




Pharmaceutics 2021, 13(9), 1509; https://doi.org/10.3390/pharmaceutics13091509 - 18 Sep 2021
Cited by 2 | Viewed by 3559
Abstract
►
Show Figures
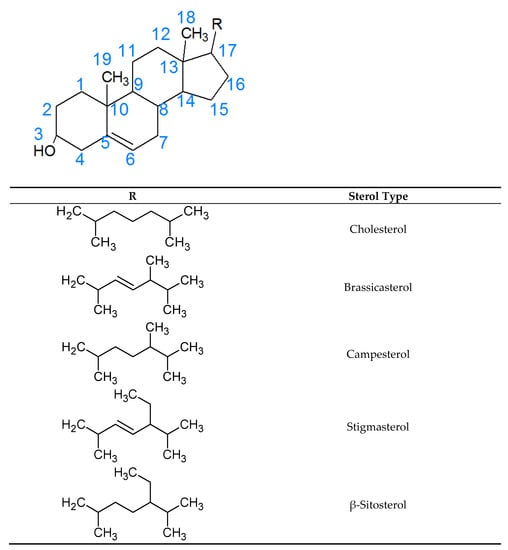
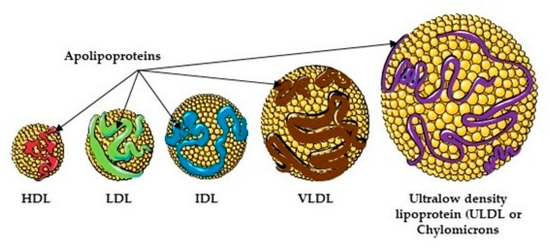
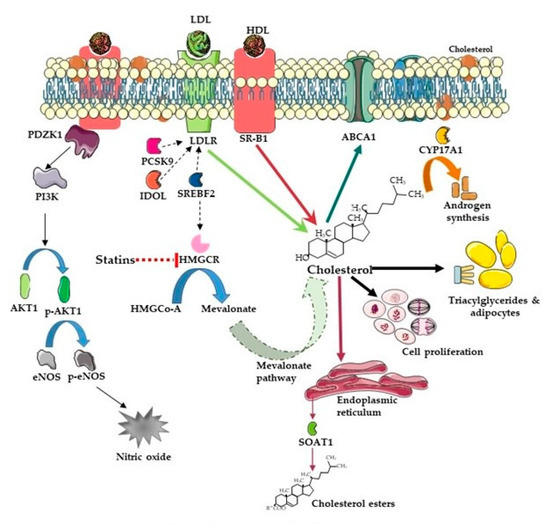
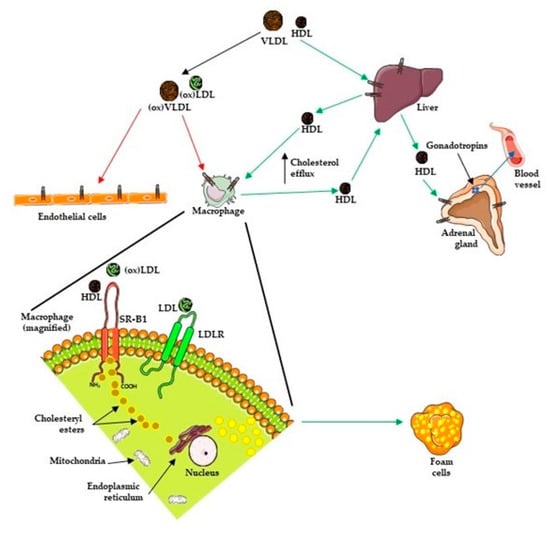
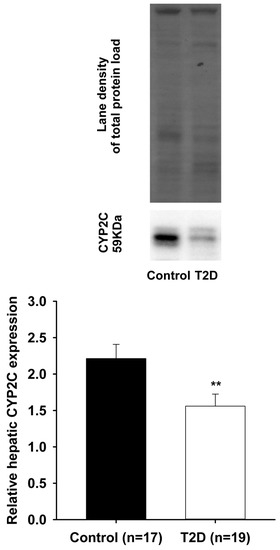
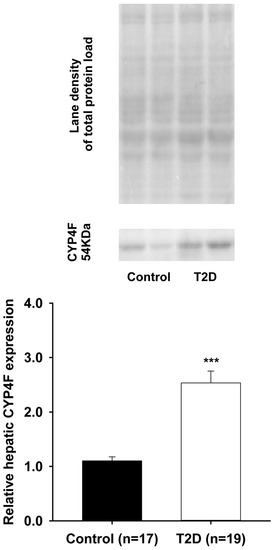
There have been several studies that have linked elevated scavenger receptor class b type 1 (SR-B1) expression and activity to the development and progression of castration-resistant prostate cancer (CRPC). SR-B1 facilitates the influx of cholesterol to the cell from lipoproteins in systemic circulation.
[...] Read more.
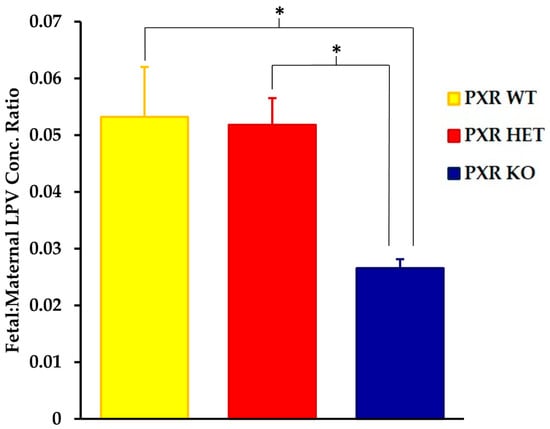
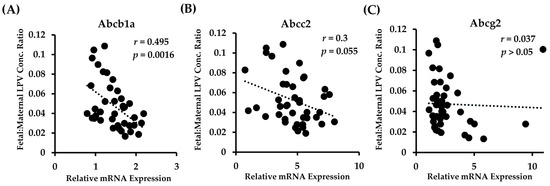
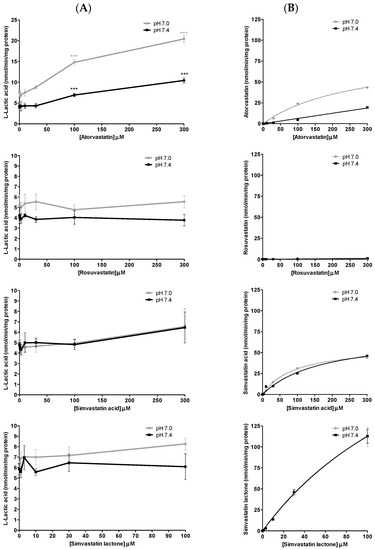
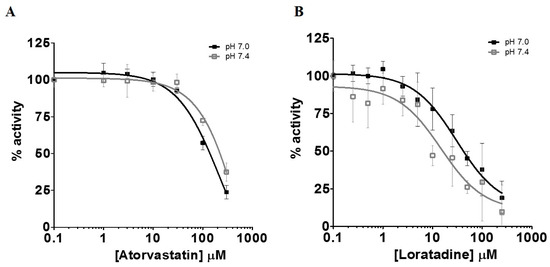
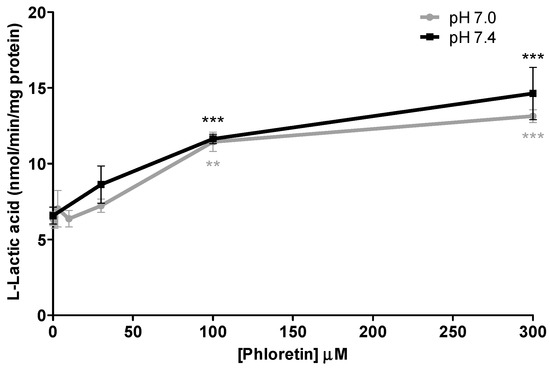
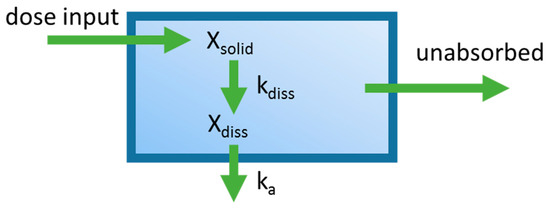
There have been several studies that have linked elevated scavenger receptor class b type 1 (SR-B1) expression and activity to the development and progression of castration-resistant prostate cancer (CRPC). SR-B1 facilitates the influx of cholesterol to the cell from lipoproteins in systemic circulation. This influx of cholesterol may be important for many cellular functions, including the synthesis of androgens. Castration-resistant prostate cancer tumors can synthesize androgens de novo to supplement the loss of exogenous sources often induced by androgen deprivation therapy. Silencing of SR-B1 may impact the ability of prostate cancer cells, particularly those of the castration-resistant state, to maintain the intracellular supply of androgens by removing a supply of cholesterol. SR-B1 expression is elevated in CRPC models and has been linked to poor survival of patients. The overarching belief has been that cholesterol modulation, through either synthesis or uptake inhibition, will impact essential signaling processes, impeding the proliferation of prostate cancer. The reduction in cellular cholesterol availability can impede prostate cancer proliferation through both decreased steroid synthesis and steroid-independent mechanisms, providing a potential therapeutic target for the treatment of prostate cancer. In this article, we discuss and highlight the work on SR-B1 as a potential novel drug target for CRPC management.
Full article
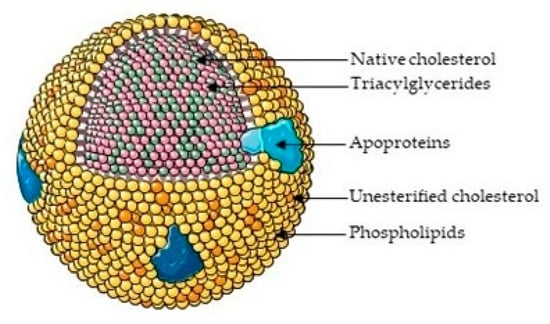
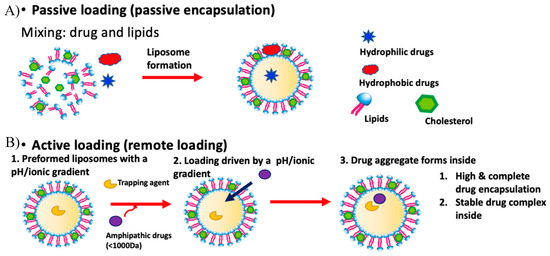
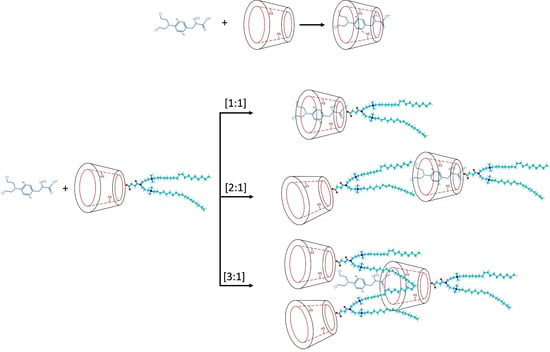
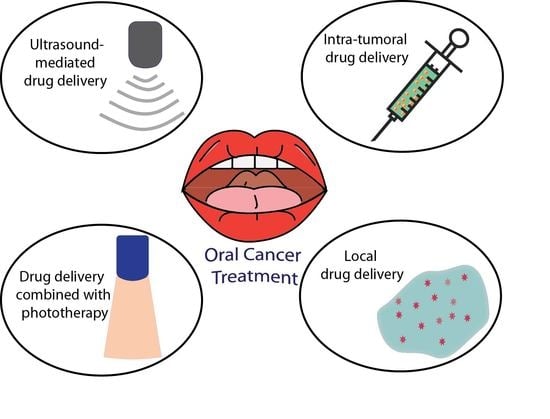
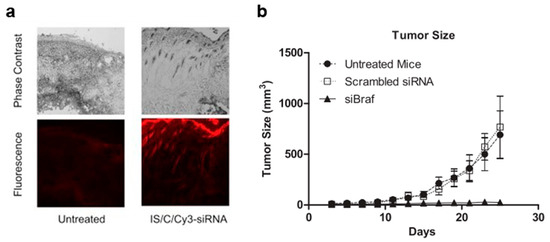
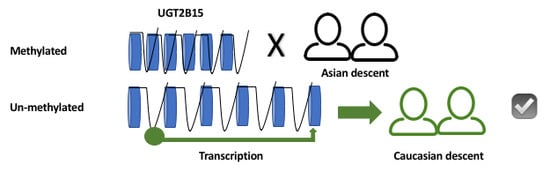
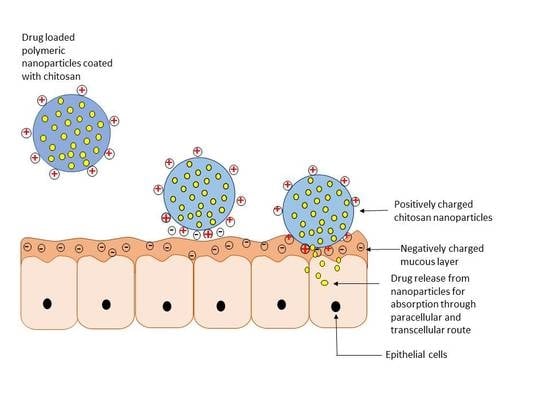
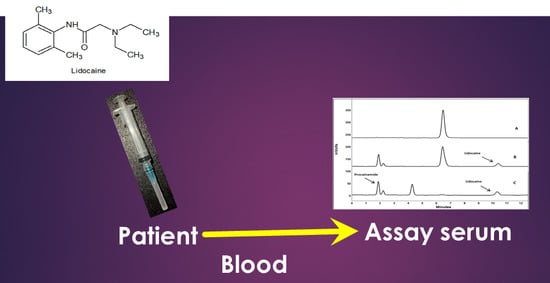
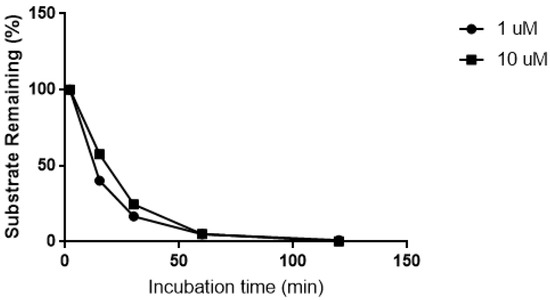
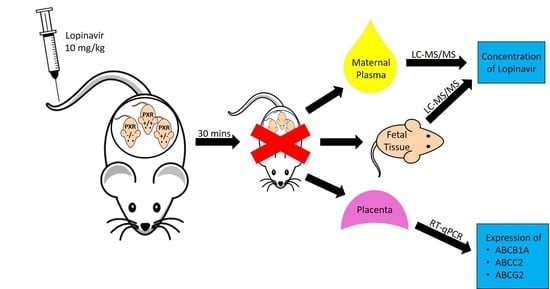
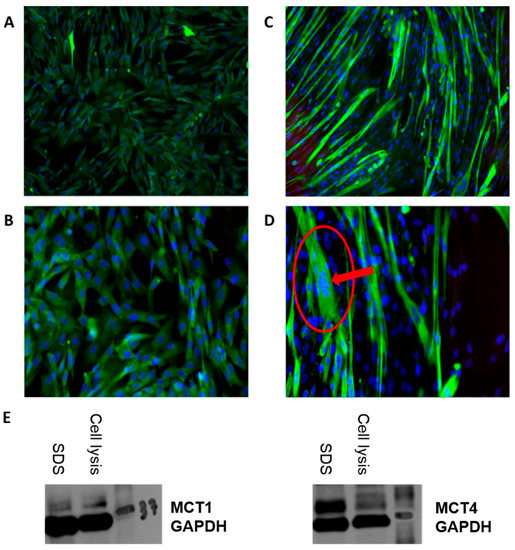
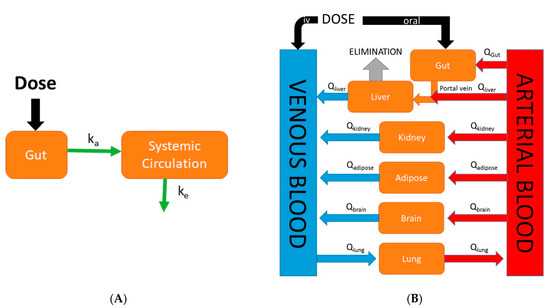
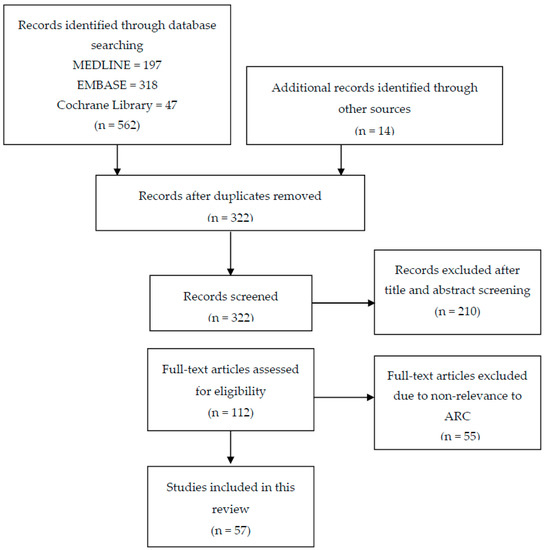
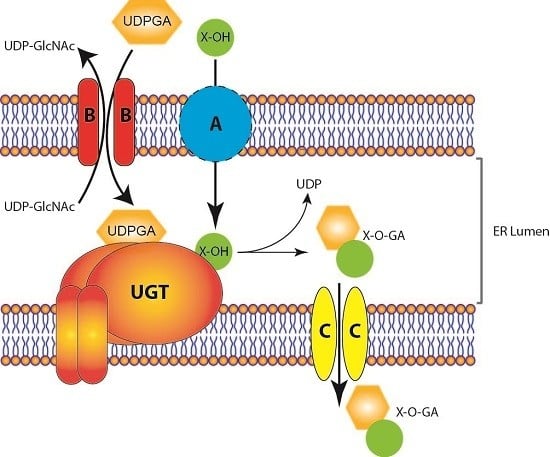
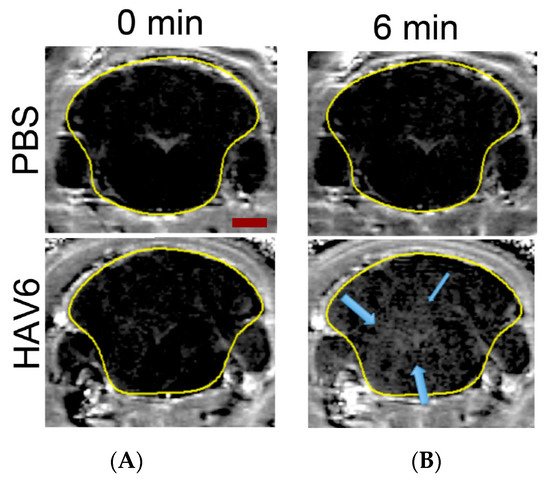
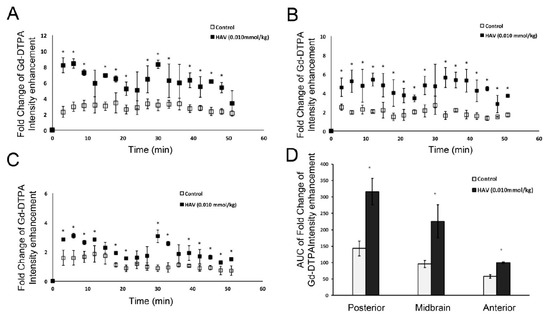
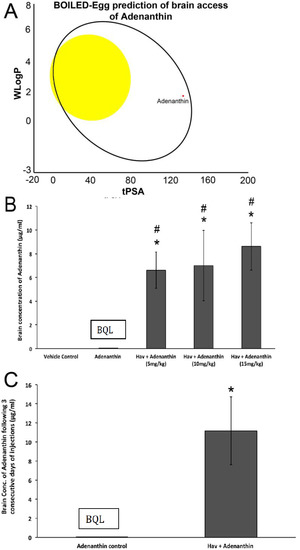
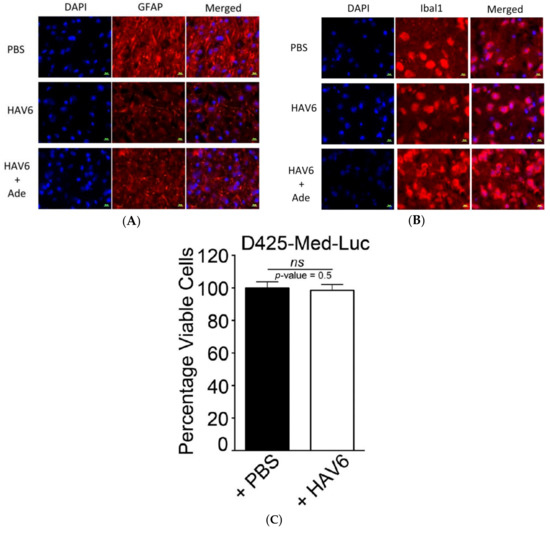
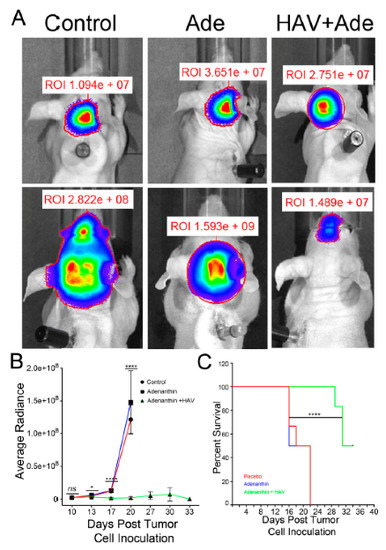
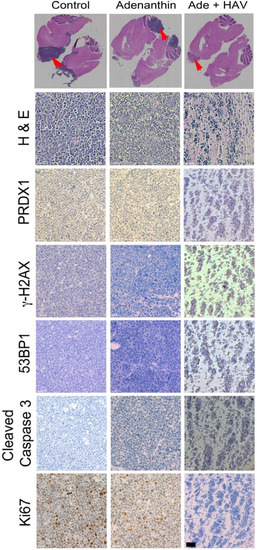
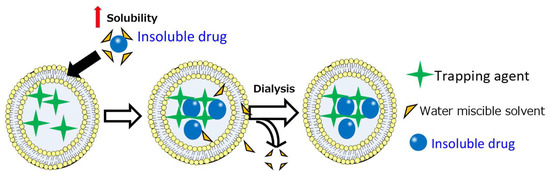
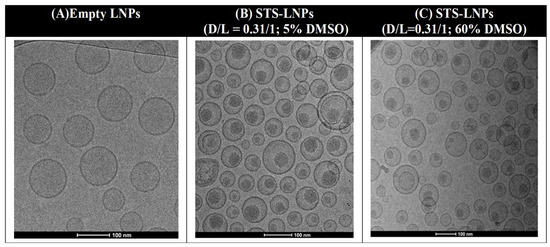
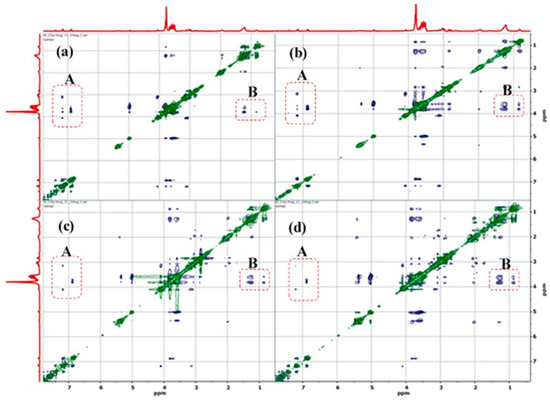
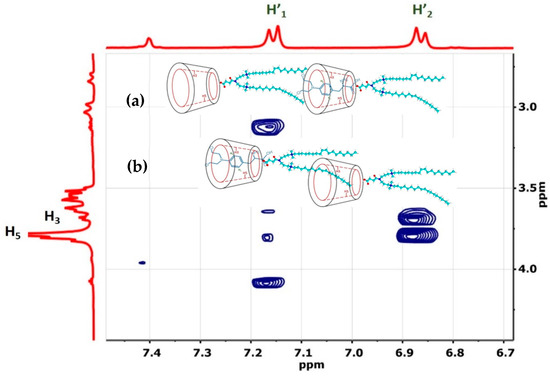
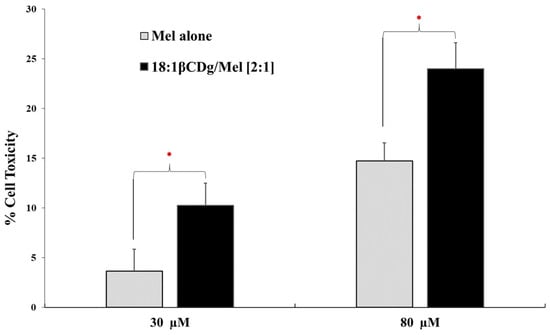
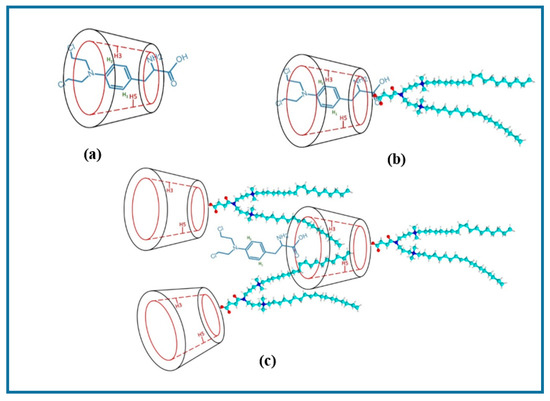
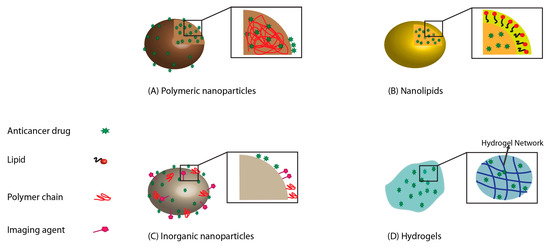
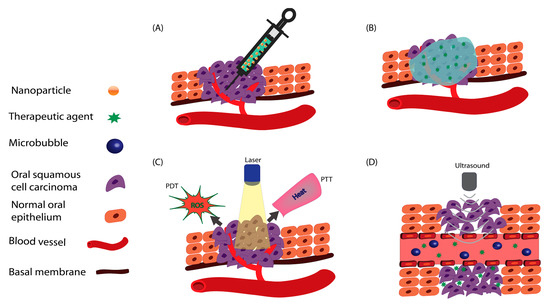
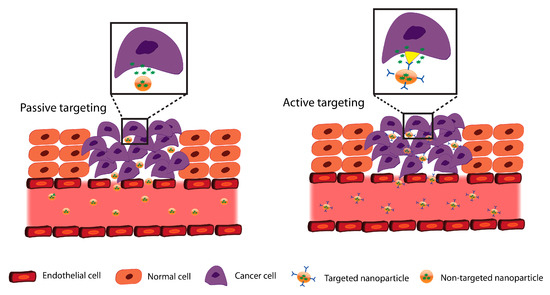
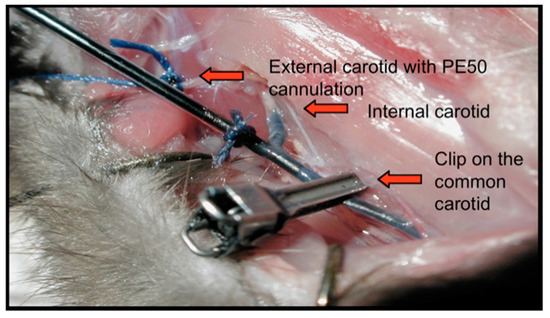
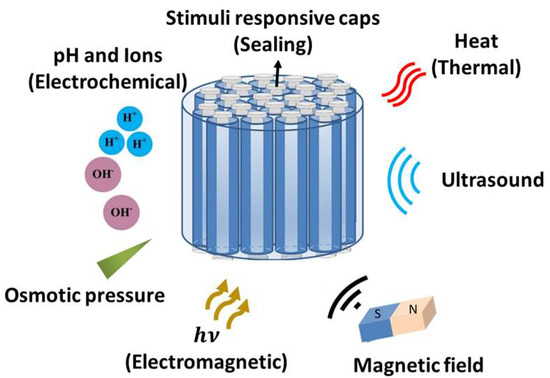
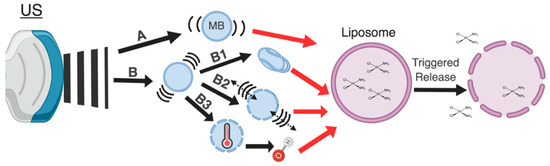
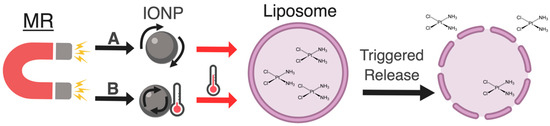
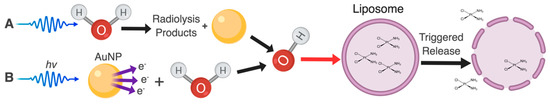
Figure 1




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}