A High-Performance Liquid Chromatography Assay Method for the Determination of Lidocaine in Human Serum


Abstract
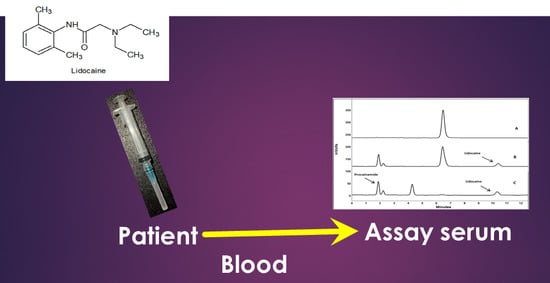
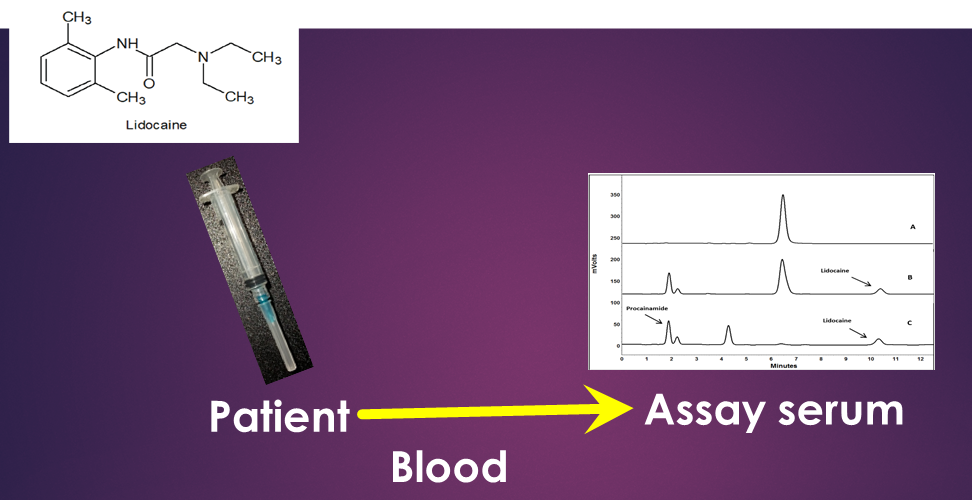
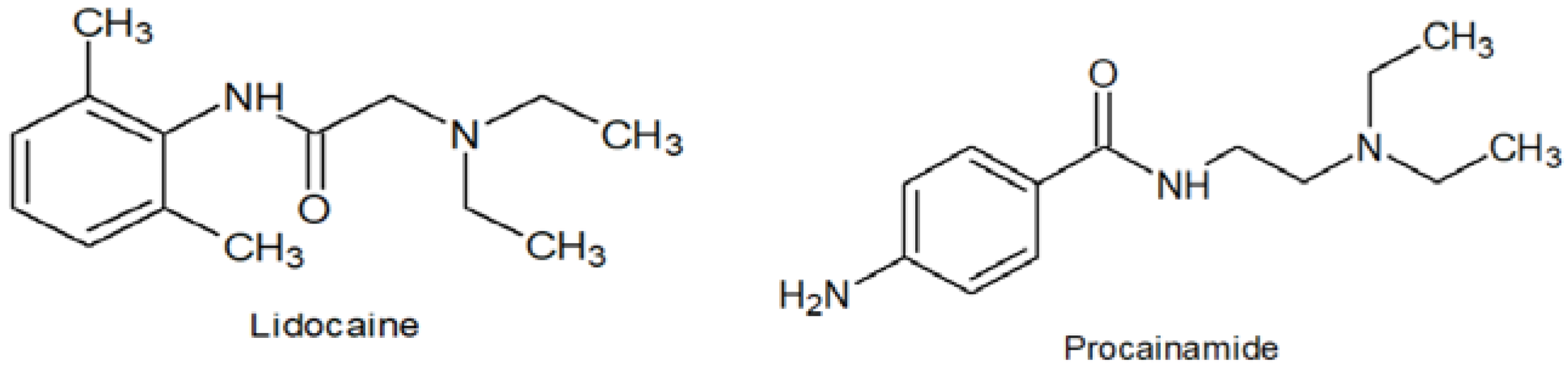
:
1. Introduction
2. Materials and Methods
2.1. Instrumentation and Chromatographic Conditions
2.2. Standard and Stock Solutions
2.3. Extraction Procedure
2.4. Recovery
2.5. Calibration, Accuracy and Validation
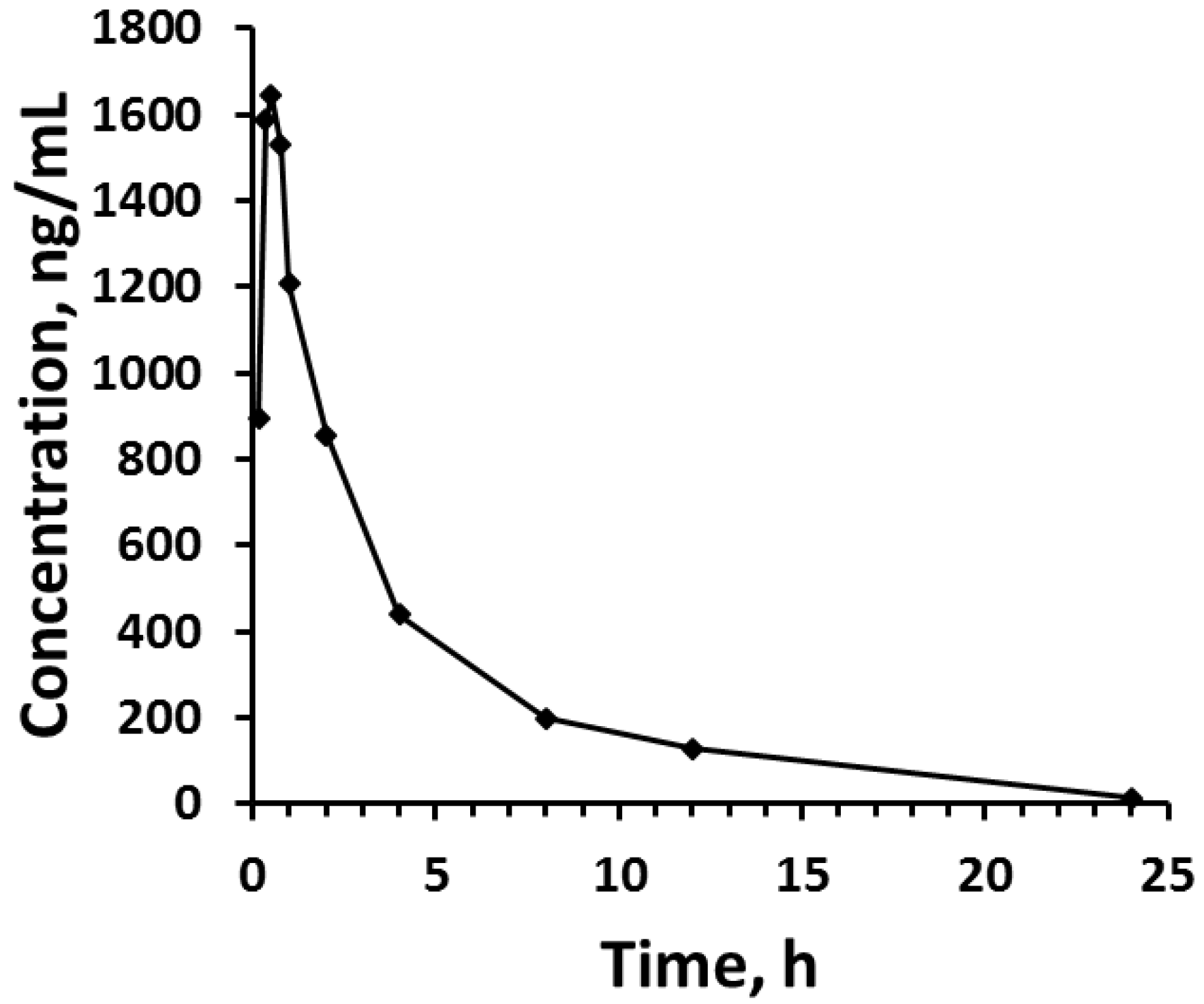
2.6. Applicability
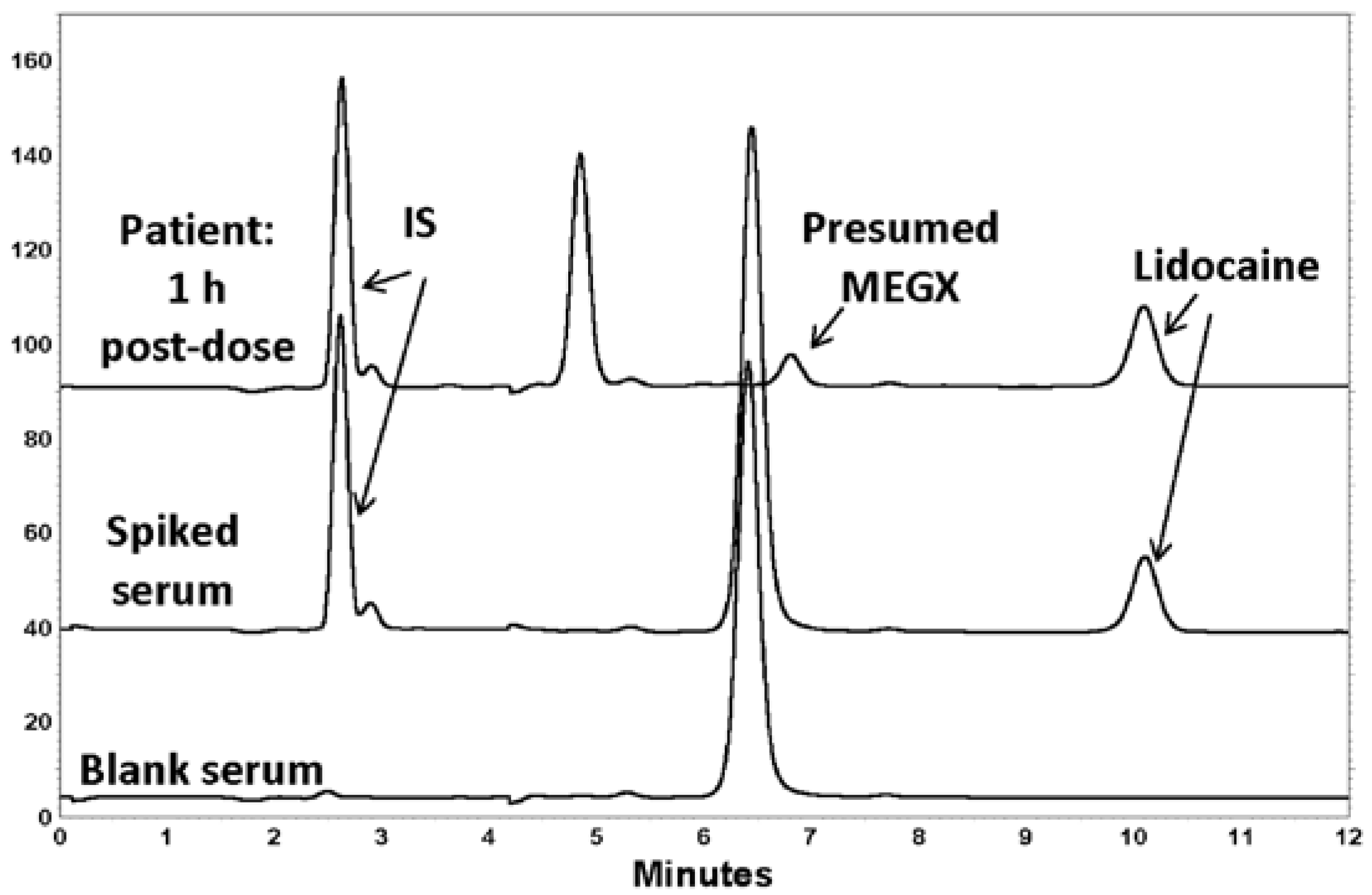
3. Results
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| CV% | Coefficient of variation expressed as percent |
| LLQ | Lower limit of quantitation |
| SPE | Solid phase extraction |
| GC-MS | Gas chromatography-mass spectrometry |
| ppt | Precipitation |
| IS | Internal standard |
| LLE | Liquid-liquid extraction |
| UV | Ultraviolet detection |
| NS | Not specified or unclear |
References
- Benowitz, N.L.; Meister, W. Clinical pharmacokinetics of lignocaine. Clin. Pharmacokinet. 1978, 3, 177–201. [Google Scholar] [CrossRef] [PubMed]
- Tremont-Lukats, I.; Teixeira, G.; Backonja, M. Systemic administration of local anesthetic agents to relieve neuropathic pain. Anesth. Analg. 2005, 101, 1736–1737. [Google Scholar]
- World Health Organization. WHO Model List of Essential Medicines: 20th List. March 2017. Available online: http://www.who.int/medicines/publications/essentialmedicines/20th_EML2017_FINAL_amendedAug2017.pdf?ua=1 (accessed on 15 November 2017).
- Ohshima, T.; Takayasu, T. Simultaneous determination of local anesthetics including ester-type anesthetics in human plasma and urine by gas chromatography-mass spectrometry with solid-phase extraction. J. Chromatogr. B Biomed. Sci. Appl. 1999, 726, 185–194. [Google Scholar] [CrossRef]
- Piwowarska, J.; Kuczynska, J.; Pachecka, J. Liquid chromatographic method for the determination of lidocaine and monoethylglycine xylidide in human serum containing various concentrations of bilirubin for the assessment of liver function. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2004, 805, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Liao, L.; Zuo, Z.; Yan, Y.; Yang, L.; Fu, Q.; Chen, Y.; Hou, J. Simultaneous determination of nikethamide and lidocaine in human blood and cerebrospinal fluid by high performance liquid chromatography. J. Pharm. Biomed. Anal. 2007, 43, 1757–1762. [Google Scholar] [CrossRef] [PubMed]
- Tam, Y.K.; Tawfik, S.R.; Ke, J.; Coutts, R.T.; Gray, M.R.; Wyse, D.G. High-performance liquid chromatography of lidocaine and nine of its metabolites in human plasma and urine. J. Chromatogr. 1987, 423, 199–206. [Google Scholar] [CrossRef]
- Lotfi, H.; Debord, J.; Dreyfuss, M.F.; Marquet, P.; Ben Rhaiem, M.; Feiss, P.; Lachâtre, G. Simultaneous determination of lidocaine and bupivacaine in human plasma: Application to pharmacokinetics. Ther. Drug Monit. 1997, 19, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.W.; Jiao, Z.; Zhong, M.K.; Shi, X.J.; Zhang, J.; Li, Z.D.; Cui, X.Y. Simultaneous determination of procaine, lidocaine, ropivacaine, tetracaine and bupivacaine in human plasma by high-performance liquid chromatography. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2010, 878, 1185–1189. [Google Scholar] [CrossRef] [PubMed]
- Bhusal, P.; Sharma, M.; Harrison, J.; Procter, G.; Andrews, G.; Jones, D.S.; Hill, A.G.; Svirskis, D. Development, validation and application of a stability indicating HPLC method to quantify lidocaine from polyethylene-co-vinyl acetate (EVA) matrices and biological fluids. J. Chromatogr. Sci. 2017, 55, 832–838. [Google Scholar] [CrossRef] [PubMed]
- Nation, R.L.; Peng, G.W.; Chiou, W.L. High-performance liquid chromatographic method for the simultaneous determination of lidocaine and its N-dealkylated metabolites in plasma. J. Chromatogr. 1979, 162, 466–473. [Google Scholar] [CrossRef]
- Sintov, A.; Siden, R.; Levy, R.J. Sensitive high-performance liquid chromatographic assay using 9-fluorenylmethylchloroformate for monitoring controlled-release lidocaine in plasma. J. Chromatogr. 1989, 496, 335–344. [Google Scholar] [CrossRef]
- Kihara, S.; Miyabe, M.; Kakiuchi, Y.; Takahashi, S.; Fukuda, T.; Kohda, Y.; Toyooka, H. Plasma concentrations of lidocaine and its principal metabolites during continuous epidural infusion of lidocaine with or without epinephrine. Reg. Anesth. Pain Med. 1999, 24, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Dusci, L.J.; Hackett, L.P. Simultaneous determination of lidocaine, mexiletine, disopyramide, and quinidine in plasma by high performance liquid chromatography. J. Anal. Toxicol. 1985, 9, 67–70. [Google Scholar] [CrossRef] [PubMed]
- Ter Weijden, E.; Van den Broek, M.; Ververs, F. Easy and fast LC–MS/MS determination of lidocaine and MEGX in plasma for therapeutic drug monitoring in neonates with seizures. J. Chromatogr. B 2012, 881, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Bo, L.D.; Mazzucchelli, P.; Marzo, A. Highly sensitive bioassay of lidocaine in human plasma by high-performance liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 1999, 854, 3–11. [Google Scholar] [PubMed]
- Ochoa-Aranda, E.; Esteve-Romero, J.; Rambla-Alegre, M.; Martinavarro-Domnguez, A.; Marcos-Toms, J.V.; Bose, D. Monitoring disopyramide, lidocaine, and quinidine by micellar liquid chromatography. J. AOAC Int. 2011, 94, 537–542. [Google Scholar] [PubMed]
- Guideline on Bioanalytical Method Validation. Available online: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2011/08/WC500109686.pdf (accessed on 15 November 2017).
- Roos, R.W.; Lau-Cam, C.A. General reversed-phase high-performance liquid chromatographic method for the separation of drugs using triethylamine as a competing base. J. Chromatogr. 1986, 370, 403–418. [Google Scholar] [CrossRef]
- Ruiz-Angel, M.; Torres-Lapasió, J.; Carda-Broch, S.; García-Alvarez-Coque, M. Improvement of peak shape and separation performance of β-blockers in conventional reversed-phase columns using solvent modifiers. J. Chromatogr. Sci. 2003, 41, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Wang, Q.; Shi, K.; Liu, F.; Liu, L.; Ni, J.; Fang, X.; Xu, X. The Effects of Lidocaine Used in Sciatic Nerve on the Pharmacodynamics and Pharmacokinetics of Ropivacaine in Sciatic Nerve Combined with Lumbar Plexus Blockade: A Double-Blind, Randomized Study. Basic Clin. Pharmacol. Toxicol. 2013, 112, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Bargetzi, M.J.; Aoyama, T.; Gonzalez, F.J.; Meyer, U.A. Lidocaine metabolism in human liver microsomes by cytochrome P450IIIA4. Clin. Pharmacol. Ther. 1989, 46, 521–527. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Volume of Specimen (mL) | Validated LLQ (ng/mL) | Type of Human Matrix | Sample Preparation Method | Analytical Column | Detection Method | References |
|---|---|---|---|---|---|---|
| 0.2 | 50 | Plasma | SPE | DB-1 | GC–MS | [4] |
| 0.5 | 200 | Serum | LLE | C8 | UV | [5] |
| 0.1 | 680 | Plasma | Protein ppt | C18 | UV | [6] |
| 0.5 | 200 | Plasma | LLE | C18 | UV | [7] |
| 1 | 10 | Serum | LLE | C18 | UV | [8] |
| 0.5 | 50 | Plasma | LLE | C18 | UV | [9] |
| 0.1 | 400 | Plasma | Protein ppt | C18 | UV | [10] |
| 0.5 | 100 | Plasma | LLE | Phenyl | UV | [11] |
| 0.5–1 | 25 | Plasma | LLE | C18 | Fluorescence of derivative | [12] |
| 0.25 | NS | Plasma | NS | C18 | UV | [13] |
| 1 | 1000 | Plasma | LLE | C18 | UV | [14] |
| 0.01 | 200 | Plasma | Protein ppt | C18 | LC-MS/MS | [15] |
| 1 | 0.2 | Plasma | LLE | C18 | LC-MS/MS | [16] |
| 1 | 20 | Serum | None | C18 | UV | [17] |
| 0.25 | 43 | Serum | LLE | C18 | UV | Current method |
| Nominal Concentration of Lidocaine *, ng/mL | Intraday | Interday | ||||
|---|---|---|---|---|---|---|
| Mean ± SD ng/mL (CV%) | Mean ± SD ng/mL | CV% | Error% | |||
| 43.3 | 45.9 ± 4.54 (9.90) | 38.5 ± 5.37 (14.0) | 40.8 ± 6.05 (14.8) | 41.7 ± 5.32 | 12.9 | −3.57 |
| 216 | 2158 ± 11.5 (5.35) | 220 ± 9.60 (4.35) | 226 ± 26.6 (11.8) | 221 ± 15.9 | 7.16 | 1.99 |
| 433 | 379 ± 28.7 (7.56) | 395 ± 25.6 (6.46) | 407 ± 38.5 (9.47) | 394 ± 30.9 | 7.83 | −8.97 |
| 1731 | 1489 ± 60.5 (4.06) | 1704 ± 43.3 (2.53) | 1635 ± 47.1 (2.87) | 1609 ± 50.3 | 3.16 | −7.02 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al Nebaihi, H.M.; Primrose, M.; Green, J.S.; Brocks, D.R. A High-Performance Liquid Chromatography Assay Method for the Determination of Lidocaine in Human Serum. Pharmaceutics 2017, 9, 52. https://doi.org/10.3390/pharmaceutics9040052
Al Nebaihi HM, Primrose M, Green JS, Brocks DR. A High-Performance Liquid Chromatography Assay Method for the Determination of Lidocaine in Human Serum. Pharmaceutics. 2017; 9(4):52. https://doi.org/10.3390/pharmaceutics9040052
Chicago/Turabian StyleAl Nebaihi, Hamdah M., Matthew Primrose, James S. Green, and Dion R. Brocks. 2017. "A High-Performance Liquid Chromatography Assay Method for the Determination of Lidocaine in Human Serum" Pharmaceutics 9, no. 4: 52. https://doi.org/10.3390/pharmaceutics9040052