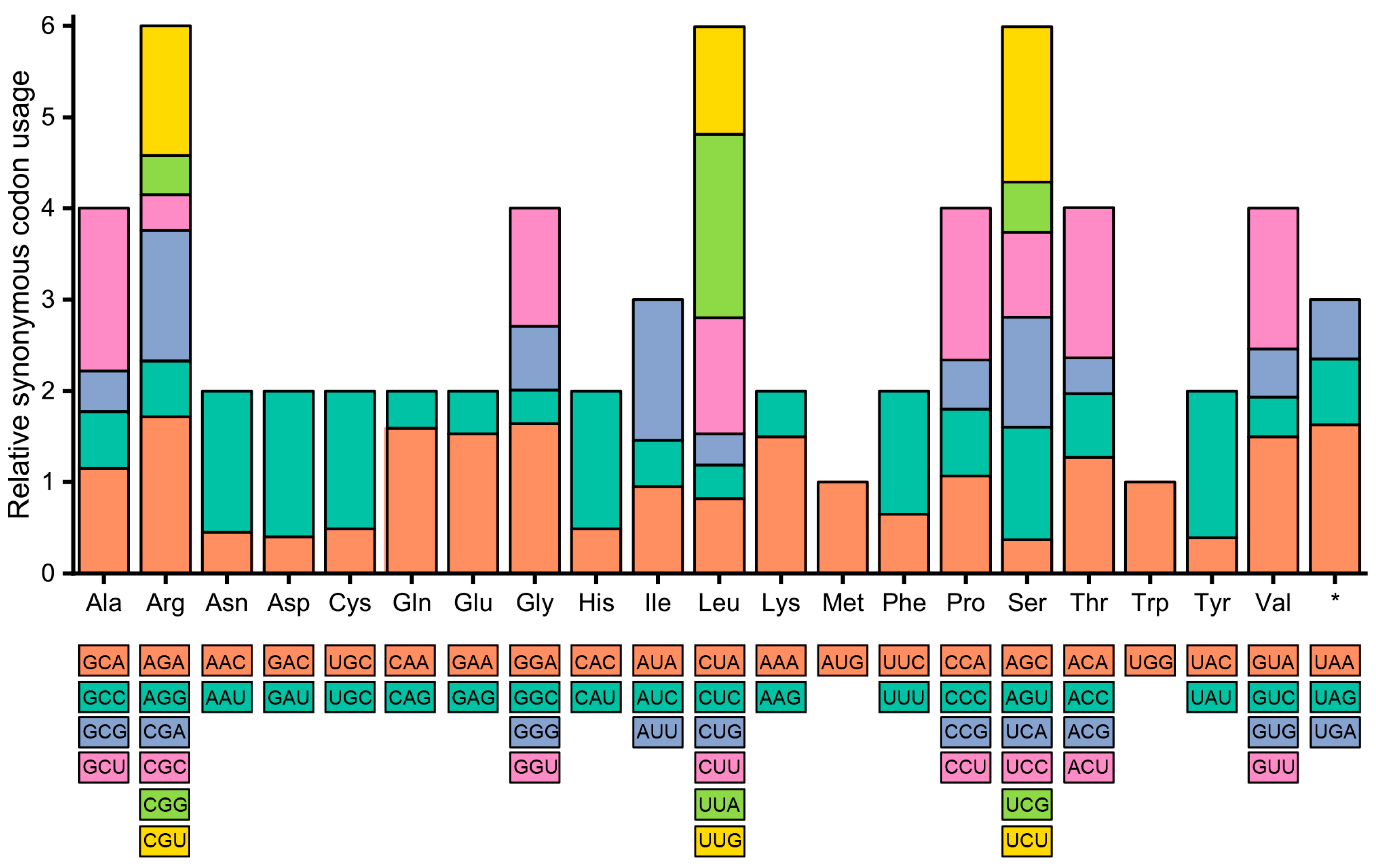
3.2. Codon Usage Bias of A. sessilis and A. philoxeroides cp Genomes
3.3. SSRs and Long Repeated Sequences
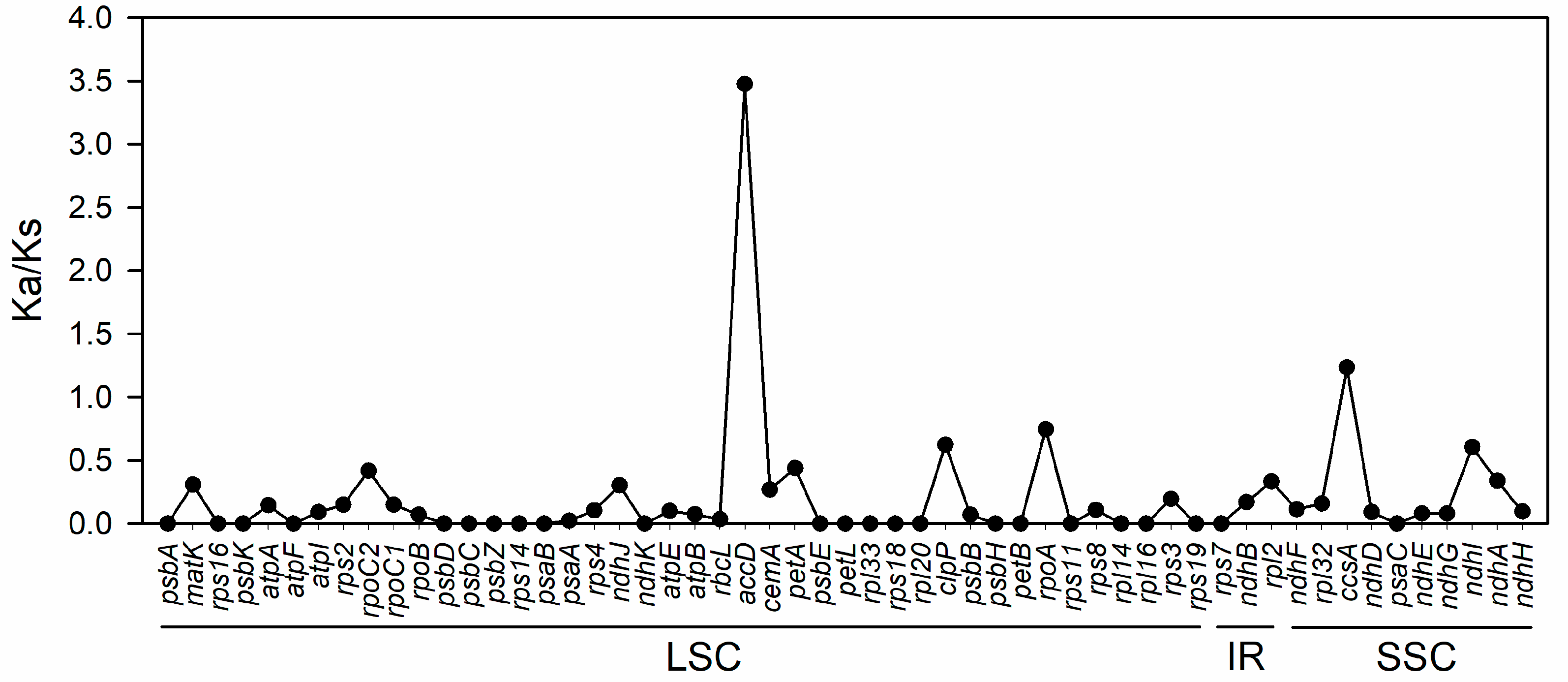
3.4. Divergence Hotspots and Ka/Ks
3.5. Comparison of Chloroplast Genomes in the Amaranthaceae Family
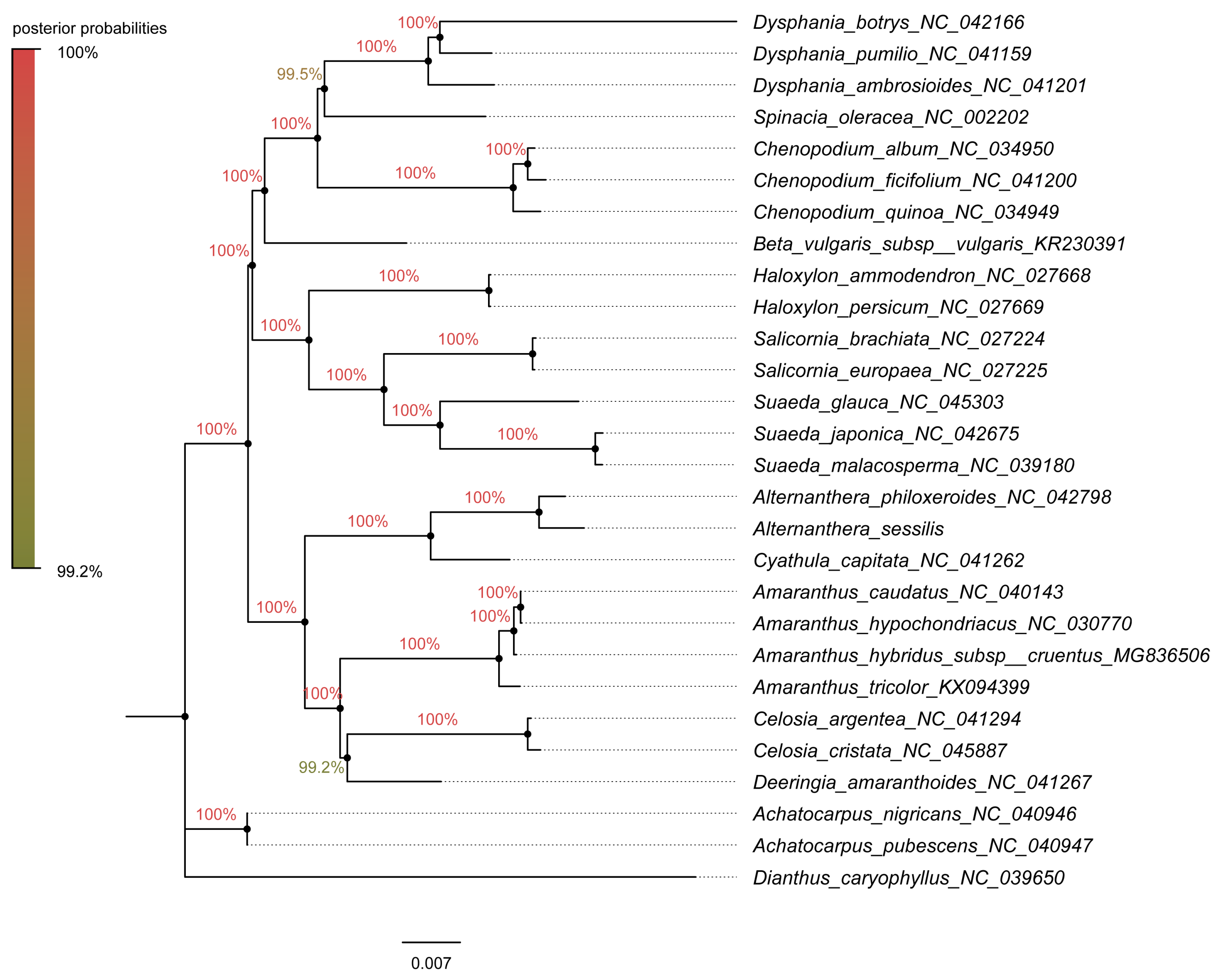
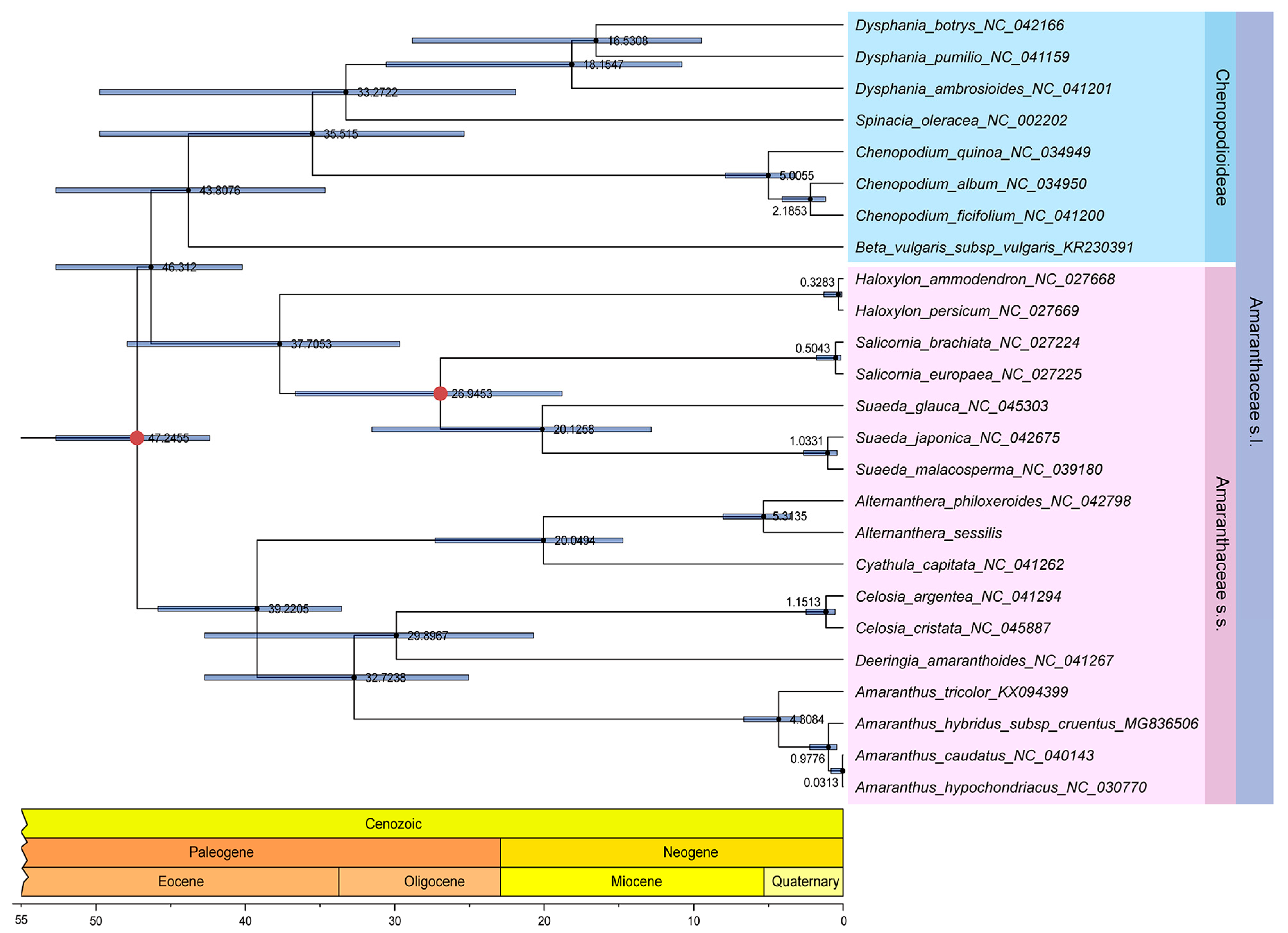
3.6. Phylogenetic Analysis and Estimation of Divergence Time
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Julien, M.H.; Skarratt, B.; Maywald, G. Potential geographical distribution of alligator weed and its biological control by Agasicles hygrophila. J. Aquat. Plant Mang. 1995, 33, 55–60. [Google Scholar]
- Coulson, J.R. Biological Control of Alligatorweed, 1959–1972: A Review and Evaluation; US Department of Agriculture, Agricultural Research Service: Washington, DC, USA, 1977. [Google Scholar]
- Pan, X.-Y. Invasive Alternanthera philoxeroides: Biology, ecology and management. Acta Phytotaxon. Sin. 2007, 45, 884–900. [Google Scholar] [CrossRef]
- Global Invasive Species Database (2024) Species Profile: Alternanthera sessilis. Available online: http://www.iucngisd.org/gisd/speciesname/Alternanthera+sessilis (accessed on 22 April 2024).
- Fan, S.; Yu, D.; Liu, C. The invasive plant Alternanthera philoxeroides was suppressed more intensively than its native congener by a native generalist: Implications for the biotic resistance hypothesis. PLoS ONE 2013, 8, e83619. [Google Scholar] [CrossRef]
- Baucom, R.S.; Holt, J.S. Weeds of agricultural importance: Bridging the gap between evolutionary ecology and crop and weed science. New Phytol. 2009, 184, 741–743. [Google Scholar] [CrossRef]
- Neve, P.; Barney, J.N.; Buckley, Y.; Cousens, R.D.; Graham, S.; Jordan, N.R.; Lawton-Rauh, A.; Liebman, M.; Mesgaran, M.B.; Schut, M.; et al. Reviewing research priorities in weed ecology, evolution and management: A horizon scan. Weed Res. 2018, 58, 250–258. [Google Scholar] [CrossRef]
- Vigueira, C.C.; Olsen, K.M.; Caicedo, A.L. The red queen in the corn: Agricultural weeds as models of rapid adaptive evolution. Heredity 2013, 110, 303–311. [Google Scholar] [CrossRef]
- Chu, S.; Cong, S.; Li, R.; Hou, Y. Host range of Herpetogramma basalis (Lepidoptera: Crambidae), a biological control agent for the invasive weed Alternanthera philoxeroides (Centrospermae: Amaranthaceae) in China. J. Insect Sci. 2019, 19, 1–7. [Google Scholar] [CrossRef]
- Sun, Y.; Ding, J.; Frye, M.J. Effects of resource availability on tolerance of herbivory in the invasive Alternanthera philoxeroides and the native Alternanthera sessilis. Weed Res. 2010, 50, 527–536. [Google Scholar] [CrossRef]
- Qin, H.; Guo, W.; Li, X. Density-dependent interactions between the nematode Meloidogyne incognita and the biological control agent Agasicles hygrophila on invasive Alternanthera philoxeroides and its native congener Alternantera sessilis. BioControl 2021, 66, 837–848. [Google Scholar] [CrossRef]
- Manoharan, B.; Qi, S.S.; Dhandapani, V.; Chen, Q.; Rutherford, S.; Wan, J.S.; Jegadeesan, S.; Yang, H.Y.; Li, Q.; Li, J.; et al. Gene expression profiling reveals enhanced defense responses in an invasive weed compared to its native congener during pathogenesis. Int. J. Mol. Sci. 2019, 20, 4916. [Google Scholar] [CrossRef]
- Chen, Y.; Zhou, Y.; Yin, T.F.; Liu, C.X.; Luo, F.L. The invasive wetland plant Alternanthera philoxeroides shows a higher tolerance to waterlogging than its native Congener Alternanthera sessilis. PLoS ONE 2013, 8, e81456. [Google Scholar] [CrossRef]
- Wang, T.; Hu, J.; Miao, L.; Yu, D.; Liu, C. The invasive stoloniferous clonal plant Alternanthera philoxeroides outperforms its co-occurring non-invasive functional counterparts in heterogeneous soil environments—Invasion implications. Sci. Rep. 2016, 6, 38036. [Google Scholar] [CrossRef] [PubMed]
- Gao, L. Comparisons of morphological variation and cellular osmotic potential adjustment between invasive species Alternanthera philoxeroides and its native congener A. sessilis under different water treatments. Plant Sci. 2015, 33, 195–202. [Google Scholar] [CrossRef]
- You, W.; Li, N.; Zhang, J.; Song, A.; Du, D. The plant invader Alternanthera philoxeroides benefits from clonal integration more than its native co-genus in response to patch contrast. Plants 2023, 12, 2371. [Google Scholar] [CrossRef]
- Lu, Y.; Yao, J. Chloroplasts at the crossroad of photosynthesis, pathogen infection and plant defense. Int. J. Mol. Sci. 2018, 19, 3900. [Google Scholar] [CrossRef] [PubMed]
- Zecca, G.; Abbott, J.R.; Sun, W.B.; Spada, A.; Sala, F.; Grassi, F. The timing and the mode of evolution of wild grapes (Vitis). Mol. Phylogenet Evol. 2012, 62, 736–747. [Google Scholar] [CrossRef]
- Li, C.; Cai, C.; Tao, Y.; Sun, Z.; Jiang, M.; Chen, L.; Li, J. Variation and evolution of the whole chloroplast genomes of Fragaria spp. (Rosaceae). Front. Plant Sci. 2021, 12, 754209. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.Z.; Li, Z.Y.; Jin, X.H. DNA barcoding of invasive plants in China: A resource for identifying invasive plants. Mol. Ecol. Resour. 2018, 18, 128–136. [Google Scholar] [CrossRef]
- Viljoen, E.; Odeny, D.A.; Coetzee, M.P.A.; Berger, D.K.; Rees, D.J.G. Application of chloroplast phylogenomics to resolve species relationships within the plant genus Amaranthus. J. Mol. Evol. 2018, 86, 216–239. [Google Scholar] [CrossRef]
- Doorduin, L.; Gravendeel, B.; Lammers, Y.; Ariyurek, Y.; Chin, A.W.T.; Vrieling, K. The complete chloroplast genome of 17 individuals of pest species Jacobaea vulgaris: SNPs, microsatellites and barcoding markers for population and phylogenetic studies. DNA Res. 2011, 18, 93–105. [Google Scholar] [CrossRef]
- Su, Y.; Huang, L.; Wang, Z.; Wang, T. Comparative chloroplast genomics between the invasive weed Mikania micrantha and its indigenous congener Mikania cordata: Structure variation, identification of highly divergent regions, divergence time estimation, and phylogenetic analysis. Mol. Phylogenet. Evol. 2018, 126, 181–195. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.S.; Kim, J.H.; Kim, C.S.; Mejias, J.A.; Kim, S.C. Sow thistle chloroplast genomes: Insights into the plastome evolution and relationship of two weedy species, Sonchus asper and Sonchus oleraceus (Asteraceae). Genes 2019, 10, 881. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Liu, J.; Yu, J.; Wang, L.; Zhou, S. Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. PLoS ONE 2012, 7, e35071. [Google Scholar] [CrossRef] [PubMed]
- Daniell, H.; Lin, C.S.; Yu, M.; Chang, W.J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 2016, 17, 134. [Google Scholar] [CrossRef] [PubMed]
- Duan, R.-Y.; Xiang, G.-H.; Luo, Y.-C. The complete chloroplast genome of the invasive alligator weed Alternanthera philoxeroides (Caryophyllales: Amaranthaceae). Mitochondrial DNA Part B 2019, 4, 1345–1346. [Google Scholar] [CrossRef]
- Laslett, D.; Canback, B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004, 32, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq—Versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef]
- Subramanian, S. Nearly neutrality and the evolution of codon usage bias in eukaryotic genomes. Genetics 2008, 178, 2429–2432. [Google Scholar] [CrossRef]
- Qi, Y.; Xu, W.; Xing, T.; Zhao, M.; Li, N.; Yan, L.; Xia, G.; Wang, M. Synonymous codon usage bias in the plastid genome is unrelated to gene structure and shows evolutionary heterogeneity. Evol. Bioinform. 2015, 11, 65–77. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Shaw, J.; Lickey, E.B.; Beck, J.T.; Farmer, S.B.; Liu, W.; Miller, J.; Siripun, K.C.; Winder, C.T.; Schilling, E.E.; Small, R.L. The tortoise and the hare II: Relative utility of 21 noncoding chloroplast DNA sequences for phylogenetic analysis. Am. J. Bot. 2005, 92, 142–166. [Google Scholar] [CrossRef]
- Shaw, J.; Lickey, E.B.; Schilling, E.E.; Small, R.L. Comparison of whole chloroplast genome sequences to choose noncoding regions for phylogenetic studies in angiosperms: The tortoise and the hare III. Am. J. Bot. 2007, 94, 275–288. [Google Scholar] [CrossRef]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Munch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sanchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sanchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Guisinger, M.M.; Kuehl, J.V.; Boore, J.L.; Jansen, R.K. Extreme reconfiguration of plastid genomes in the angiosperm family Geraniaceae: Rearrangements, repeats, and codon usage. Mol. Biol. Evol. 2011, 28, 583–600. [Google Scholar] [CrossRef] [PubMed]
- Amiryousefi, A.; Hyvonen, J.; Poczai, P. IRscope: An online program to visualize the junction sites of chloroplast genomes. Bioinformatics 2018, 34, 3030–3031. [Google Scholar] [CrossRef] [PubMed]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Jakovlic, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Hedges, S.B.; Marin, J.; Suleski, M.; Paymer, M.; Kumar, S. Tree of life reveals clock-like speciation and diversification. Mol. Biol. Evol. 2015, 32, 835–845. [Google Scholar] [CrossRef] [PubMed]
- Palmer, J.D.; Nugent, J.M.; Herbon, L.A. Unusual structure of geranium chloroplast DNA: A triple-sized inverted repeat, extensive gene duplications, multiple inversions, and two repeat families. Proc. Natl. Acad. Sci. USA 1987, 84, 769–773. [Google Scholar] [CrossRef]
- Wolfe, K.H.; Morden, C.W.; Palmer, J.D. Function and evolution of a minimal plastid genome from a nonphotosynthetic parasitic plant. Proc. Natl. Acad. Sci. USA 1992, 89, 10648–10652. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.-M.; Wang, J.; Feng, L.; Liu, S.; Pang, H.; Qi, L.; Li, J.; Sun, Y.; Qiao, W.; Zhang, L.; et al. Inferring the evolutionary mechanism of the chloroplast genome size by comparing whole-chloroplast genome sequences in seed plants. Sci. Rep. 2017, 7, 1555. [Google Scholar] [CrossRef]
- Timmis, J.N.; Ayliffe, M.A.; Huang, C.Y.; Martin, W. Endosymbiotic gene transfer: Organelle genomes forge eukaryotic chromosomes. Nat. Rev. Genet. 2004, 5, 123–135. [Google Scholar] [CrossRef]
- Bock, R.; Timmis, J.N. Reconstructing evolution: Gene transfer from plastids to the nucleus. Bioessays 2008, 30, 556–566. [Google Scholar] [CrossRef]
- Yao, G.; Jin, J.J.; Li, H.T.; Yang, J.B.; Mandala, V.S.; Croley, M.; Mostow, R.; Douglas, N.A.; Chase, M.W.; Christenhusz, M.J.M.; et al. Plastid phylogenomic insights into the evolution of Caryophyllales. Mol. Phylogenet. Evol. 2019, 134, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Fleischmann, T.T.; Scharff, L.B.; Alkatib, S.; Hasdorf, S.; Schottler, M.A.; Bock, R. Nonessential plastid-encoded ribosomal proteins in tobacco: A developmental role for plastid translation and implications for reductive genome evolution. Plant Cell 2011, 23, 3137–3155. [Google Scholar] [CrossRef] [PubMed]
- Gantt, J.S.; Baldauf, S.L.; Calie, P.J.; Weeden, N.F.; Palmer, J.D. Transfer of rpl22 to the nucleus greatly preceded its loss from the chloroplast and involved the gain of an intron. EMBO J. 1991, 10, 3073–3078. [Google Scholar] [CrossRef] [PubMed]
- Ehrnthaler, M.; Scharff, L.B.; Fleischmann, T.T.; Hasse, C.; Ruf, S.; Bock, R. Synthetic lethality in the tobacco plastid ribosome and its rescue at elevated growth temperatures. Plant Cell 2014, 26, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Goulding, S.E.; Olmstead, R.G.; Morden, C.W.; Wolfe, K.H. Ebb and flow of the chloroplast inverted repeat. Mol. Gen. Genet. 1996, 252, 195–206. [Google Scholar] [CrossRef]
- Li, X.; Yang, J.B.; Wang, H.; Song, Y.; Corlett, R.T.; Yao, X.; Li, D.Z.; Yu, W.B. Plastid NDH pseudogenization and gene loss in a recently derived lineage from the largest hemiparasitic plant genus Pedicularis (Orobanchaceae). Plant Cell Physiol. 2021, 62, 971–984. [Google Scholar] [CrossRef] [PubMed]
- Wakasugi, T.; Tsudzuki, J.; Ito, S.; Nakashima, K.; Tsudzuki, T.; Sugiura, M. Loss of all ndh genes as determined by sequencing the entire chloroplast genome of the black pine Pinus thunbergii. Proc. Natl. Acad. Sci. USA 1994, 91, 9794–9798. [Google Scholar] [CrossRef] [PubMed]
- Martin, W.; Stoebe, B.; Goremykin, V.; Hapsmann, S.; Hasegawa, M.; Kowallik, K.V. Gene transfer to the nucleus and the evolution of chloroplasts. Nature 1998, 393, 162–165. [Google Scholar] [CrossRef]
- Schmitz-Linneweber, C.; Maier, R.M.; Alcaraz, J.P.; Cottet, A.; Herrmann, R.G.; Mache, R. The plastid chromosome of spinach (Spinacia oleracea): Complete nucleotide sequence and gene organization. Plant Mol. Biol. 2001, 45, 307–315. [Google Scholar] [CrossRef]
- Bergthorsson, U.; Adams, K.L.; Thomason, B.; Palmer, J.D. Widespread horizontal transfer of mitochondrial genes in flowering plants. Nature 2003, 424, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Pang, X.; Liu, H.; Wu, S.; Yuan, Y.; Li, H.; Dong, J.; Liu, Z.; An, C.; Su, Z.; Li, B. Species identification of Oaks (Quercus L., Fagaceae) from gene to genome. Int. J. Mol. Sci. 2019, 20, 5940. [Google Scholar] [CrossRef] [PubMed]
- Schwenkert, S.; Legen, J.; Takami, T.; Shikanai, T.; Herrmann, R.G.; Meurer, J. Role of the low-molecular-weight subunits PetL, PetG, and PetN in assembly, stability, and dimerization of the cytochrome b6f complex in tobacco. Plant Physiol. 2007, 144, 1924–1935. [Google Scholar] [CrossRef] [PubMed]
- Wakasugi, T.; Tsudzuki, T.; Sugiura, M. The genomics of land plant chloroplasts: Gene content and alteration of genomic information by RNA editing. Photosynth. Res. 2001, 70, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Kode, V.; Mudd, E.A.; Iamtham, S.; Day, A. The tobacco plastid accD gene is essential and is required for leaf development. Plant J. 2005, 44, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-l.; Nie, L.-y.; Deng, S.-w.; Duan, L.; Wang, Z.-f.; Charboneau, J.L.M.; Ho, B.-C.; Chen, H.-f. Characterization of Firmiana danxiaensis plastomes and comparative analysis of Firmiana: Insight into its phylogeny and evolution. BMC Genom. 2024, 25, 203. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Merchant, S. The Plastid-encoded ccsA Gene is required for heme attachment to chloroplast c-type cytochromes (*). J. Biol. Chem. 1996, 271, 4632–4639. [Google Scholar] [CrossRef]
- Dong, W.-L.; Wang, R.-N.; Zhang, N.-Y.; Fan, W.-B.; Fang, M.-F.; Li, Z.-H. Molecular evolution of chloroplast genomes of orchid opecies: Insights into phylogenetic relationship and adaptive evolution. Int. J. Mol. Sci. 2018, 19, 716. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-H.; Yang, J.; Cho, M.-S.; Stuessy, T.F.; Crawford, D.J.; Kim, S.-C. Chloroplast genome provides insights into molecular evolution and species relationship of fleabanes (Erigeron: Tribe Astereae, Asteraceae) in the Juan Fernández Islands, Chile. Plants 2024, 13, 612. [Google Scholar] [CrossRef]
- Sheikh-Assadi, M.; Naderi, R.; Kafi, M.; Fatahi, R.; Salami, S.A.; Shariati, V. Complete chloroplast genome of Lilium ledebourii (Baker) Boiss and its comparative analysis: Lights into selective pressure and adaptive evolution. Sci. Rep. 2022, 12, 9375. [Google Scholar] [CrossRef]
- Wang, J.-H.; Moore, M.J.; Wang, H.; Zhu, Z.-X.; Wang, H.-F. Plastome evolution and phylogenetic relationships among Malvaceae subfamilies. Gene 2021, 765, 145103. [Google Scholar] [CrossRef] [PubMed]
- The Angiosperm Phylogeny Group; Chase, M.W.; Christenhusz, M.J.M.; Fay, M.F.; Byng, J.W.; Judd, W.S.; Soltis, D.E.; Mabberley, D.J.; Sennikov, A.N.; Soltis, P.S.; et al. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 2016, 181, 1–20. [Google Scholar] [CrossRef]
- Sánchez-Del Pino, I.; Motley, T.J.; Borsch, T. Molecular phylogenetics of Alternanthera (Gomphrenoideae, Amaranthaceae): Resolving a complex taxonomic history caused by different interpretations of morphological characters in a lineage with C4 and C3-C4 intermediate species. Bot. J. Linn. Soc. 2012, 169, 493–517. [Google Scholar] [CrossRef]
- Sage, R.F.; Sage, T.L.; Pearcy, R.W.; Borsch, T. The taxonomic distribution of C4 photosynthesis in Amaranthaceae sensu stricto. Am. J. Bot. 2007, 94, 1992–2003. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Genus | GenBank Number | Genome Size (bp) | LSC (bp) | SSC (bp) | IR (bp) | GC (%) | tRNA Gene Number | rRNA Gene Number | Protein-Coding Gene Number | Total Gene Number |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Alternanthera sessilis | Alternanthera | PP239384 | 151,935 | 84,449 | 17,298 | 25,095 | 36.3 | 37 | 8 | 83 | 128 |
| Alternanthera philoxeroides | Alternanthera | NC_042798 | 152,255 | 84,670 | 17,319 | 25,133 | 36.4 | 35 | 8 | 83 | 126 |
| Amaranthus tricolor | Alternanthera | KX094399 | 150,027 | 83,735 | 17,600 | 24,346 | 36.6 | 33 | 8 | 89 | 130 |
| Amaranthus hypochondriacus cultivar Plainsman | Amaranthus | NC_030770 | 150,518 | 83,873 | 17,941 | 24,352 | 36.6 | 31 | 8 | 73 | 112 |
| Amaranthus caudatus | Amaranthus | NC_040143 | 150,523 | 83,878 | 17,941 | 24,352 | 36.6 | 37 | 8 | 84 | 129 |
| Amaranthus hybridus subsp. PI566897 | Amaranthus | MG836506 | 150,757 | 84,101 | 17,954 | 24,351 | 36.6 | 37 | 8 | 84 | 129 |
| Celosia argentea | Celosia | NC_041294 | 153,673 | 85,140 | 17,711 | 25,411 | 36.7 | 37 | 8 | 85 | 130 |
| Celosia cristata | Celosia | NC_045887 | 153,472 | 84,975 | 17,681 | 25,408 | 36.7 | 36 | 8 | 73 | 117 |
| Cyathula capitata | Cyathula | NC_041262 | 151,557 | 83,350 | 17,129 | 25,539 | 36.4 | 37 | 8 | 84 | 129 |
| Deeringia amaranthoides | Deeringia | NC_041267 | 155,108 | 86,065 | 18,305 | 25,369 | 36.8 | 35 | 8 | 84 | 127 |
| Function of Genes | Category of Genes | Gene Name |
|---|---|---|
| Self-replication | Large subunit of ribosome | rpl2 a, rpl14, rpl16 b, rpl20, rpl22(ses), rpl23 a(phi), rpl32, rpl33, rpl36 |
| DNA-dependent RNA polymerase | rpoA, rpoB, rpoC1 b, rpoC2 | |
| Ribosomal RNA genes | rrn4.5 a, rrn5 a, rrn16 a, rrn23 a | |
| Small subunit of ribosome | rps2, rps3, rps4, rps7 a, rps8, rps11, rps12 abe, rps14, rps15(ses), rps16 b, rps18, rps19 d(ses), rps19 a(phi) | |
| Transfer RNA genes | trnA-UGC ab, trnC-GCA, trnD-GUC, trnE-UUC, trnF-GAA, trnG-GCC(ses), trnH-GUG, trnI-CAU a, trnI-GAU ab, trnK-UUU b, trnL-CAA a, trnL-UAA b, trnL-UAG, trnM-CAU(ses), trnM-CAU a(phi), trnN-GUU a, trnP-UGG, trnQ-UUG, trnR-ACG a, trnR-UCU, trnS-CGA b(ses), trnS-GCU, trnS-GGA, trnS-UGA, trnT-GGU, trnT-UGU, trnV-GAC a, trnV-UAC b, trnW-CCA, trnY-GUA, trnfM-CAU(ses) | |
| Genes for photosynthesis | Subunits of ATP synthase | atpA, atpB, atpE, atpF b, atpH, atpI |
| Subunits of NADH dehydrogenase | ndhA b(ses), ndhA(phi), ndhB ab, ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK | |
| Subunits of photosystem I | psaA, psaB, psaC, psaI, psaJ | |
| Subunits of photosystem II | psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbN, psbT, psbZ | |
| Subunits of cytochrome | petA, petB b, petD b, petG, petL, petN | |
| Large subunit of Rubisco | rbcL | |
| Other genes | Subunit of acetyl-CoA-carboxylase | accD |
| C-type cytochrome synthesis gene | ccsA | |
| Envelop membrane protein | cemA | |
| ATP-dependent protease subunit p gene | clpP c | |
| Maturase | matK | |
| Translation initiation factor | infA | |
| Genes of unknown function | Conserved open reading frames | ycf1 d(ses), ycf15 ad(ses), ycf1, ycf2 a, ycf3 c, ycf4 |
| Gene | Location | Intron Number | Exon I | Intron I | Exon II | Intron II | Exon III | CDS Length |
|---|---|---|---|---|---|---|---|---|
| trnK-UUU | LSC | 1 | 37 | 2538 | 35 | 72 | ||
| rps16 | LSC | 1 | 42 | 911 | 213 | 255 | ||
| trnS-CGA | LSC | 1 | 31 | 713 | 60 | 91 | ||
| atpF | LSC | 1 | 148 | 800 | 410 | 558 | ||
| rpoC1 | LSC | 1 | 435 | 771 | 1602 | 2037 | ||
| ycf3 | LSC | 2 | 124 | 764 | 230 | 774 | 153 | 507 |
| trnL-UAA | LSC | 1 | 35 | 650 | 50 | 85 | ||
| trnV-UAC | LSC | 1 | 38 | 595 | 35 | 73 | ||
| rps12 | IRa | 114 | - | 232 | 538 | 26 | 372 | |
| clpP | LSC | 2 | 69 | 600 | 293 | 875 | 226 | 588 |
| petB | LSC | 1 | 6 | 769 | 642 | 648 | ||
| petD | LSC | 1 | 8 | 774 | 475 | 483 | ||
| rpl16 | LSC | 1 | 9 | 1055 | 402 | 411 | ||
| ndhB | IRb | 1 | 778 | 667 | 758 | 1536 | ||
| rps12 | IRb | 232 | - | 26 | 538 | 114 | 372 | |
| trnI-GAU | IRb | 1 | 37 | 946 | 35 | 72 | ||
| trnA-UGC | IRb | 1 | 38 | 825 | 36 | 74 | ||
| ndhA | SSC | 1 | 556 | 959 | 539 | 1095 | ||
| trnA-UGC | IRa | 1 | 38 | 825 | 36 | 74 | ||
| trnI-GAU | IRa | 1 | 37 | 946 | 35 | 72 | ||
| ndhB | IRa | 1 | 778 | 667 | 758 | 1536 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Zhao, X.; Chen, Q.; Yang, J.; Hu, J.; Jia, D.; Ma, R. Complete Chloroplast Genome of Alternanthera sessilis and Comparative Analysis with Its Congeneric Invasive Weed Alternanthera philoxeroides. Genes 2024, 15, 544. https://doi.org/10.3390/genes15050544
Wang Y, Zhao X, Chen Q, Yang J, Hu J, Jia D, Ma R. Complete Chloroplast Genome of Alternanthera sessilis and Comparative Analysis with Its Congeneric Invasive Weed Alternanthera philoxeroides. Genes. 2024; 15(5):544. https://doi.org/10.3390/genes15050544
Chicago/Turabian StyleWang, Yuanxin, Xueying Zhao, Qianhui Chen, Jun Yang, Jun Hu, Dong Jia, and Ruiyan Ma. 2024. "Complete Chloroplast Genome of Alternanthera sessilis and Comparative Analysis with Its Congeneric Invasive Weed Alternanthera philoxeroides" Genes 15, no. 5: 544. https://doi.org/10.3390/genes15050544