
Inhibition of Scavenger Receptor Class B Type 1 (SR-B1) Expression and Activity as a Potential Novel Target to Disrupt Cholesterol Availability in Castration-Resistant Prostate Cancer

Abstract
:1. Background and Focus of This Perspective
Methods Used to Review the Literature
2. Cholesterol
Lipoproteins and Transport
Cholesterol and Prostate Cancer (PCa)
3. Intratumoral Androgen Synthesis in Castrate-Resistant Prostate Cancer
4. Effect of a Class of Drugs That Inhibit Cholesterol Synthesis on Prostate Cancer (PCa)
5. SR-B1
5.1. Transcriptional Regulation
5.2. SR-B1 Function
5.3. Inhibitors of SR-B1
5.4. SR-B1 and PCa
6. Stress and Autophagy
7. Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Litwin, M.S.; Tan, H.J. The Diagnosis and Treatment of Prostate Cancer: A Review. JAMA 2017, 317, 2532–2542. [Google Scholar] [CrossRef]
- Teo, Y.M.; Rathkopf, D.E.; Kantoff, P. Treatment of Advanced Prostate Cancer. Annu. Rev. Med. 2019, 70, 479–499. [Google Scholar] [CrossRef] [PubMed]
- Mansinho, A.; Macedo, D.; Fernandes, I.; Costa, L. Castration-Resistant Prostate Cancer: Mechanisms, Targets and Treatment. Prostate Cancer 2019, 1126, 117–133. [Google Scholar]
- Crowley, F.; Sterpi, M.; Buckley, C.; Margetich, L.; Handa, S.; Dovey, Z. A Review of the Pathophysiological Mechanisms Underlying Castration-resistant Prostate Cancer. Res. Rep. Urol. 2021, 13, 457–472. [Google Scholar] [CrossRef] [PubMed]
- Vellky, J.E.; Ricke, W.A. Development and prevalence of castration-resistant prostate cancer subtypes. Neoplasia 2020, 22, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Sumanasuriya, S.; De Bono, J. Treatment of Advanced Prostate Cancer—A Review of Current Therapies and Future Promise. Cold Spring Harb. Perspect. Med. 2018, 8, a030635. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; He, B. Androgen Receptor Signaling in the Development of Castration-Resistant Prostate Cancer. Front. Oncol. 2019, 9, 858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montgomery, R.B.; Mostaghel, E.A.; Vessella, R.; Hess, D.L.; Kalhorn, T.F.; Higano, C.S.; True, L.D.; Nelson, P.S. Maintenance of Intratumoral Androgens in Metastatic Prostate Cancer: A Mechanism for Castration-Resistant Tumor Growth. Cancer Res. 2008, 68, 4447–4454. [Google Scholar] [CrossRef] [Green Version]
- Locke, J.A.; Guns, E.S.; Lubik, A.A.; Adomat, H.H.; Hendy, S.C.; Wood, C.A.; Ettinger, S.L.; Gleave, M.E.; Nelson, C.C. Androgen Levels Increase by Intratumoral de novo Steroidogenesis during Progression of Castration-Resistant Prostate Cancer. Cancer Res. 2008, 68, 6407–6415. [Google Scholar] [CrossRef] [Green Version]
- Fontana, F.; Limonta, P. Dissecting the Hormonal Signaling Landscape in Castration-Resistant Prostate Cancer. Cells 2021, 10, 1133. [Google Scholar] [CrossRef]
- Jung, M.E.; Ouk, S.; Yoo, D.; Sawyers, C.L.; Chen, C.; Tran, C.; Wongvipat, J. Structure-Activity Relationship for Thiohydantoin Androgen Receptor Antagonists for Castration-Resistant Prostate Cancer (CRPC). J. Med. Chem. 2010, 53, 2779–2796. [Google Scholar] [CrossRef] [Green Version]
- Narayanan, R. Therapeutic targeting of the androgen receptor (AR) and AR variants in prostate cancer. Asian J. Urol. 2020, 7, 271–283. [Google Scholar] [CrossRef] [PubMed]
- Potter, G.A.; Barrie, S.E.; Jarman, M.; Rowlands, M.G. Novel Steroidal Inhibitors of Human Cytochrome P450(17-Alpha) (17-Alpha-Hydroxylase-C-17,C-20-Lyase)—Potential Agents for the Treatment of Prostatic-Cancer. J. Med. Chem. 1995, 38, 2463–2471. [Google Scholar] [CrossRef]
- Duarte, C.; Jimeno, A.; Kessler, E. Abiraterone acetate to treat metastatic castration-resistant prostate cancer in combination with prednisone. Drugs Today 2019, 55, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Mitsiades, N.; Kaochar, S. Androgen receptor signaling inhibitors: Post-chemotherapy, pre-chemotherapy and now in castration-sensitive prostate cancer. Endocr. Relat. Cancer 2021, 28, T19–T38. [Google Scholar] [CrossRef] [PubMed]
- Velho, P.I.; Bastos, D.A.; Antonarakis, E.S. New approaches to targeting the androgen receptor pathway in prostate cancer. Clin. Adv. Hematol. Oncol. 2021, 19, 228–240. [Google Scholar] [PubMed]
- Small, E.J. Redefining Hormonal Therapy for Advanced Prostate Cancer: Results from the LATITUDE and STAMPEDE Studies. Cancer Cell 2017, 32, 392. [Google Scholar] [CrossRef] [PubMed]
- Mori, K.; Miura, N.; Mostafaei, H.; Quhal, F.; Motlagh, R.S.; Pradere, B.; Kimura, S.; Kimura, T.; Egawa, S.; Briganti, A.; et al. Sequential therapy of abiraterone and enzalutamide in castration-resistant prostate cancer: A systematic review and meta-analysis. Prostate Cancer Prostatic Dis. 2020, 23, 539–548. [Google Scholar] [CrossRef]
- Fizazi, K.; Kramer, G.; Eymard, J.-C.; Sternberg, C.N.; de Bono, J.; Castellano, D.; Tombal, B.; Wülfing, C.; Liontos, M.; Carles, J.; et al. Quality of life in patients with metastatic prostate cancer following treatment with cabazitaxel versus abiraterone or enzalutamide (CARD): An analysis of a randomised, multicentre, open-label, phase 4 study. Lancet Oncol. 2020, 21, 1513–1525. [Google Scholar] [CrossRef]
- Presicce, F.; Giacinti, S.; Bassanelli, M.; Tubaro, A. Castration-resistance prostate cancer: What is in the pipeline? Minerva Urol. Nefrol. 2018, 70, 22–41. [Google Scholar]
- Watson, P.A.; Arora, V.K.; Sawyers, C.L. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Rev. Cancer 2015, 15, 701–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snow, O.; Lallous, N.; Singh, K.; Lack, N.; Rennie, P.; Cherkasov, A. Androgen receptor plasticity and its implications for prostate cancer therapy. Cancer Treat. Rev. 2019, 81, 101871. [Google Scholar] [CrossRef]
- Zhang, T.; Karsh, L.I.; Nissenblatt, M.J.; Canfield, S.E. Androgen Receptor Splice Variant, AR-V7, as a Biomarker of Resistance to Androgen Axis-Targeted Therapies in Advanced Prostate Cancer. Clin. Genitourin. Cancer 2020, 18, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Dalrymple, S.L.; Coleman, I.; Zheng, S.L.; Xu, J.; Hooper, J.E.; Antonarakis, E.S.; De Marzo, A.M.; Meeker, A.K.; Nelson, P.S.; et al. Role of androgen receptor splice variant-7 (AR-V7) in prostate cancer resistance to 2nd-generation androgen receptor signaling inhibitors. Oncogene 2020, 39, 6935–6949. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.; Chen, Y.; Fedor, H.L.; et al. AR-V7 and Resistance to Enzalutamide and Abiraterone in Prostate Cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharp, A.; Coleman, I.; Yuan, W.; Sprenger, C.; Dolling, D.; Rodrigues, D.N.; Russo, J.W.; Figueiredo, I.; Bertan, C.; Seed, G.; et al. Androgen receptor splice variant-7 expression emerges with castration resistance in prostate cancer. J. Clin. Investig. 2019, 129, 192–208. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, K.T.; Huitema, A.D.R.; Chau, C.H.; Figg, W.D. Resistance to second-generation androgen receptor antagonists in prostate cancer. Nat. Rev. Urol. 2021, 18, 209–226. [Google Scholar] [CrossRef]
- Labrecque, M.; Coleman, I.M.; Brown, L.G.; True, L.D.; Kollath, L.; Lakely, B.; Nguyen, H.M.; Yang, Y.C.; Gil Da Costa, R.M.; Kaipainen, A.; et al. Molecular profiling stratifies diverse phenotypes of treatment-refractory metastatic castration-resistant prostate cancer. J. Clin. Investig. 2019, 129, 4492–4505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beltran, H.; Prandi, D.; Mosquera, J.M.; Benelli, M.; Puca, L.; Cyrta, J.; Marotz, C.; Giannopoulou, E.; Chakravarthi, B.V.; Varambally, S.; et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med. 2016, 22, 298–305. [Google Scholar] [CrossRef] [Green Version]
- Obinata, D.; Lawrence, M.G.; Takayama, K.; Choo, N.; Risbridger, G.P.; Takahashi, S.; Inoue, S. Recent Discoveries in the Androgen Receptor Pathway in Castration-Resistant Prostate Cancer. Front. Oncol. 2020, 10, 581515. [Google Scholar] [CrossRef]
- Moll, J.M.; Kumagai, J.; van Royen, M.; Teubel, W.J.; Van Soest, R.J.; French, P.J.; Homma, Y.; Jenster, G.; De Wit, R.; Van Weerden, W.M. A bypass mechanism of abiraterone-resistant prostate cancer: Accumulating CYP17A1 substrates activate androgen receptor signaling. Prostate 2019, 79, 937–948. [Google Scholar] [CrossRef] [Green Version]
- Taplin, M.-E.; RajeshKumar, B.; Halabi, S.; Werner, C.P.; Woda, B.A.; Picus, J.; Stadler, W.; Hayes, D.F.; Kantoff, P.W.; Vogelzang, N.J.; et al. Androgen Receptor Mutations in Androgen-Independent Prostate Cancer: Cancer and Leukemia Group B Study 9663. J. Clin. Oncol. 2003, 21, 2673–2678. [Google Scholar] [CrossRef] [PubMed]
- Nacusi, L.P.; Tindall, D.J. Androgen receptor abnormalities in castration-recurrent prostate cancer. Expert Rev. Endocrinol. Metab. 2009, 4, 417–422. [Google Scholar] [CrossRef] [Green Version]
- Cai, C.; Chen, S.; Ng, P.; Bubley, G.J.; Nelson, P.S.; Mostaghel, E.A.; Marck, B.; Matsumoto, A.M.; Simon, N.; Wang, H.; et al. Intratumoral de Novo Steroid Synthesis Activates Androgen Receptor in Castration-Resistant Prostate Cancer and Is Upregulated by Treatment with CYP17A1 Inhibitors. Cancer Res. 2011, 71, 6503–6513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchanan, G.; Irvine, R.A.; Coetzee, G.A.; Tilley, W. Contribution of the Androgen Receptor to Prostate Cancer Predisposition and Progression. Cancer Metastasis Rev. 2001, 20, 207–223. [Google Scholar] [CrossRef] [PubMed]
- Attard, G.; Reid, A.H.M.; Auchus, R.J.; Hughes, B.A.; Cassidy, A.M.; Thompson, E.; Oommen, N.B.; Folkerd, E.; Dowsett, M.; Arlt, W.; et al. Clinical and Biochemical Consequences of CYP17A1 Inhibition with Abiraterone Given with and without Exogenous Glucocorticoids in Castrate Men with Advanced Prostate Cancer. J. Clin. Endocrinol. Metab. 2012, 97, 507–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vis, A.N.; Schröder, F.H. Key targets of hormonal treatment of prostate cancer. Part 1: The androgen receptor and steroidogenic pathways. BJU Int. 2009, 104, 438–448. [Google Scholar] [CrossRef]
- Vis, A.N.; Schröder, F.H. Key targets of hormonal treatment of prostate cancer. Part 2: The androgen receptor and 5α-reductase. BJU Int. 2009, 104, 1191–1197. [Google Scholar] [CrossRef]
- Deb, S.; Chin, M.Y.; Pham, S.; Adomat, H.; Hurtado-Coll, A.; Gleave, M.E.; Guns, E.S.T. Steroidogenesis in Peripheral and Transition Zones of Human Prostate Cancer Tissue. Int. J. Mol. Sci. 2021, 22, 487. [Google Scholar] [CrossRef]
- Cortes, V.A.; Busso, D.; Maiz, A.; Arteaga, A.; Nervi, F.; Rigotti, A. Physiological and pathological implications of cholesterol. Front. Biosci. 2014, 19, 416–428. [Google Scholar] [CrossRef] [Green Version]
- Iso, H.; Ikeda, A.; Inoue, M.; Sato, S.; Tsugane, S.; JPHC Study Group. Serum cholesterol levels in relation to the incidence of cancer: The JPHC study cohorts. Int. J. Cancer 2009, 125, 2679–2686. [Google Scholar] [CrossRef]
- Pelton, K.; Freeman, M.R.; Solomon, K.R. Cholesterol and prostate cancer. Curr. Opin. Pharmacol. 2012, 12, 751–759. [Google Scholar] [CrossRef] [Green Version]
- Saranyutanon, S.; Deshmukh, S.K.; Dasgupta, S.; Pai, S.; Singh, S.; Singh, A.P. Cellular and Molecular Progression of Prostate Cancer: Models for Basic and Preclinical Research. Cancers 2020, 12, 2651. [Google Scholar] [CrossRef]
- Bott, S.R.J.; Ng, K.L. (Eds.) Prostate Cancer; Exon Publications: Brisbane, Australia, 2021. [Google Scholar]
- Pisano, C.; Tucci, M.; Di Stefano, R.F.; Turco, F.; Scagliotti, G.V.; Di Maio, M.; Buttigliero, C. Interactions between androgen receptor signaling and other molecular pathways in prostate cancer progression: Current and future clinical implications. Crit. Rev. Oncol. Hematol. 2021, 157, 103185. [Google Scholar] [CrossRef]
- Aurilio, G.; Cimadamore, A.; Mazzucchelli, R.; Lopez-Beltran, A.; Verri, E.; Scarpelli, M.; Massari, F.; Cheng, L.; Santoni, M.; Montironi, R. Androgen Receptor Signaling Pathway in Prostate Cancer: From Genetics to Clinical Applications. Cells 2020, 9, 2653. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Yang, H.; Song, B.-L. Mechanisms and regulation of cholesterol homeostasis. Nat. Rev. Mol. Cell Biol. 2020, 21, 225–245. [Google Scholar] [CrossRef] [PubMed]
- Schade, D.S.; Shey, L.; Eaton, R.P. Cholesterol Review: A Metabolically Important Molecule. Endocr. Pract. 2020, 26, 1514–1523. [Google Scholar] [CrossRef] [PubMed]
- Ryu, S.; Howland, A.; Song, B.; Youn, C.; Song, P.I. Scavenger Receptor Class A to E Involved in Various Cancers. Chonnam Med. J. 2020, 56, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Huff, T.; Boyd, B.; Jialal, I. Physiology, Cholesterol; StatPearls: Treasure Island, FL, USA, 2021. [Google Scholar]
- Ference, A.B.; Kastelein, J.J.P.; Catapano, A.L. Lipids and Lipoproteins in 2020. JAMA 2020, 324, 595–596. [Google Scholar] [CrossRef]
- Feingold, K.R.; Grunfeld, C. Introduction to Lipids and Lipoproteins; Endotext: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Servier medical art. Available online: http://smart.servier.com/ (accessed on 1 June 2021).
- Gomaraschi, M. Role of Lipoproteins in the Microenvironment of Hormone-Dependent Cancers. Trends Endocrinol. Metab. 2020, 31, 256–268. [Google Scholar] [CrossRef]
- Charlton-Menys, V.; Durrington, P.N. Human cholesterol metabolism and therapeutic molecules. Exp. Physiol. 2008, 93, 27–42. [Google Scholar] [CrossRef]
- Parthasarathy, S.; Raghavamenon, A.; Garelnabi, M.O.; Santanam, N. Oxidized low-density lipoprotein. Methods Mol. Biol. 2010, 610, 403–417. [Google Scholar] [PubMed] [Green Version]
- Zhou, L.; Li, C.; Gao, L.; Wang, A. High-density lipoprotein synthesis and metabolism. Mol. Med. Rep. 2015, 12, 4015–4021. [Google Scholar] [CrossRef] [Green Version]
- Luo, D.-X.; Cao, D.-L.; Xiong, Y.; Peng, X.-H.; Liao, D.-F. A novel model of cholesterol efflux from lipid-loaded cells. Acta Pharmacol. Sin. 2010, 31, 1243–1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotani, K.; Sekine, Y.; Ishikawa, S.; Ikpot, I.Z.; Suzuki, K.; Remaley, A.T. High-Density Lipoprotein and Prostate Cancer: An Overview. J. Epidemiol. 2013, 23, 313–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, T.; Zhu, N.; Shi, Y.; Wang, Y.; Zhang, C.; Deng, C.; Liao, D.; Qin, L. Targeting HDL in tumor microenvironment: New hope for cancer therapy. J. Cell. Physiol. 2021. [Google Scholar] [CrossRef]
- Liang, C.-Z.; Fan, Y.-D.; Wang, J.; Xu, L.-F.; Liu, C.; Huang, T. Identifying the role of apolipoprotein A-I in prostate cancer. Asian J. Androl. 2021, 23, 400–408. [Google Scholar] [CrossRef]
- White, C.P. On the occurrence of crystals in tumours. J. Pathol. Bacteriol. 1909, 13, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Swyer, G.I.M. The cholesterol content of normal and enlarged prostates. Cancer Res. 1942, 2, 372–375. [Google Scholar]
- Pearce, M.L.; Dayton, S. Incidence of Cancer in Men on a Diet High in Polyunsaturated Fat. Lancet 1971, 297, 464–467. [Google Scholar] [CrossRef]
- Muldoon, M.F.; Manuck, S.B.; Matthews, K.A. Lowering cholesterol concentrations and mortality: A quantitative review of primary prevention trials. BMJ 1990, 301, 309–314. [Google Scholar] [CrossRef] [Green Version]
- Yusuf, S.; Wittes, J.; Friedman, L. Overview of results of randomized clinical trials in heart disease. II. Unstable angina, heart failure, primary prevention with aspirin, and risk factor modification. JAMA 1988, 260, 2259–2263. [Google Scholar] [CrossRef]
- Solomon, K.R.; Freeman, M.R. The Complex Interplay Between Cholesterol and Prostate Malignancy. Urol. Clin. N. Am. 2011, 38, 243–259. [Google Scholar] [CrossRef] [Green Version]
- Bravi, F.; Scotti, L.; Bosetti, C.; Talamini, R.; Negri, E.; Montella, M.; Franceschi, S.; La Vecchia, C. Self-reported history of hypercholesterolaemia and gallstones and the risk of prostate cancer. Ann. Oncol. 2006, 17, 1014–1017. [Google Scholar] [CrossRef]
- Platz, E.A.; Clinton, S.K.; Giovannucci, E. Association between plasma cholesterol and prostate cancer in the PSA era. Int. J. Cancer 2008, 123, 1693–1698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batty, G.D.; Kivimaki, M.; Clarke, R.; Smith, G.D.; Shipley, M.J. Modifiable risk factors for prostate cancer mortality in London: Forty years of follow-up in the Whitehall study. Cancer Causes Control. 2011, 22, 311–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yannucci, J.; Manola, J.; Garnick, M.B.; Bhat, G.; Bubley, G.J. The Effect of Androgen Deprivation Therapy on Fasting Serum Lipid and Glucose Parameters. J. Urol. 2006, 176, 520–525. [Google Scholar] [CrossRef] [PubMed]
- Mohamedali, H.Z.; Breunis, H.; Timilshina, N.; Alibhai, S.M. Changes in blood glucose and cholesterol levels due to androgen deprivation therapy in men with non-metastatic prostate cancer. Can. Urol. Assoc. J. 2011, 5, 28–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, W.; Gao, S.; Barrett, D.; Ahmed, M.; Han, D.; Macoska, J.A.; He, H.H.; Cai, C. Reactivation of androgen receptor-regulated lipid biosynthesis drives the progression of castration-resistant prostate cancer. Oncogene 2018, 37, 710–721. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.R.; Finkelstein, J.S.; McGovern, F.J.; Zietman, A.L.; Fallon, M.A.; Schoenfeld, D.A.; Kantoff, P.W. Changes in body composition during androgen deprivation therapy for prostate cancer. J. Clin. Endocrinol. Metab. 2002, 87, 599–603. [Google Scholar] [CrossRef] [PubMed]
- Dockery, F.; Bulpitt, C.J.; Agarwal, S.; Donaldson, M.; Rajkumar, C. Testosterone suppression in men with prostate cancer leads to an increase in arterial stiffness and hyperinsulinaemia. Clin. Sci. 2003, 104, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Eri, L.M.; Urdal, P.; Bechensteen, A.G. Effects of the luteinizing hormone-releasing hormone agonist leuprolide on lipoproteins, fibrinogen and plasminogen activator inhibitor in patients with benign prostatic hyperplasia. J. Urol. 1995, 154, 100–104. [Google Scholar] [CrossRef]
- Torimoto, K.; Samma, S.; Kagebayashi, Y.; Chihara, Y.; Tanaka, N.; Hirayama, A.; Fujimoto, K.; Hirao, Y. The Effects of Androgen Deprivation Therapy on Lipid Metabolism and Body Composition in Japanese Patients with Prostate Cancer. Jpn. J. Clin. Oncol. 2011, 41, 577–581. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.M.; Kam, S.C. Metabolic effects of androgen deprivation therapy. Korean J. Urol. 2015, 56, 12–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thysell, E.; Surowiec, I.; Hörnberg, E.; Crnalic, S.; Widmark, A.; Johansson, A.I.; Stattin, P.; Bergh, A.; Moritz, T.; Antti, H.; et al. Metabolomic Characterization of Human Prostate Cancer Bone Metastases Reveals Increased Levels of Cholesterol. PLoS ONE 2010, 5, e14175. [Google Scholar] [CrossRef] [Green Version]
- Hirano, H.; Ide, H.; Lu, Y.; Inoue, Y.; Okada, H.; Horie, S. Impact of Pretreatment Total Cholesterol Level Is Associated With Metastasis of Prostate Cancer. Am. J. Men’s Health 2020, 14. [Google Scholar] [CrossRef]
- Llaverias, G.; Danilo, C.; Wang, Y.; Witkiewicz, A.K.; Daumer, K.; Lisanti, M.P.; Frank, P. A Western-Type Diet Accelerates Tumor Progression in an Autochthonous Mouse Model of Prostate Cancer. Am. J. Pathol. 2010, 177, 3180–3191. [Google Scholar] [CrossRef]
- Zhuang, L.; Kim, J.; Adam, R.M.; Solomon, K.R.; Freeman, M.R. Cholesterol targeting alters lipid raft composition and cell survival in prostate cancer cells and xenografts. J. Clin. Investig. 2005, 115, 959–968. [Google Scholar] [CrossRef] [Green Version]
- Solomon, K.R.; Pelton, K.; Boucher, K.; Joo, J.; Tully, C.; Zurakowski, D.; Schaffner, C.P.; Kim, J.; Freeman, M.R. Ezetimibe Is an Inhibitor of Tumor Angiogenesis. Am. J. Pathol. 2009, 174, 1017–1026. [Google Scholar] [CrossRef] [Green Version]
- Mostaghel, E.A.; Solomon, K.R.; Pelton, K.; Freeman, M.R.; Montgomery, R.B. Impact of Circulating Cholesterol Levels on Growth and Intratumoral Androgen Concentration of Prostate Tumors. PLoS ONE 2012, 7, e30062. [Google Scholar] [CrossRef]
- Dłubek, J.; Rysz, J.; Jabłonowski, Z.; Gluba-Brzózka, A.; Franczyk, B. The Correlation between Lipid Metabolism Disorders and Prostate Cancer. Curr. Med. Chem. 2021, 28, 2048–2061. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.; Balk, S.P. Intratumoral androgen biosynthesis in prostate cancer pathogenesis and response to therapy. Endocr.-Relat. Cancer 2011, 18, R175–R182. [Google Scholar] [CrossRef] [Green Version]
- Deb, S.; Pham, S.; Ming, D.-S.; Chin, M.Y.; Adomat, H.; Hurtado-Coll, A.; Gleave, M.E.; Guns, E.S.T. Characterization of Precursor-Dependent Steroidogenesis in Human Prostate Cancer Models. Cancers 2018, 10, 343. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Hughes-Fulford, M. Human prostate cancer cells lack feedback regulation of low-density lipoprotein receptor and its regulator, SREBP2. Int. J. Cancer 2001, 91, 41–45. [Google Scholar] [CrossRef]
- Schörghofer, D.; Kinslechner, K.; Preitschopf, A.; Schütz, B.; Röhrl, C.; Hengstschläger, M.; Stangl, H.; Mikula, M. The HDL receptor SR-BI is associated with human prostate cancer progression and plays a possible role in establishing androgen independence. Reprod. Biol. Endocrinol. 2015, 13, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Li-Beisson, Y.; Carrière, F. Biogenesis and fate of lipid droplets. Biochimie 2020, 169, 1–2. [Google Scholar] [CrossRef]
- Lee, B.; Taylor, M.; Robinet, P.; Smith, J.D.; Schweitzer, J.; Sehayek, E.; Falzarano, S.M.; Magi-Galluzzi, C.; Klein, E.A.; Ting, A.H. Dysregulation of Cholesterol Homeostasis in Human Prostate Cancer through Loss of ABCA1. Cancer Res. 2013, 73, 1211–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murtola, T.J.; Syvälä, H.; Pennanen, P.; Bläuer, M.; Solakivi, T.; Ylikomi, T.; Tammela, T.L.J. The Importance of LDL and Cholesterol Metabolism for Prostate Epithelial Cell Growth. PLoS ONE 2012, 7, e39445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekine, Y.; Demosky, S.J.; Stonik, J.A.; Furuya, Y.; Koike, H.; Suzuki, K.; Remaley, A.T. High-Density Lipoprotein Induces Proliferation and Migration of Human Prostate Androgen–Independent Cancer Cells by an ABCA1-Dependent Mechanism. Mol. Cancer Res. 2010, 8, 1284–1294. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Wang, Y.; Hao, S.; Qin, Y.; Wu, Y. Knockdown of sterol O-acyltransferase 1 (SOAT1) suppresses SCD1-mediated lipogenesis and cancer procession in prostate cancer. Prostaglandins Other Lipid Mediat. 2021, 153, 106537. [Google Scholar] [CrossRef]
- Zhang, D.; Lagace, T.A.; Garuti, R.; Zhao, Z.; McDonald, M.; Horton, J.D.; Cohen, J.C.; Hobbs, H.H. Binding of Proprotein Convertase Subtilisin/Kexin Type 9 to Epidermal Growth Factor-like Repeat A of Low Density Lipoprotein Receptor Decreases Receptor Recycling and Increases Degradation. J. Biol. Chem. 2007, 282, 18602–18612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahboobnia, K.; Pirro, M.; Marini, E.; Grignani, F.; Bezsonov, E.E.; Jamialahmadi, T.; Sahebkar, A. PCSK9 and cancer: Rethinking the link. Biomed. Pharmacother. 2021, 140, 111758. [Google Scholar] [CrossRef] [PubMed]
- Klein-Szanto, A.J.; Bassi, D.E. Proprotein convertase inhibition: Paralyzing the cell’s master switches. Biochem. Pharmacol. 2017, 140, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Jernberg, E.; Bergh, A.; Wikström, P. Clinical relevance of androgen receptor alterations in prostate cancer. Endocr. Connect. 2017, 6, R146–R161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alaupovic, P.; Heinonen, T.; Shurzinske, L.; Black, D.M. Effect of a new HMG-CoA reductase inhibitor, atorvastatin, on lipids, apolipoproteins and lipoprotein particles in patients with elevated serum cholesterol and triglyceride levels. Atherosclerosis 1997, 133, 123–133. [Google Scholar] [CrossRef]
- Olsson, A.G.; Pears, J.; McKellar, J.; Mizan, J.; Raza, A. Effect of rosuvastatin on low-density lipoprotein cholesterol in patients with hypercholesterolemia. Am. J. Cardiol. 2001, 88, 504–508. [Google Scholar] [CrossRef]
- Giugliano, R.P.; Desai, N.R.; Kohli, P.; Rogers, W.J.; Somaratne, R.; Huang, F.; Liu, T.; Mohanavelu, S.; Hoffman, E.B.; McDonald, S.T.; et al. Efficacy, safety, and tolerability of a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 in combination with a statin in patients with hypercholesterolaemia (LAPLACE-TIMI 57): A randomised, placebo-controlled, dose-ranging, phase 2 study. Lancet 2012, 380, 2007–2017. [Google Scholar] [CrossRef] [Green Version]
- Chan, J.C.Y.; Piper, D.E.; Cao, Q.; Liu, D.; King, C.; Wang, W.; Tang, J.; Liu, Q.; Higbee, J.; Xia, Z.; et al. A proprotein convertase subtilisin/kexin type 9 neutralizing antibody reduces serum cholesterol in mice and nonhuman primates. Proc. Natl. Acad. Sci. USA 2009, 106, 9820–9825. [Google Scholar] [CrossRef] [Green Version]
- McCague, R.; Rowlands, M.G.; Barrie, S.E.; Houghton, J. Inhibition of enzymes of estrogen and androgen biosynthesis by esters of 4-pyridylacetic acid. J. Med. Chem. 1990, 33, 3050–3055. [Google Scholar] [CrossRef]
- Baigent, C.; Keech, A.C.; Kearney, P.; Blackwell, L.; Buck, G.; Pollicino, C.; Kirby, A.; Sourjina, T.; Peto, R.; Collins, R.; et al. Efficacy and safety of cholesterol-lowering treatment: Prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 2005, 366, 1267–1278. [Google Scholar] [CrossRef] [PubMed]
- Cholesterol Treatment Trialists’ (CTT) Collaboration; Baigent, C.; Blackwell, L.; Emberson, J.; Holland, L.E.; Reith, C.; Bhala, N.; Peto, R.; Barnes, E.H.; Keech, A.; et al. Efficacy and safety of more intensive lowering of LDL cholesterol: A meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet 2010, 376, 1670–1681. [Google Scholar] [PubMed] [Green Version]
- Cholesterol Treatment Trialists’ (CTT) Collaboration; Fulcher, J.; O’Connell, R.; Voysey, M.; Emberson, J.; Blackwell, L.; Mihaylova, B.; Simes, J.; Collins, R.; Kirby, A.; et al. Efficacy and safety of LDL-lowering therapy among men and women: Meta-analysis of individual data from 174,000 participants in 27 randomised trials. Lancet 2015, 385, 1397–1405. [Google Scholar] [PubMed]
- Barbalata, C.I.; Tefas, L.R.; Achim, M.; Tomuta, I.; Porfire, A.S. Statins in risk-reduction and treatment of cancer. World J. Clin. Oncol. 2020, 11, 573–588. [Google Scholar] [CrossRef] [PubMed]
- Stancu, C.S.; Sima, A. Statins: Mechanism of action and effects. J. Cell. Mol. Med. 2001, 5, 378–387. [Google Scholar] [CrossRef]
- Feingold, K.R. Cholesterol Lowering Drugs; Endotext: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Ferri, N.; Corsini, A. Clinical Pharmacology of Statins: An Update. Curr. Atheroscler. Rep. 2020, 22, 1–9. [Google Scholar] [CrossRef]
- Ikonen, E. Cellular cholesterol trafficking and compartmentalization. Nat. Rev. Mol. Cell Biol. 2008, 9, 125–138. [Google Scholar] [CrossRef]
- Rached, F.; Santos, R.D. The Role of Statins in Current Guidelines. Curr. Atheroscler. Rep. 2020, 22, 1–11. [Google Scholar] [CrossRef]
- Demierre, M.-F.; Higgins, P.D.R.; Gruber, S.B.; Hawk, E.T.; Lippman, S.M. Statins and cancer prevention. Nat. Rev. Cancer 2005, 5, 930–942. [Google Scholar] [CrossRef]
- Papadopoulos, G.; Delakas, D.; Nakopoulou, L.; Kasimatis, T. Statins and prostate cancer: Molecular and clinical aspects. Eur. J. Cancer 2011, 47, 819–830. [Google Scholar] [CrossRef]
- Alfaqih, M.A.; Allott, E.H.; Hamilton, R.J.; Freeman, M.R.; Freedland, S.J. The current evidence on statin use and prostate cancer prevention: Are we there yet? Nat. Rev. Urol. 2017, 14, 107–119. [Google Scholar] [PubMed] [Green Version]
- Mucci, L.A.; Stampfer, M.J. Mounting Evidence for Prediagnostic Use of Statins in Reducing Risk of Lethal Prostate Cancer. J. Clin. Oncol. 2014, 32, 1–2. [Google Scholar] [CrossRef]
- Hatano, K.; Fujita, K.; Nonomura, N. Application of Anti-Inflammatory Agents in Prostate Cancer. J. Clin. Med. 2020, 9, 2680. [Google Scholar] [CrossRef] [PubMed]
- Hoque, A.; Chen, H.; Xu, X.-C. Statin Induces Apoptosis and Cell Growth Arrest in Prostate Cancer Cells. Cancer Epidemiol. Biomark. Prev. 2008, 17, 88–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, M.; Hart, C.A.; Tawadros, T.; Ramani, V.; Sangar, V.; Lau, M.; Clarke, N. The differential effects of statins on the metastatic behaviour of prostate cancer. Br. J. Cancer 2012, 106, 1689–1696. [Google Scholar] [CrossRef] [Green Version]
- Ingersoll, M.A.; Miller, D.R.; Martinez, O.; Wakefield, C.B.; Hsieh, K.-C.; Simha, M.V.; Kao, C.-L.; Chen, H.-T.; Batra, S.K.; Lin, M.-F. Statin derivatives as therapeutic agents for castration-resistant prostate cancer. Cancer Lett. 2016, 383, 94–105. [Google Scholar] [CrossRef] [Green Version]
- Neuwirt, H.; Bouchal, J.; Kharaishvili, G.; Ploner, C.; Jöhrer, K.; Pitterl, F.; Weber, A.; Klocker, H.; Eder, I.E. Cancer-associated fibroblasts promote prostate tumor growth and progression through upregulation of cholesterol and steroid biosynthesis. Cell Commun. Signal. 2020, 18, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Syvälä, H.; Pennanen, P.; Bläuer, M.; Tammela, T.L.; Murtola, T.J. Additive inhibitory effects of simvastatin and enzalutamide on androgen-sensitive LNCaP and VCaP prostate cancer cells. Biochem. Biophys. Res. Commun. 2016, 481, 46–50. [Google Scholar] [CrossRef]
- Wang, H.; Cui, X.-X.; Goodin, S.; Ding, N.; Van Doren, J.; Du, Z.; Huang, M.-T.; Liu, Y.; Cheng, X.; Dipaola, R.S.; et al. Inhibition of IL-6 expression in LNCaP prostate cancer cells by a combination of atorvastatin and celecoxib. Oncol. Rep. 2013, 31, 835–841. [Google Scholar] [CrossRef] [Green Version]
- Parikh, A.; Childress, C.; Deitrick, K.; Lin, Q.; Rukstalis, D.; Yang, W. Statin-induced autophagy by inhibition of geranylgeranyl biosynthesis in prostate cancer PC3 cells. Prostate 2010, 70, 971–981. [Google Scholar] [CrossRef]
- Zheng, X.; Cui, X.-X.; Gao, Z.; Zhao, Y.; Lin, Y.; Shih, W.J.; Huang, M.-T.; Liu, Y.; Rabson, A.; Reddy, B.; et al. Atorvastatin and Celecoxib in Combination Inhibits the Progression of Androgen-Dependent LNCaP Xenograft Prostate Tumors to Androgen Independence. Cancer Prev. Res. 2010, 3, 114–124. [Google Scholar] [CrossRef] [Green Version]
- Krycer, J.R.; Kristiana, I.; Brown, A.J. Cholesterol Homeostasis in Two Commonly Used Human Prostate Cancer Cell-Lines, LNCaP and PC-3. PLoS ONE 2009, 4, e8496. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Egger, M.; Plattner, R.; Klocker, H.; Eder, I.E. Lovastatin Causes Diminished PSA Secretion by Inhibiting AR Expression and Function in LNCaP Prostate Cancer Cells. Urology 2011, 77, 1508.e1–1508.e7. [Google Scholar] [CrossRef]
- Graaf, M.R.; Beiderbeck, A.B.; Egberts, T.; Richel, D.J.; Guchelaar, H.-J. The Risk of Cancer in Users of Statins. J. Clin. Oncol. 2004, 22, 2388–2394. [Google Scholar] [CrossRef] [PubMed]
- Friis, S.; Poulsen, A.H.; Johnsen, S.P.; McLaughlin, J.K.; Fryzek, J.P.; Dalton, S.O.; Sørensen, H.T.; Olsen, J.H. Cancer risk among statin users: A population-based cohort study. Int. J. Cancer 2005, 114, 643–647. [Google Scholar] [CrossRef]
- Shannon, J.; Tewoderos, S.; Garzotto, M.; Beer, T.M.; Derenick, R.; Palma, A.; Farris, P.E. Statins and Prostate Cancer Risk: A Case-Control Study. Am. J. Epidemiol. 2005, 162, 318–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singal, R.; Khurana, V.; Caldito, G.; Fort, C. Statins and prostate cancer risk: A large case control study in veterans. J. Clin. Oncol. 2005, 23, 1004. [Google Scholar] [CrossRef]
- Platz, E.A.; Leitzmann, M.F.; Visvanathan, K.; Rimm, E.B.; Stampfer, M.J.; Willett, W.C.; Giovannucci, E. Statin Drugs and Risk of Advanced Prostate Cancer. J. Natl. Cancer Inst. 2006, 98, 1819–1825. [Google Scholar] [CrossRef] [PubMed]
- Murtola, T.J.; Tammela, T.L.; Lahtela, J.; Auvinen, A. Cholesterol-Lowering Drugs and Prostate Cancer Risk: A Population-based Case-Control Study. Cancer Epidemiol. Biomark. Prev. 2007, 16, 2226–2232. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, E.J.; Rodriguez, C.; Bain, E.B.; Wang, Y.; Thun, M.J.; Calle, E.E. Cholesterol-Lowering Drugs and Advanced Prostate Cancer Incidence in a Large U.S. Cohort. Cancer Epidemiol. Biomark. Prev. 2007, 16, 2213–2217. [Google Scholar] [CrossRef] [Green Version]
- Bonovas, S.; Filioussi, K.; Sitaras, N.M. Statin use and the risk of prostate cancer: A metaanalysis of 6 randomized clinical trials and 13 observational studies. Int. J. Cancer 2008, 123, 899–904. [Google Scholar] [CrossRef] [PubMed]
- Breau, R.H.; Karnes, R.J.; Jacobson, D.J.; McGree, M.E.; Jacobsen, S.J.; Nehra, A.; Lieber, M.M.; Sauver, J.L.S. The Association Between Statin Use and the Diagnosis of Prostate Cancer in a Population Based Cohort. J. Urol. 2010, 184, 494–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murtola, T.J.; Tammela, T.L.; Määttänen, L.; Huhtala, H.; Platz, E.A.; Ala-Opas, M.; Stenman, U.-H.; Auvinen, A. Prostate cancer and PSA among statin users in the Finnish prostate cancer screening trial. Int. J. Cancer 2010, 127, 1650–1659. [Google Scholar] [CrossRef]
- Farwell, W.R.; D’Avolio, L.W.; Scranton, R.E.; Lawler, E.V.; Gaziano, J.M. Statins and Prostate Cancer Diagnosis and Grade in a Veterans Population. J. Natl. Cancer Inst. 2011, 103, 885–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, N.; Klein, E.A.; Li, J.; Moussa, A.S.; Jones, J.S. Statin Use and Risk of Prostate Cancer in a Population of Men Who Underwent Biopsy. J. Urol. 2011, 186, 86–90. [Google Scholar] [CrossRef]
- Bansal, D.; Undela, K.; D’Cruz, S.; Schifano, F. Statin Use and Risk of Prostate Cancer: A Meta-Analysis of Observational Studies. PLoS ONE 2012, 7, e46691. [Google Scholar] [CrossRef]
- Jespersen, C.G.; Nørgaard, M.; Friis, S.; Skriver, C.; Borre, M. Statin use and risk of prostate cancer: A Danish population-based case-control study, 1997–2010. Cancer Epidemiol. 2014, 38, 42–47. [Google Scholar] [CrossRef]
- Lustman, A.; Nakar, S.; Cohen, A.D.; Vinker, S. Statin use and incident prostate cancer risk: Does the statin brand matter? A population-based cohort study. Prostate Cancer Prostatic Dis. 2014, 17, 6–9. [Google Scholar] [CrossRef]
- Van Rompay, M.I.; Solomon, K.R.; Nickel, J.C.; Ranganathan, G.; Kantoff, P.W.; McKinlay, J.B. Prostate cancer incidence and mortality among men using statins and non-statin lipid-lowering medications. Eur. J. Cancer 2019, 112, 118–126. [Google Scholar] [CrossRef]
- Allott, E.H.; Ebot, E.M.; Stopsack, K.H.; Gonzalez-Feliciano, A.G.; Markt, S.C.; Wilson, K.M.; Ahearn, T.U.; Gerke, T.; Downer, M.K.; Rider, J.R.; et al. Statin Use Is Associated with Lower Risk of PTEN-Null and Lethal Prostate Cancer. Clin. Cancer Res. 2020, 26, 1086–1093. [Google Scholar] [CrossRef]
- Kantor, E.D.; Lipworth, L.; Fowke, J.H.; Giovannucci, E.L.; Mucci, L.A.; Signorello, L.B. Statin use and risk of prostate cancer: Results from the Southern Community Cohort Study. Prostate 2015, 75, 1384–1393. [Google Scholar] [CrossRef] [Green Version]
- Park, H.S.; Schoenfeld, J.D.; Mailhot, R.B.; Shive, M.; Hartman, R.I.; Ogembo, R.; Mucci, L.A. Statins and prostate cancer recurrence following radical prostatectomy or radiotherapy: A systematic review and meta-analysis. Ann. Oncol. 2013, 24, 1427–1434. [Google Scholar] [CrossRef] [PubMed]
- Geybels, M.S.; Wright, J.L.; Holt, S.K.; Kolb, S.; Feng, Z.; Stanford, J.L. Statin Use in Relation to Prostate Cancer Outcomes in a Population-based Patient Cohort Study. Prostate 2013, 73, 1214–1222. [Google Scholar] [CrossRef] [Green Version]
- Harshman, L.C.; Wang, X.; Nakabayashi, M.; Xie, W.; Valenca, L.B.; Werner, L.; Yu, Y.; Kantoff, A.M.; Sweeney, C.J.; Mucci, L.A.; et al. Statin Use at the Time of Initiation of Androgen Deprivation Therapy and Time to Progression in Patients with Hormone-Sensitive Prostate Cancer. JAMA Oncol. 2015, 1, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Marcella, S.W.; David, A.; Ohman-Strickland, P.A.; Carson, J.; Rhoads, G.G. Statin use and fatal prostate cancer: A matched case-control study. Cancer 2012, 118, 4046–4052. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.F.; Nordestgaard, B.G.; Bojesen, S.E. Statin Use and Reduced Cancer-Related Mortality. N. Engl. J. Med. 2012, 367, 1792–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grytli, H.H.; Fagerland, M.W.; Fosså, S.D.; Taskén, K.A. Association between use of beta-blockers and prostate cancer-specific survival: A cohort study of 3561 prostate cancer patients with high-risk or metastatic disease. Eur. Urol. 2014, 65, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Yu, O.; Eberg, M.; Benayoun, S.; Aprikian, A.; Batist, G.; Suissa, S.; Azoulay, L. Use of Statins and the Risk of Death in Patients With Prostate Cancer. J. Clin. Oncol. 2014, 32, 5–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsen, S.B.; Dehlendorff, C.; Skriver, C.; Dalton, S.O.; Jespersen, C.G.; Borre, M.; Brasso, K.; Nørgaard, M.; Johansen, C.; Sørensen, H.T.; et al. Postdiagnosis Statin Use and Mortality in Danish Patients with Prostate Cancer. J. Clin. Oncol. 2017, 35, 3290–3297. [Google Scholar] [CrossRef]
- Friedman, G.D.; Flick, E.D.; Udaltsova, N.; Chan Pharm, D.J.; Quesenberry, C.P.; Habel, L. Screening statins for possible carcinogenic risk: Up to 9 years of follow-up of 361 859 recipients. Pharmacoepidemiol. Drug Saf. 2008, 17, 27–36. [Google Scholar] [CrossRef]
- Kuoppala, J.; Lamminpää, A.; Pukkala, E. Statins and cancer: A systematic review and meta-analysis. Eur. J. Cancer 2008, 44, 2122–2132. [Google Scholar] [CrossRef]
- Smeeth, L.; Douglas, I.; Hall, A.J.; Hubbard, R.; Evans, S. Effect of statins on a wide range of health outcomes: A cohort study validated by comparison with randomized trials. Br. J. Clin. Pharmacol. 2009, 67, 99–109. [Google Scholar] [CrossRef] [Green Version]
- Haukka, J.; Sankila, R.; Klaukka, T.; Lonnqvist, J.; Niskanen, L.; Tanskanen, A.; Wahlbeck, K.; Tiihonen, J. Incidence of cancer and statin usage-Record linkage study. Int. J. Cancer 2010, 126, 279–284. [Google Scholar] [CrossRef]
- Hippisley-Cox, J.; Coupland, C. Unintended effects of statins in men and women in England and Wales: Population based cohort study using the QResearch database. BMJ 2010, 340, c2197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobs, E.J.; Newton, C.C.; Thun, M.J.; Gapstur, S.M. Long-term Use of Cholesterol-Lowering Drugs and Cancer Incidence in a Large United States Cohort. Cancer Res. 2011, 71, 1763–1771. [Google Scholar] [CrossRef] [Green Version]
- Chan, J.M.; Litwack-Harrison, S.; Bauer, S.; Daniels, N.A.; Wilt, T.J.; Shannon, J.; Bauer, D.C. Statin Use and Risk of Prostate Cancer in the Prospective Osteoporotic Fractures in Men (MrOS) Study. Cancer Epidemiol. Biomark. Prev. 2012, 21, 1886–1888. [Google Scholar] [CrossRef] [Green Version]
- Freedland, S.J.; Hamilton, R.; Gerber, L.; Banez, L.L.; Moreira, D.; Andriole, G.L.; Rittmaster, R.S. Statin use and risk of prostate cancer and high-grade prostate cancer: Results from the REDUCE study. Prostate Cancer Prostatic Dis. 2013, 16, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Platz, E.A.; Tangen, C.M.; Goodman, P.J.; Till, C.; Parnes, H.L.; Figg, W.D.; Albanes, D.; Neuhouser, M.L.; Klein, E.A.; Lucia, M.S.; et al. Statin Drug Use is Not Associated with Prostate Cancer Risk in Men Who are Regularly Screened. J. Urol. 2014, 192, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Fowke, J.H.; Motley, S.S.; Barocas, D.A.; Cookson, M.S.; Concepcion, R.; Byerly, S.; Smith, J.A. The associations between statin use and prostate cancer screening, prostate size, high-grade prostatic intraepithelial neoplasia (PIN), and prostate cancer. Cancer Causes Control. 2011, 22, 417–426. [Google Scholar] [CrossRef] [Green Version]
- Coogan, P.F.; Kelly, J.P.; Strom, B.L.; Rosenberg, L. Statin and NSAID use and prostate cancer risk. Pharmacoepidemiol. Drug Saf. 2010, 19, 752–755. [Google Scholar] [CrossRef] [PubMed]
- Boudreau, D.M.; Yu, O.; Buist, D.S.M.; Miglioretti, D.L. Statin use and prostate cancer risk in a large population-based setting. Cancer Causes Control. 2008, 19, 767–774. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.-C.; Ho, S.-C.; Chiu, H.-F.; Yang, C.-Y. Statins increase the risk of prostate cancer: A population-based case-control study. Prostate 2011, 71, 1818–1824. [Google Scholar] [CrossRef]
- Flick, E.D.; Habel, L.A.; Chan, K.A.; Van Den Eeden, S.K.; Quinn, V.P.; Haque, R.; Orav, E.J.; Seeger, J.D.; Sadler, M.C.; Quesenberry, C.P., Jr.; et al. Statin use and risk of prostate cancer in the California Men’s Health Study cohort. Cancer Epidemiol. Biomark. Prev. 2007, 16, 2218–2225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanai, Y.; Kosaka, T.; Hongo, H.; Oya, M. Clinically complete response to abiraterone acetate in a patient with metastatic castration-resistant prostate cancer. IJU Case Rep. 2019, 2, 187–189. [Google Scholar] [CrossRef] [PubMed]
- Di Lorenzo, G.; Sonpavde, G.; Pond, G.; Lucarelli, G.; Rossetti, S.; Facchini, G.; Scagliarini, S.; Cartenì, G.; Federico, P.; Daniele, B.; et al. Statin Use and Survival in Patients with Metastatic Castration-resistant Prostate Cancer Treated with Abiraterone Acetate. Eur. Urol. Focus 2018, 4, 874–879. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Pang, L.; Hu, X.; Wang, W.; Xu, B.; Zhang, X.; Liu, L. The effect of statins on advanced prostate cancer patients with androgen deprivation therapy or abiraterone/enzalutamide: A systematic review and meta-analysis. J. Clin. Pharm. Ther. 2020, 45, 488–495. [Google Scholar] [CrossRef] [PubMed]
- Gordon, J.A.; Buonerba, C.; Pond, G.; Crona, D.; Gillessen, S.; Lucarelli, G.; Rossetti, S.; Dorff, T.; Artale, S.; Locke, J.A.; et al. Statin use and survival in patients with metastatic castration-resistant prostate cancer treated with abiraterone or enzalutamide after docetaxel failure: The international retrospective observational STABEN study. Oncotarget 2018, 9, 19861–19873. [Google Scholar] [CrossRef] [PubMed]
- Gordon, J.A.; Midha, A.; Szeitz, A.; Ghaffari, M.; Adomat, H.H.; Guo, Y.; Klassen, T.L.; Guns, E.S.; Wasan, K.M.; Cox, M.E. Oral simvastatin administration delays castration-resistant progression and reduces intratumoral steroidogenesis of LNCaP prostate cancer xenografts. Prostate Cancer Prostatic Dis. 2016, 19, 21–27. [Google Scholar] [CrossRef]
- Twiddy, A.L.; Leon, C.G.; Wasan, K.M. Cholesterol as a Potential Target for Castration-Resistant Prostate Cancer. Pharm. Res. 2011, 28, 423–437. [Google Scholar] [CrossRef]
- Kim, J.H.; Cox, M.E.; Wasan, K.M. Effect of simvastatin on castration-resistant prostate cancer cells. Lipids Health Dis. 2014, 13, 56. [Google Scholar] [CrossRef] [Green Version]
- Zani, I.A.; Stephen, S.L.; Mughal, N.A.; Russell, D.; Homer-Vanniasinkam, S.; Wheatcroft, S.B.; Ponnambalam, S. Scavenger Receptor Structure and Function in Health and Disease. Cells 2015, 4, 178–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landschulz, K.T.; Pathak, R.K.; Rigotti, A.; Krieger, M.; Hobbs, H.H. Regulation of scavenger receptor, class B, type I, a high density lipoprotein receptor, in liver and steroidogenic tissues of the rat. J. Clin. Investig. 1996, 98, 984–995. [Google Scholar] [CrossRef] [Green Version]
- Rigotti, A.; Edelman, E.R.; Seifert, P.; Iqbal, S.N.; DeMattos, R.B.; Temel, R.E.; Krieger, M.; Williams, D.L. Regulation by Adrenocorticotropic Hormone of the in Vivo Expression of Scavenger Receptor Class B Type I (SR-BI), a High Density Lipoprotein Receptor, in Steroidogenic Cells of the Murine Adrenal Gland. J. Biol. Chem. 1996, 271, 33545–33549. [Google Scholar] [CrossRef] [Green Version]
- Canton, J.; Neculai, D.; Grinstein, S. Scavenger receptors in homeostasis and immunity. Nat. Rev. Immunol. 2013, 13, 621–634. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.-J.; Azhar, S.; Kraemer, F. SR-B1: A Unique Multifunctional Receptor for Cholesterol Influx and Efflux. Annu. Rev. Physiol. 2018, 80, 95–116. [Google Scholar] [CrossRef] [PubMed]
- Krieger, M. Charting the Fate of the “Good Cholesterol”: Identification and Characterization of the High-Density Lipoprotein Receptor SR-BI. Annu. Rev. Biochem. 1999, 68, 523–558. [Google Scholar] [CrossRef]
- Rigotti, A.; Miettinen, H.E.; Krieger, M. The Role of the High-Density Lipoprotein Receptor SR-BI in the Lipid Metabolism of Endocrine and Other Tissues. Endocr. Rev. 2003, 24, 357–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gwynne, J.T.; Mahaffee, D.D. Rat adrenal uptake and metabolism of high density lipoprotein cholesteryl ester. J. Biol. Chem. 1989, 264, 8141–8150. [Google Scholar] [CrossRef]
- Gwynne, J.T.; Mahaffee, D.; Brewer, H.B.; Ney, R.L. Adrenal cholesterol uptake from plasma lipoproteins: Regulation by corticotropin. Proc. Natl. Acad. Sci. USA 1976, 73, 4329–4333. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.W.; Azhar, S.; Kraemer, F.B. ACTH Regulation of Adrenal SR-B1. Front. Endocrinol. 2016, 7, 42. [Google Scholar] [CrossRef] [Green Version]
- Shen, W.-J.; Hu, J.; Hu, Z.; Kraemer, F.B.; Azhar, S. Scavenger Receptor class B type I (SR-BI): A versatile receptor with multiple functions and actions. Metabolism 2014, 63, 875–886. [Google Scholar] [CrossRef] [PubMed]
- Gwynne, J.T.; Hess, B.; Hughes, T.; Rountree, R.; Mahaffee, D. The Role of Serum High Density Lipoproteins in Adrenal Steroidogenesis. Endocr. Res. 1985, 10, 411–430. [Google Scholar] [CrossRef]
- Gaälman, C.; Angelin, B.; Rudling, M. Prolonged Stimulation of the Adrenals by Corticotropin Suppresses Hepatic Low-Density Lipoprotein and High-Density Lipoprotein Receptors and Increases Plasma Cholesterol. Endocrinology 2002, 143, 1809–1816. [Google Scholar] [CrossRef]
- Beaven, S.W.; Tontonoz, P. Nuclear Receptors in Lipid Metabolism: Targeting the Heart of Dyslipidemia. Annu. Rev. Med. 2006, 57, 313–329. [Google Scholar] [CrossRef] [PubMed]
- Cao, G.P.; Garcia, C.K.; Wyne, K.L.; Schultz, R.A.; Parker, K.L.; Hobbs, H.H. Structure and localization of the human gene encoding SR-BI/CLA-1—Evidence for transcriptional control by steroidogenic factor 1. J. Biol. Chem. 1997, 272, 33068–33076. [Google Scholar] [CrossRef] [Green Version]
- Parker, K.L. The roles of steroidogenic factor 1 in endocrine development and function. Mol. Cell. Endocrinol. 1998, 145, 15–20. [Google Scholar] [CrossRef]
- Lekstrom-Himes, J.; Xanthopoulos, K.G. Biological Role of the CCAAT/Enhancer-binding Protein Family of Transcription Factors. J. Biol. Chem. 1998, 273, 28545–28548. [Google Scholar] [CrossRef] [Green Version]
- Vickers, K.C.; Rodriguez, A. Human scavenger receptor class B type I variants, lipid traits, and cardiovascular disease. Circ. Cardiovasc. Genet. 2014, 7, 735–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christianson, M.; Yates, M. Scavenger receptor class B type 1 gene polymorphisms and female fertility. Curr. Opin. Endocrinol. Diabetes Obes. 2012, 19, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Valacchi, G.; Sticozzi, C.; Lim, Y.; Pecorelli, A. Scavenger receptor class B type I: A multifunctional receptor. Ann. N. Y. Acad. Sci. 2011, 1229, E1–E7. [Google Scholar] [CrossRef]
- Ma, B.; Jia, J.; Wang, X.; Zhang, R.; Niu, S.; Ni, L.; Di, X.; Liu, C. Differential roles of Scavenger receptor class B type I: A protective molecule and a facilitator of atherosclerosis (Review). Mol. Med. Rep. 2020, 22, 2599–2604. [Google Scholar] [CrossRef]
- Zheng, Z.; Ai, J.; Li, X.-A. Scavenger receptor class B type I and immune dysfunctions. Curr. Opin. Endocrinol. Diabetes Obes. 2014, 21, 121–128. [Google Scholar] [CrossRef]
- Shen, W.-J.; Asthana, S.; Kraemer, F.B.; Azhar, S. Thematic Review Series: Lipid Transfer Proteins Scavenger receptor B type 1: Expression, molecular regulation, and cholesterol transport function. J. Lipid Res. 2018, 59, 1114–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thi, V.L.D.; Dreux, M.; Cosset, F.-L. Scavenger receptor class B type I and the hypervariable region-1 of hepatitis C virus in cell entry and neutralisation. Expert Rev. Mol. Med. 2011, 13, e13. [Google Scholar] [CrossRef]
- Westhaus, S.; Deest, M.; Nguyen, A.T.; Stanke, F.; Heckl, D.; Costa, R.; Schambach, A.; Manns, M.P.; Berg, T.; Vondran, F.W.; et al. Scavenger receptor class B member 1 (SCARB1) variants modulate hepatitis C virus replication cycle and viral load. J. Hepatol. 2017, 67, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.C.; Wan, L.; Yan, Q.; Wang, X.; Zhang, J.; Yang, X.; Zhang, Y.; Fan, C.; Li, D.; Deng, Y.; et al. HDL-scavenger receptor B type 1 facilitates SARS-CoV-2 entry. Nat. Metab. 2020, 2, 1391–1400. [Google Scholar] [CrossRef]
- Thuahnai, S.T.; Lund-Katz, S.; Williams, D.L.; Phillips, M.C. Scavenger receptor class B, type I-mediated uptake of various lipids into cells. Influence of the nature of the donor particle interaction with the receptor. J. Biol. Chem. 2001, 276, 43801–43808. [Google Scholar] [CrossRef] [Green Version]
- Glass, C.; Pittman, R.C.; Weinstein, D.B.; Steinberg, D. Dissociation of tissue uptake of cholesterol ester from that of apoprotein A-I of rat plasma high density lipoprotein: Selective delivery of cholesterol ester to liver, adrenal, and gonad. Proc. Natl. Acad. Sci. USA 1983, 80, 5435–5439. [Google Scholar] [CrossRef] [Green Version]
- Silver, D.L.; Wang, N.; Xiao, X.; Tall, A.R. High Density Lipoprotein (HDL) Particle Uptake Mediated by Scavenger Receptor Class B Type 1 Results in Selective Sorting of HDL Cholesterol from Protein and Polarized Cholesterol Secretion. J. Biol. Chem. 2001, 276, 25287–25293. [Google Scholar] [CrossRef] [Green Version]
- Gillard, B.K.; Bassett, G.R., Jr.; Gotto, A.M.; Rosales, C.; Pownall, H. Scavenger receptor B1 (SR-B1) profoundly excludes high density lipoprotein (HDL) apolipoprotein AII as it nibbles HDL-cholesteryl ester. J. Biol. Chem. 2017, 292, 8864–8873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irene, G.-R.; César, M.; Fernando, C.; Ana, C. SR-B1, a Key Receptor Involved in the Progression of Cardiovascular Disease: A Perspective from Mice and Human Genetic Studies. Biomedicines 2021, 9, 612. [Google Scholar] [CrossRef] [PubMed]
- Neculai, D.; Schwake, M.; Ravichandran, M.; Zunke, F.; Collins, R.F.; Peters, J.; Neculai, M.; Plumb, J.; Loppnau, P.; Pizarro, J.C.; et al. Structure of LIMP-2 provides functional insights with implications for SR-BI and CD36. Nature 2013, 504, 172–176. [Google Scholar] [CrossRef]
- Yancey, P.G.; de la Llera-Moya, M.; Swarnakar, S.; Monzo, P.; Klein, S.M.; Connelly, M.A.; Johnson, W.J.; Williams, D.L.; Rothblat, G.H. High Density Lipoprotein Phospholipid Composition Is a Major Determinant of the Bi-directional Flux and Net Movement of Cellular Free Cholesterol Mediated by Scavenger Receptor BI. J. Biol. Chem. 2000, 275, 36596–36604. [Google Scholar] [CrossRef] [Green Version]
- Reaven, E.; Chen, Y.D.; Spicher, M.; Azhar, S. Morphological evidence that high density lipoproteins are not internalized by steroid-producing cells during in situ organ perfusion. J. Clin. Investig. 1984, 74, 1384–1397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azhar, S.; Reaven, E. Scavenger receptor class BI and selective cholesteryl ester uptake: Partners in the regulation of steroidogenesis. Mol. Cell. Endocrinol. 2002, 195, 1–26. [Google Scholar] [CrossRef]
- Mineo, C.; Yuhanna, I.S.; Quon, M.; Shaul, P.W. High Density Lipoprotein-induced Endothelial Nitric-oxide Synthase Activation Is Mediated by Akt and MAP Kinases. J. Biol. Chem. 2003, 278, 9142–9149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, P.; Mulay, V.; Darabi, M.; Chan, K.C.; Heeren, J.; Pol, A.; Lambert, G.; Rye, K.-A.; Enrich, C.; Grewal, T. Ras/Mitogen-activated Protein Kinase (MAPK) Signaling Modulates Protein Stability and Cell Surface Expression of Scavenger Receptor SR-BI. J. Biol. Chem. 2011, 286, 23077–23092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grewal, T.; de Diego, I.; Kirchhoff, M.F.; Tebar, F.; Heeren, J.; Rinninger, F.; Enrich, C. High Density Lipoprotein-induced Signaling of the MAPK Pathway Involves Scavenger Receptor Type BI-mediated Activation of Ras. J. Biol. Chem. 2003, 278, 16478–16481. [Google Scholar] [CrossRef] [Green Version]
- Al-Jarallah, A.; Trigatti, B.L. A role for the scavenger receptor, class B type I in high density lipoprotein dependent activation of cellular signaling pathways. Biochim. Biophys. Acta 2010, 1801, 1239–1248. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-A.; Titlow, W.B.; Jackson, B.A.; Giltiay, N.; Nikolova-Karakashian, M.; Uittenbogaard, A.; Smart, E.J. High Density Lipoprotein Binding to Scavenger Receptor, Class B, Type I Activates Endothelial Nitric-oxide Synthase in a Ceramide-dependent Manner. J. Biol. Chem. 2002, 277, 11058–11063. [Google Scholar] [CrossRef] [Green Version]
- Mineo, C.; Shaul, P.W. HDL stimulation of endothelial nitric oxide synthase—A novel mechanism of HDL action. Trends Cardiovasc. Med. 2003, 13, 226–231. [Google Scholar] [CrossRef]
- Yuhanna, I.S.; Zhu, Y.; Cox, B.E.; Hahner, L.D.; Osborne-Lawrence, S.; Lu, P.; Marcel, Y.L.; Anderson, R.G.; Mendelsohn, M.E.; Hobbs, H.H.; et al. High-density lipoprotein binding to scavenger receptor-BI activates endothelial nitric oxide synthase. Nat. Med. 2001, 7, 853–857. [Google Scholar] [CrossRef]
- Förstermann, U.; Xia, N.; Li, H. Roles of Vascular Oxidative Stress and Nitric Oxide in the Pathogenesis of Atherosclerosis. Circ. Res. 2017, 120, 713–735. [Google Scholar] [CrossRef] [PubMed]
- Tousoulis, D.; Kampoli, A.-M.; Papageorgiou, N.; Stefanadis, C. The Role of Nitric Oxide on Endothelial Function. Curr. Vasc. Pharmacol. 2012, 10, 4–18. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-A.; Guo, L.; Dressman, J.L.; Asmis, R.; Smart, E.J. A Novel Ligand-independent Apoptotic Pathway Induced by Scavenger Receptor Class B, Type I and Suppressed by Endothelial Nitric-oxide Synthase and High Density Lipoprotein. J. Biol. Chem. 2005, 280, 19087–19096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nofer, J.R.; van der Giet, M.; Tölle, M.; Wolinska, I.; von Wnuck Lipinski, K.; Baba, H.A.; Tietge, U.J.; Gödecke, A.; Ishii, I.; Kleuser, B.; et al. HDL induces NO-dependent vasorelaxation via the lysophospholipid receptor S1P3. J. Clin. Investig. 2004, 113, 569–581. [Google Scholar] [CrossRef]
- Murata, N.; Sato, K.; Kon, J.; Tomura, H.; Yanagita, M.; Kuwabara, A.; Ui, M.; Okajima, F. Interaction of sphingosine 1-phosphate with plasma components, including lipoproteins, regulates the lipid receptor-mediated actions. Biochem. J. 2000, 352, 809–815. [Google Scholar] [CrossRef]
- Igarashi, J.; Michel, T. Sphingosine 1-phosphate and isoform-specific activation of phosphoinositide 3-kinase beta. Evidence for divergence and convergence of receptor-regulated endothelial nitric-oxide synthase signaling pathways. J. Biol. Chem. 2001, 276, 36281–36288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorbek, G.; Lewinska, M.; Rozman, D. Cytochrome P450s in the synthesis of cholesterol and bile acids—From mouse models to human diseases. FEBS J. 2012, 279, 1516–1533. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Yancey, P.G.; Su, Y.R.; Babaev, V.R.; Zhang, Y.; Fazio, S.; Linton, M.F. Inactivation of Macrophage Scavenger Receptor Class B Type I Promotes Atherosclerotic Lesion Development in Apolipoprotein E–Deficient Mice. Circulation 2003, 108, 2258–2263. [Google Scholar] [CrossRef] [Green Version]
- Tao, H.; Yancey, P.G.; Babaev, V.R.; Blakemore, J.L.; Zhang, Y.; Ding, L.; Fazio, S.; Linton, M.F. Macrophage SR-BI mediates efferocytosis via Src/PI3K/Rac1 signaling and reduces atherosclerotic lesion necrosis. J. Lipid Res. 2015, 56, 1449–1460. [Google Scholar] [CrossRef] [Green Version]
- Kruth, H.S. Macrophage foam cells and atherosclerosis. Front. Biosci. 2001, 6, D429–D455. [Google Scholar] [CrossRef] [PubMed]
- Tall, R.A.; Costet, P.; Wang, N. Regulation and mechanisms of macrophage cholesterol efflux. J. Clin. Investig. 2002, 110, 899–904. [Google Scholar] [CrossRef] [PubMed]
- Yancey, P.G.; Bortnick, A.E.; Kellner-Weibel, G.; De La Llera-Moya, M.; Phillips, M.C.; Rothblat, G.H. Importance of Different Pathways of Cellular Cholesterol Efflux. Arter. Thromb. Vasc. Biol. 2003, 23, 712–719. [Google Scholar] [CrossRef]
- von Eckardstein, A. Cholesterol efflux from macrophages and other cells. Curr. Opin. Lipidol. 1996, 7, 308–319. [Google Scholar] [CrossRef]
- Ross, R. Atherosclerosis is an inflammatory disease. Am. Heart J. 1999, 138, S419–S420. [Google Scholar] [CrossRef]
- Nieland, T.J.F.; Shaw, J.; Jaipuri, F.A.; Duffner, J.L.; Koehler, A.N.; Banakos, S.; Zannis, V.I.; Kirchhausen, T.; Krieger, M. Identification of the Molecular Target of Small Molecule Inhibitors of HDL Receptor SR-BI Activity. Biochemistry 2008, 47, 460–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieland, T.J.F.; Penman, M.; Dori, L.; Krieger, M.; Kirchhausen, T. Nonlinear partial differential equations and applications: Discovery of chemical inhibitors of the selective transfer of lipids mediated by the HDL receptor SR-BI. Proc. Natl. Acad. Sci. USA 2002, 99, 15422–15427. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Romer, K.A.; Nieland, T.J.F.; Xu, S.; Saenz-Vash, V.; Penman, M.; Yesilaltay, A.; Carr, S.A.; Krieger, M. Exoplasmic cysteine Cys384 of the HDL receptor SR-BI is critical for its sensitivity to a small-molecule inhibitor and normal lipid transport activity. Proc. Natl. Acad. Sci. USA 2011, 108, 12243–12248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieland, T.J.F.; Chroni, A.; Fitzgerald, M.L.; Maliga, Z.; Zannis, V.; Kirchhausen, T.; Krieger, M. Cross-inhibition of SR-BI- and ABCA1-mediated cholesterol transport by the small molecules BLT-4 and glyburide. J. Lipid Res. 2004, 45, 1256–1265. [Google Scholar] [CrossRef] [Green Version]
- Masson, D.; Koseki, M.; Ishibashi, M.; Larson, C.J.; Miller, S.G.; King, B.D.; Tall, A.R. Increased HDL cholesterol and apoA-I in humans and mice treated with a novel SR-BI inhibitor. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 2054–2060. [Google Scholar] [CrossRef] [Green Version]
- Rowe, I.A.; Tully, D.C.D.C.; Armstrong, M.J.; Parker, R.; Guo, K.; Barton, D.; Morse, G.D.G.D.; Venuto, C.S.C.S.; Ogilvie, C.B.; Hedegaard, D.L.; et al. Effect of scavenger receptor class B type I antagonist ITX5061 in patients with hepatitis C virus infection undergoing liver transplantation. Liver Transplant. 2016, 22, 287–297. [Google Scholar] [CrossRef] [Green Version]
- Sulkowski, M.S.; Kang, M.; Matining, R.; Wyles, D.; Johnson, V.A.; Morse, G.D.; Amorosa, V.; Bhattacharya, D.; Coughlin, K.; Wong-Staal, F.; et al. Safety and Antiviral Activity of the HCV Entry Inhibitor ITX5061 in Treatment-Naive HCV-Infected Adults: A Randomized, Double-Blind, Phase 1b Study. J. Infect. Dis. 2014, 209, 658–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishizawa, T.; Kitayama, K.; Wakabayashi, K.; Yamada, M.; Uchiyama, M.; Abe, K.; Ubukata, N.; Inaba, T.; Oda, T.; Amemiya, Y. A novel compound, R-138329, increases plasma HDL cholesterol via inhibition of scavenger receptor BI-mediated selective lipid uptake. Atherosclerosis 2007, 194, 300–308. [Google Scholar] [CrossRef] [PubMed]
- Dockendorff, C.; Faloon, P.W.; Yu, M.; Youngsaye, W.; Penman, M.; Nieland, T.J.F.; Nag, P.P.; Lewis, T.; Pu, J.; Bennion, M.; et al. Indolinyl-Thiazole Based Inhibitors of Scavenger Receptor-BI (SR-BI)-Mediated Lipid Transport. ACS Med. Chem. Lett. 2015, 6, 375–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faloon, P.W.; Dockendorff, C.; Youngsaye, W.; Yu, M.; Nag, P.P.; Lewis, T.A.; Bennion, M.; Paterson, C.; Lam, G.; Dandapani, S.; et al. A Small Molecule Inhibitor of Scavenger Receptor BI-mediated Lipid Uptake—Probe 1. In Probe Reports from the NIH Molecular Libraries Program; National Center for Biotechnology Information: Bethesda, MD, USA, 2010. [Google Scholar]
- Dockendorff, C.; Faloon, P.W.; Germain, A.; Yu, M.; Youngsaye, W.; Nag, P.P.; Bennion, M.; Penman, M.; Nieland, T.J.F.; Dandapani, S.; et al. Discovery of bisamide-heterocycles as inhibitors of scavenger receptor BI (SR-BI)-mediated lipid uptake. Bioorganic Med. Chem. Lett. 2015, 25, 2594–2598. [Google Scholar] [CrossRef] [Green Version]
- Dockendorff, C.; Faloon, P.W.; Pu, J.; Yu, M.; Johnston, S.; Bennion, M.; Penman, M.; Nieland, T.J.; Dandapani, S.; Perez, J.R.; et al. Benzo-fused lactams from a diversity-oriented synthesis (DOS) library as inhibitors of scavenger receptor BI (SR-BI)-mediated lipid uptake. Bioorgan. Med. Chem. Lett. 2015, 25, 2100–2105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Liu, Y.; Jin, H.; Pan, S.; Qian, Y.; Huang, C.; Zeng, Y.; Luo, Q.; Zeng, M.; Zhang, Z. Scavenger Receptor B1 is a Potential Biomarker of Human Nasopharyngeal Carcinoma and Its Growth is Inhibited by HDL-mimetic Nanoparticles. Theranostics 2013, 3, 477–486. [Google Scholar] [CrossRef]
- Julovi, S.M.; Xue, A.; Le, T.N.T.; Gill, A.J.; Bulanadi, J.C.; Patel, M.; Waddington, L.J.; Rye, K.-A.; Moghaddam, M.J.; Smith, R.C. Apolipoprotein A-II Plus Lipid Emulsion Enhance Cell Growth via SR-B1 and Target Pancreatic Cancer In Vitro and In Vivo. PLoS ONE 2016, 11, e0151475. [Google Scholar] [CrossRef] [Green Version]
- McMahon, K.M.; Foit, L.; Angeloni, N.L.; Giles, F.J.; Gordon, L.I.; Thaxton, C.S. Synthetic High-Density Lipoprotein-Like Nanoparticles as Cancer Therapy. Cancer Treat. Res. 2015, 166, 129–150. [Google Scholar] [CrossRef] [Green Version]
- Berney, E.; Sabnis, N.; Panchoo, M.; Raut, S.; Dickerman, R.; Lacko, A.G. The SR-B1 Receptor as a Potential Target for Treating Glioblastoma. J. Oncol. 2019, 2019, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Leon, C.G.; Locke, J.A.; Adomat, H.H.; Etinger, S.L.; Twiddy, A.L.; Neumann, R.D.; Nelson, C.C.; Guns, E.S.; Wasan, K.M. Alterations in cholesterol regulation contribute to the production of intratumoral androgens during progression to castration-resistant prostate cancer in a mouse xenograft model. Prostate 2010, 70, 390–400. [Google Scholar] [CrossRef] [PubMed]
- Twiddy, A.L.; Cox, M.E.; Wasan, K.M. Knockdown of scavenger receptor Class B Type I reduces prostate specific antigen secretion and viability of prostate cancer cells. Prostate 2012, 72, 955–965. [Google Scholar] [CrossRef]
- Gordon, J.A.; Noble, J.W.; Midha, A.; Derakhshan, F.; Wang, G.; Adomat, H.H.; Guns, E.S.T.; Lin, Y.-Y.; Ren, S.; Collins, C.C.; et al. Upregulation of Scavenger Receptor B1 Is Required for Steroidogenic and Nonsteroidogenic Cholesterol Metabolism in Prostate Cancer. Cancer Res. 2019, 79, 3320–3331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, R.; Mui, E.; Loveridge, C.; Repiscak, P.; Ahmad, I.; Hamdy, F.C.; Leung, H.Y. Targeting cholesterol transport in castration-resistant prostate cancer. Clin. Cancer Res. 2018, 24, 85. [Google Scholar]
- Traughber, C.A.; Opoku, E.; Brubaker, G.; Major, J.; Lu, H.; Lorkowski, S.W.; Neumann, C.; Hardaway, A.; Chung, Y.-M.; Gulshan, K.; et al. Uptake of high-density lipoprotein by scavenger receptor class B type 1 is associated with prostate cancer proliferation and tumor progression in mice. J. Biol. Chem. 2020, 295, 8252–8261. [Google Scholar] [CrossRef] [PubMed]
- Massie, C.; Lynch, A.; Ramos-Montoya, A.; Boren, J.; Stark, R.; Fazli, L.; Warren, A.; Scott, H.; Madhu, B.; Sharma, N.; et al. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J. 2011, 30, 2719–2733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Culig, Z.; Santer, F.R. Androgen receptor signaling in prostate cancer. Cancer Metastasis Rev. 2014, 33, 413–427. [Google Scholar] [CrossRef]
- Giunchi, F.; Fiorentino, M.; Loda, M. The Metabolic Landscape of Prostate Cancer. Eur. Urol. Oncol. 2019, 2, 28–36. [Google Scholar] [CrossRef]
- Rocchi, P.; So, A.; Kojima, S.; Signaevsky, M.; Beraldi, E.; Fazli, L.; Hurtado-Coll, A.; Yamanaka, K.; Gleave, M. Heat shock protein 27 increases after androgen ablation and plays a cytoprotective role in hormone-refractory prostate cancer. Cancer Res. 2004, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rocchi, P.; Jugpal, P.; So, A.; Sinneman, S.; Ettinger, S.; Fazli, L.; Nelson, C.; Gleave, M. Small interference RNA targeting heat-shock protein 27 inhibits the growth of prostatic cell lines and induces apoptosis via caspase-3 activation in vitro. BJU Int. 2006, 98, 1082–1089. [Google Scholar] [CrossRef]
- Zoubeidi, A.; Zardan, A.; Beraldi, E.; Fazli, L.; Sowery, R.; Rennie, P.; Nelson, C.; Gleave, M. Cooperative Interactions between Androgen Receptor (AR) and Heat-Shock Protein 27 Facilitate AR Transcriptional Activity. Cancer Res. 2007, 67, 10455–10465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoubeidi, A.; Zardan, A.; Wiedmann, R.M.; Locke, J.; Beraldi, E.; Fazli, L.; Gleave, M.E. Hsp27 Promotes Insulin-Like Growth Factor-I Survival Signaling in Prostate Cancer via p90Rsk-Dependent Phosphorylation and Inactivation of BAD. Cancer Res. 2010, 70, 2307–2317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiota, M.; Bishop, J.L.; Nip, K.M.; Zardan, A.; Takeuchi, A.; Cordonnier, T.; Beraldi, E.; Bazov, J.; Fazli, L.; Chi, K.; et al. Hsp27 Regulates Epithelial Mesenchymal Transition, Metastasis, and Circulating Tumor Cells in Prostate Cancer. Cancer Res. 2013, 73, 3109–3119. [Google Scholar] [CrossRef] [Green Version]
- Al Nakouzi, N.; Wang, C.K.; Beraldi, E.; Jager, W.; Ettinger, S.; Fazli, L.; Nappi, L.; Bishop, J.; Zhang, F.; Chauchereau, A.; et al. Clusterin knockdown sensitizes prostate cancer cells to taxane by modulating mitosis. EMBO Mol. Med. 2016, 8, 761–778. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Kumano, M.; Beraldi, E.; Fazli, L.; Du, C.; Moore, S.; Sorensen, P.; Zoubeidi, A.; Gleave, M.E. Clusterin facilitates stress-induced lipidation of LC3 and autophagosome biogenesis to enhance cancer cell survival. Nat. Commun. 2014, 5, 5775. [Google Scholar] [CrossRef] [PubMed]
- Altman, B.; Rathmell, J.C. Metabolic Stress in Autophagy and Cell Death Pathways. Cold Spring Harb. Perspect. Biol. 2012, 4, a008763. [Google Scholar] [CrossRef] [PubMed]
- Farrow, J.M.; Yang, J.C.; Evans, C.P. Autophagy as a modulator and target in prostate cancer. Nat. Rev. Urol. 2014, 11, 508–516. [Google Scholar] [CrossRef]
- Bennett, H.L.; Stockley, J.; Fleming, J.T.; Mandal, R.; O’Prey, J.; Ryan, K.M.; Robson, C.N.; Leung, H.Y. Does androgen-ablation therapy (AAT) associated autophagy have a pro-survival effect in LNCaP human prostate cancer cells? BJU Int. 2013, 111, 672–682. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, H.G.; Yang, J.C.; Kung, H.-J.; Shi, X.-B.; Tilki, D.; Lara, P.N.; White, R.W.D.; Gao, A.C.; Evans, C.P. Targeting autophagy overcomes Enzalutamide resistance in castration-resistant prostate cancer cells and improves therapeutic response in a xenograft model. Oncogene 2014, 33, 4521–4530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, Y.; Chang, Y.; Lu, W.; Sheng, X.; Wang, S.; Xu, H.; Ma, J. Regulation of Autophagy by Glycolysis in Cancer. Cancer Manag. Res. 2020, 12, 13259–13271. [Google Scholar] [CrossRef]
- Hsu, P.P.; Kang, S.A.; Rameseder, J.; Zhang, Y.; Ottina, K.A.; Lim, D.; Peterson, T.R.; Choi, Y.; Gray, N.S.; Yaffe, M.B.; et al. The mTOR-Regulated Phosphoproteome Reveals a Mechanism of mTORC1-Mediated Inhibition of Growth Factor Signaling. Science 2011, 332, 1317–1322. [Google Scholar] [CrossRef] [Green Version]
- Mihaylova, M.M.; Shaw, R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nature 2011, 13, 1016–1023. [Google Scholar] [CrossRef]
- McInnes, K.J.; Brown, K.; Hunger, N.I.; Simpson, E.R. Regulation of LKB1 expression by sex hormones in adipocytes. Int. J. Obes. 2012, 36, 982–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemos, C.; Schulze, V.K.; Baumgart, S.J.; Nevedomskaya, E.; Heinrich, T.; Lefranc, J.; Bader, B.; Christ, C.D.; Briem, H.; Kuhnke, L.P.; et al. The potent AMPK inhibitor BAY-3827 shows strong efficacy in androgen-dependent prostate cancer models. Cell. Oncol. 2021, 44, 581–594. [Google Scholar] [CrossRef] [PubMed]
- Hoyerhansen, M.; Jaattela, M. Connecting endoplasmic reticulum stress to autophagy by unfolded protein response and calcium. Cell Death Differ. 2007, 14, 1576–1582. [Google Scholar] [CrossRef]
- Metur, S.P.; Klionsky, D.J. Adaptive immunity at the crossroads of autophagy and metabolism. Cell. Mol. Immunol. 2021, 18, 1096–1105. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; He, P.; Huang, Y.; Li, Y.-F.; Lu, J.; Li, M.; Kurihara, H.; Luo, Z.; Meng, T.; Onishi, M.; et al. Selective autophagy of intracellular organelles: Recent research advances. Theranostics 2021, 11, 222–256. [Google Scholar] [CrossRef] [PubMed]
- Rashid, H.-O.; Yadav, R.K.; Kim, H.-R.; Chae, H.-J. ER stress: Autophagy induction, inhibition and selection. Autophagy 2015, 11, 1956–1977. [Google Scholar] [CrossRef]
- Röhrl, C.; Stangl, H. Cholesterol metabolism—Physiological regulation and pathophysiological deregulation by the endoplasmic reticulum. Wien. Med. Wochenschr. 2018, 168, 280–285. [Google Scholar] [CrossRef] [Green Version]
- Melamed, J.; Datta, M.W.; Becich, M.J.; Orenstein, J.M.; Dhir, R.; Silver, S.; Fidélia-Lambert, M.; Kadjacsy-Balla, A.; Macias, V.; Patel, A.; et al. The cooperative prostate cancer tissue resource: A specimen and data resource for cancer researchers. Clin. Cancer Res. 2004, 10, 4614–4621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berman, J.J.; Datta, M.; Kajdacsy-Balla, A.; Melamed, J.; Orenstein, J.; Dobbin, K.; Patel, A.; Dhir, R.; Becich, M.J. The tissue microarray data exchange specification: Implementation by the Cooperative Prostate Cancer Tissue Resource. BMC Bioinform. 2004, 5, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dash, R.C.; Robb, J.A.; Booker, D.L.; Foo, W.-C.; Witte, D.L.; Bry, L. Biospecimens and Biorepositories for the Community Pathologist. Arch. Pathol. Lab. Med. 2012, 136, 668–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, B.-R.; Simpson, R.M. Digital pathology and image analysis augment biospecimen annotation and biobank quality assurance harmonization. Clin. Biochem. 2014, 47, 274–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, J.A. Cholesterol metabolism as a target in castration-resistant prostate cancer (T). Ph.D. Thesis, University of British Columbia, Vancouver, BC, Canada, 2018. [Google Scholar]
- Liu, L.L.; Xie, N.; Sun, S.; Plymate, S.R.; Mostaghel, E.A.; Dong, X. Mechanisms of the androgen receptor splicing in prostate cancer cells. Oncogene 2014, 33, 3140–3150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, S.L.; Brauch, H.; Knutsen, T.; Johnson, B.E.; Nau, M.M.; Mitsudomi, T.; Tsai, C.M.; Whang-Peng, J.; Zbar, B.; Kaye, F.J. Molecular genetic characterization of neuroendocrine lung cancer cell lines. Anticancer Res. 1995, 15, 225–232. [Google Scholar] [PubMed]
- Kuruma, H.; Matsumoto, H.; Shiota, M.; Bishop, J.; Lamoureux, F.; Thomas, C.; Briere, D.; Los, G.; Gleave, M.; Fanjul, A.; et al. A Novel Antiandrogen, Compound 30, Suppresses Castration-Resistant and MDV3100-Resistant Prostate Cancer Growth In Vitro and In Vivo. Mol. Cancer Ther. 2013, 12, 567–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mostaghel, E.A.; Marck, B.T.; Plymate, S.R.; Vessella, R.L.; Balk, S.; Matsumoto, A.M.; Nelson, P.S.; Montgomery, R.B. Resistance to CYP17A1 Inhibition with Abiraterone in Castration-Resistant Prostate Cancer: Induction of Steroidogenesis and Androgen Receptor Splice Variants. Clin. Cancer Res. 2011, 17, 5913–5925. [Google Scholar] [CrossRef] [Green Version]
- Daniel, V.C.; Marchionni, L.; Hierman, J.S.; Rhodes, J.T.; Devereux, W.L.; Rudin, C.; Yung, R.; Parmigiani, G.; Dorsch, M.; Peacock, C.D.; et al. A Primary Xenograft Model of Small-Cell Lung Cancer Reveals Irreversible Changes in Gene Expression Imposed by Culture In vitro. Cancer Res. 2009, 69, 3364–3373. [Google Scholar] [CrossRef] [Green Version]
- Tentler, J.J.; Tan, A.C.; Weekes, C.D.; Jimeno, A.; Leong, S.; Pitts, T.M.; Arcaroli, J.J.; Messersmith, W.A.; Eckhardt, S.G. Patient-derived tumour xenografts as models for oncology drug development. Nat. Rev. Clin. Oncol. 2012, 9, 338–350. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, H.M.; Vessella, R.L.; Morrissey, C.; Brown, L.G.; Coleman, I.M.; Higano, C.S.; Mostaghel, E.A.; Zhang, X.; True, L.D.; Lam, H.-M.; et al. LuCaP Prostate Cancer Patient-Derived Xenografts Reflect the Molecular Heterogeneity of Advanced Disease and Serve as Models for Evaluating Cancer Therapeutics. Prostate 2017, 77, 654–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, A.H.; Wang, Y.; Zoubeidi, A. Patient-derived xenografts: A platform for accelerating translational research in prostate cancer. Mol. Cell. Endocrinol. 2018, 462, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Oppi, S.; Luscher, T.F.; Stein, S. Mouse Models for Atherosclerosis Research-Which Is My Line? Front. Cardiovasc. Med. 2019, 6, 46. [Google Scholar] [CrossRef] [Green Version]
- Shao, W.; Espenshade, P.J. Expanding Roles for SREBP in Metabolism. Cell Metab. 2012, 16, 414–419. [Google Scholar] [CrossRef] [Green Version]
- Subczynski, W.K.; Pasenkiewicz-Gierula, M.; Widomska, J.; Mainali, L.; Raguz, M. High Cholesterol/Low Cholesterol: Effects in Biological Membranes: A Review. Cell Biophys. 2017, 75, 369–385. [Google Scholar] [CrossRef]
- Sheng, R.; Chen, Y.; Gee, H.Y.; Stec, E.; Melowic, H.R.; Blatner, N.R.; Tun, M.P.; Kim, Y.; Källberg, M.; Fujiwara, T.; et al. Cholesterol modulates cell signaling and protein networking by specifically interacting with PDZ domain-containing scaffold proteins. Nat. Commun. 2012, 3, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Beese, C.J.; Brynjólfsdóttir, S.H.; Frankel, L.B. Selective Autophagy of the Protein Homeostasis Machinery: Ribophagy, Proteaphagy and ER-Phagy. Front. Cell Dev. Biol. 2019, 7, 373. [Google Scholar] [CrossRef] [Green Version]
- Velázquez, A.P.; Graef, M. Autophagy regulation depends on ER homeostasis controlled by lipid droplets. Autophagy 2016, 12, 1409–1410. [Google Scholar] [CrossRef] [Green Version]
- Ribas, V.; García-Ruiz, C.; Fernández-Checa, J.C. Mitochondria, cholesterol and cancer cell metabolism. Clin. Transl. Med. 2016, 5, 22. [Google Scholar] [CrossRef] [Green Version]
- Robichon, C.; Dugail, I. De novo cholesterol synthesis at the crossroads of adaptive response to extracellular stress through SREBP. Biochimie 2007, 89, 260–264. [Google Scholar] [CrossRef] [PubMed]
- DeBose-Boyd, R.A. Feedback regulation of cholesterol synthesis: Sterol-accelerated ubiquitination and degradation of HMG CoA reductase. Cell Res. 2008, 18, 609–621. [Google Scholar] [CrossRef] [Green Version]
- de Laurentiis, A.; Donovan, L.; Arcaro, A. Lipid rafts and caveolae in signaling by growth factor receptors. Open Biochem. J. 2007, 1, 12–32. [Google Scholar] [CrossRef]
- Day, K.C.; Hiles, G.L.; Kozminsky, M.; Dawsey, S.J.; Paul, A.; Broses, L.; Shah, R.; Kunja, L.P.; Hall, C.; Palanisamy, N.; et al. HER2 and EGFR Overexpression Support Metastatic Progression of Prostate Cancer to Bone. Cancer Res. 2017, 77, 74–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Yu, E. Insulin-like growth factor receptor-1 (IGF-IR) as a target for prostate cancer therapy. Cancer Metastasis Rev. 2014, 33, 607–617. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, R.; McGuinness, D.H.; McCall, P.; Underwood, M.A.; Seywright, M.; Orange, C.; Edwards, J. Upregulation of MAPK pathway is associated with survival in castrate-resistant prostate cancer. Br. J. Cancer 2011, 104, 1920–1928. [Google Scholar] [CrossRef] [PubMed]
- Siddle, K. Molecular Basis of Signaling Specificity of Insulin and IGF Receptors: Neglected Corners and Recent Advances. Front. Endocrinol. 2012, 3, 34. [Google Scholar] [CrossRef] [Green Version]
- Shanware, N.P.; Bray, K.; Abraham, R.T. The PI3K, Metabolic, and Autophagy Networks: Interactive Partners in Cellular Health and Disease. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 89–106. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Cheng, H.; Roberts, T.M.; Zhao, J.J. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug Discov. 2009, 8, 627–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molina, R.J.; Adjei, A.A. The Ras/Raf/MAPK pathway. J. Thorac. Oncol. 2006, 1, 7–9. [Google Scholar] [CrossRef] [Green Version]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef] [Green Version]
- Klymchenko, A.S.; Kreder, R. Fluorescent Probes for Lipid Rafts: From Model Membranes to Living Cells. Chem. Biol. 2014, 21, 97–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lingwood, D.; Simons, K. Detergent resistance as a tool in membrane research. Nat. Protoc. 2007, 2, 2159–2165. [Google Scholar] [CrossRef] [PubMed]
- Sajesh, B.V.; Guppy, B.J.; McManus, K.J. Synthetic Genetic Targeting of Genome Instability in Cancer. Cancers 2013, 5, 739–761. [Google Scholar] [CrossRef] [Green Version]
- Paul, J.M.; Templeton, S.D.; Baharani, A.; Freywald, A.; Vizeacoumar, F.J. Building high-resolution synthetic lethal networks: A ‘Google map’ of the cancer cell. Trends Mol. Med. 2014, 20, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Redmann, M.; Benavides, G.A.; Berryhill, T.F.; Wani, W.Y.; Ouyang, X.; Johnson, M.S.; Ravi, S.; Barnes, S.; Darley-Usmar, V.; Zhang, J. Inhibition of autophagy with bafilomycin and chloroquine decreases mitochondrial quality and bioenergetic function in primary neurons. Redox Biol. 2017, 11, 73–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- NIH. Clinical Trials Using Hydroxychloroquine. Available online: https://www.cancer.gov/about-cancer/treatment/clinical-trials/intervention/hydroxychloroquine (accessed on 1 June 2021).
- Briceño, E.; Reyes, S.; Sotelo, J. Therapy of glioblastoma multiforme improved by the antimutagenic chloroquine. Neurosurg. Focus 2003, 14, 1–6. [Google Scholar] [CrossRef]
- Sotelo, J.; Briceno, E.; Lopez-Gonzalez, M.A. Adding chloroquine to conventional treatment for glioblastoma multiforme: A randomized, double-blind, placebo-controlled trial. Ann. Intern. Med. 2006, 144, 337–343. [Google Scholar] [CrossRef]
- Briceño, E.; Calderon, A.; Sotelo, J. Institutional experience with chloroquine as an adjuvant to the therapy for glioblastoma multiforme. Surg. Neurol. 2007, 67, 388–391. [Google Scholar] [CrossRef] [PubMed]
- Rojas-Puentes, L.L.; Gonzalez-Pinedo, M.; Crismatt, A.; Ortega-Gomez, A.; Gamboa-Vignolle, C.; Nuñez-Gomez, R.; Dorantes-Gallareta, Y.; Arce-Salinas, C.; Arrieta, O. Phase II randomized, double-blind, placebo-controlled study of whole-brain irradiation with concomitant chloroquine for brain metastases. Radiat. Oncol. 2013, 8, 209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eldredge, H.B.; DeNittis, A.; DuHadaway, J.B.; Chernick, M.; Metz, R.; Prendergast, G.C. Concurrent whole brain radiotherapy and short-course chloroquine in patients with brain metastases: A pilot trial. J. Radiat. Oncol. 2013, 2, 315–321. [Google Scholar] [CrossRef]
- Kyle, R.A.; Seligman, B.R.; Wallace, H.J.; Silver, R.T.; Glidewell, O.; Holland, J.F. Mutiple myeloma resistant to melphalan (NSC-8806) treated with cyclophosphamide (NSC-26271), prednisone (NSC-10023), and chloroquine (NSC-187208). Cancer Chemother. Rep. 1975, 59, 557–562. [Google Scholar] [PubMed]
- King, M.A.; Ganley, I.; Flemington, V. Inhibition of cholesterol metabolism underlies synergy between mTOR pathway inhibition and chloroquine in bladder cancer cells. Oncogene 2016, 35, 4518–4528. [Google Scholar] [CrossRef] [Green Version]
- Hsu, S.P.; Kuo, J.; Chiang, H.-C.; Wang, H.-E.; Wang, Y.-S.; Huang, C.-C.; Huang, Y.-C.; Chi, M.-S.; Mehta, M.P.; Chi, K.-H. Temozolomide, sirolimus and chloroquine is a new therapeutic combination that synergizes to disrupt lysosomal function and cholesterol homeostasis in GBM cells. Oncotarget 2018, 9, 6883–6896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chude, C.I.; Amaravadi, R.K. Targeting Autophagy in Cancer: Update on Clinical Trials and Novel Inhibitors. Int. J. Mol. Sci. 2017, 18, 1279. [Google Scholar] [CrossRef] [Green Version]
- Jones, E.S.; Jomary, C. Clusterin. Int. J. Biochem. Cell Biol. 2002, 34, 427–431. [Google Scholar] [CrossRef]





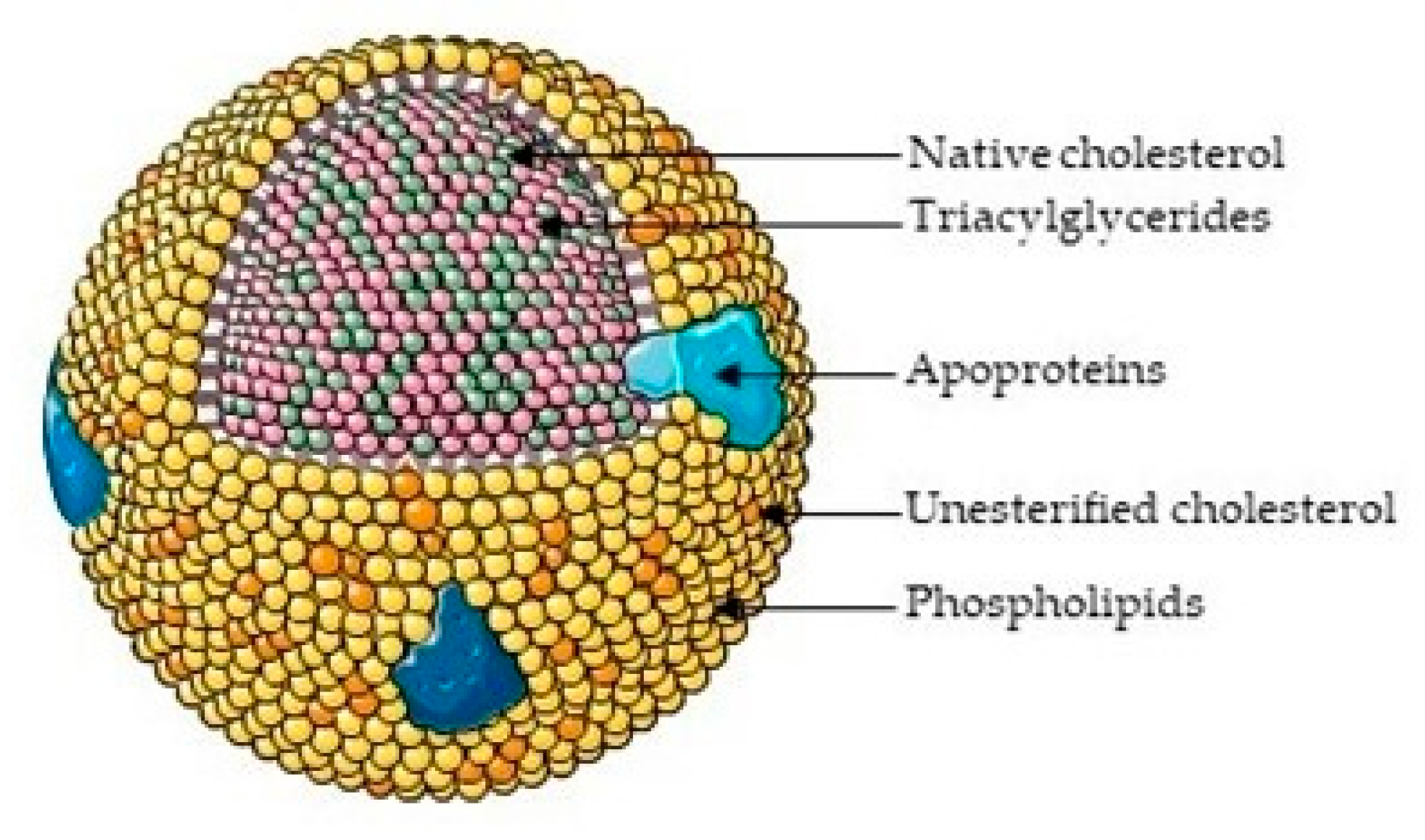{kind=link}
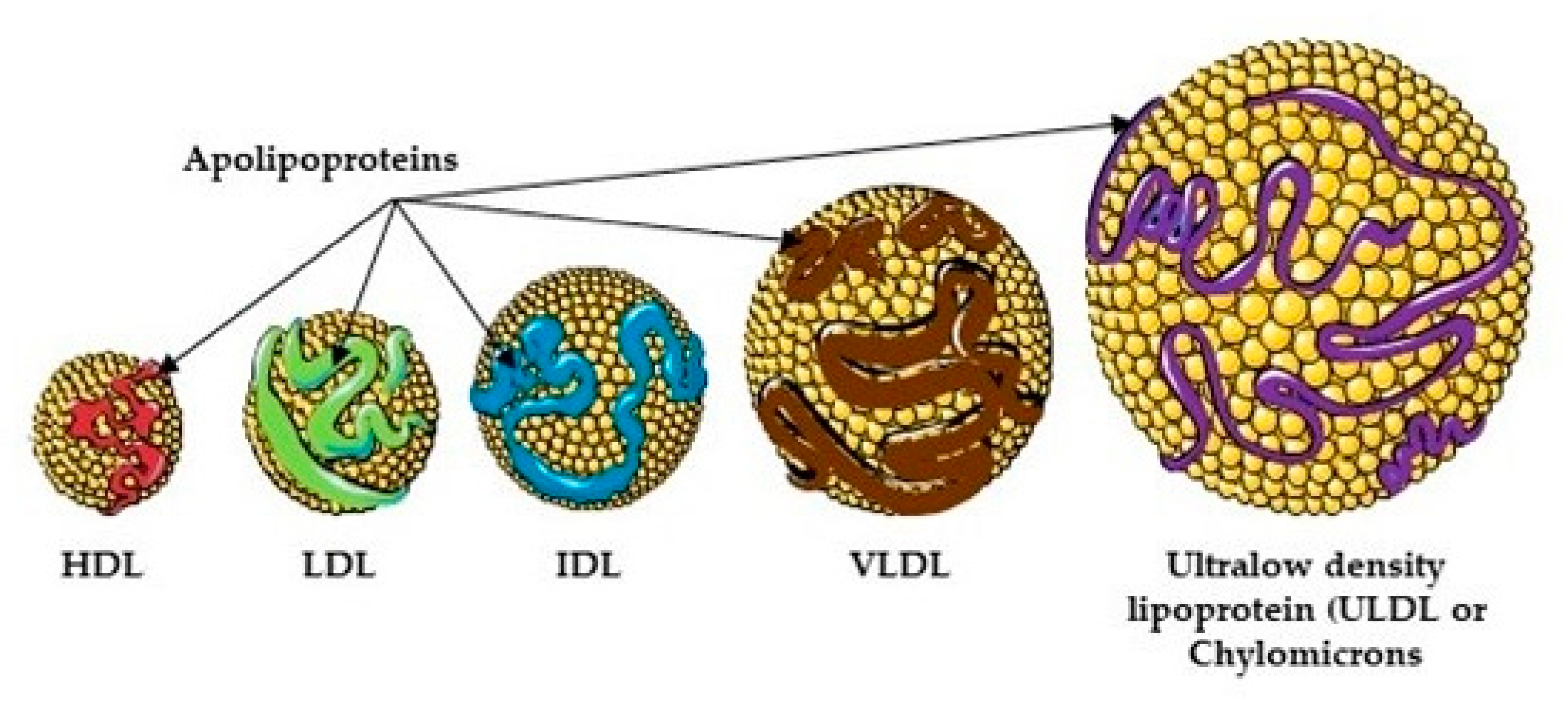{kind=link}
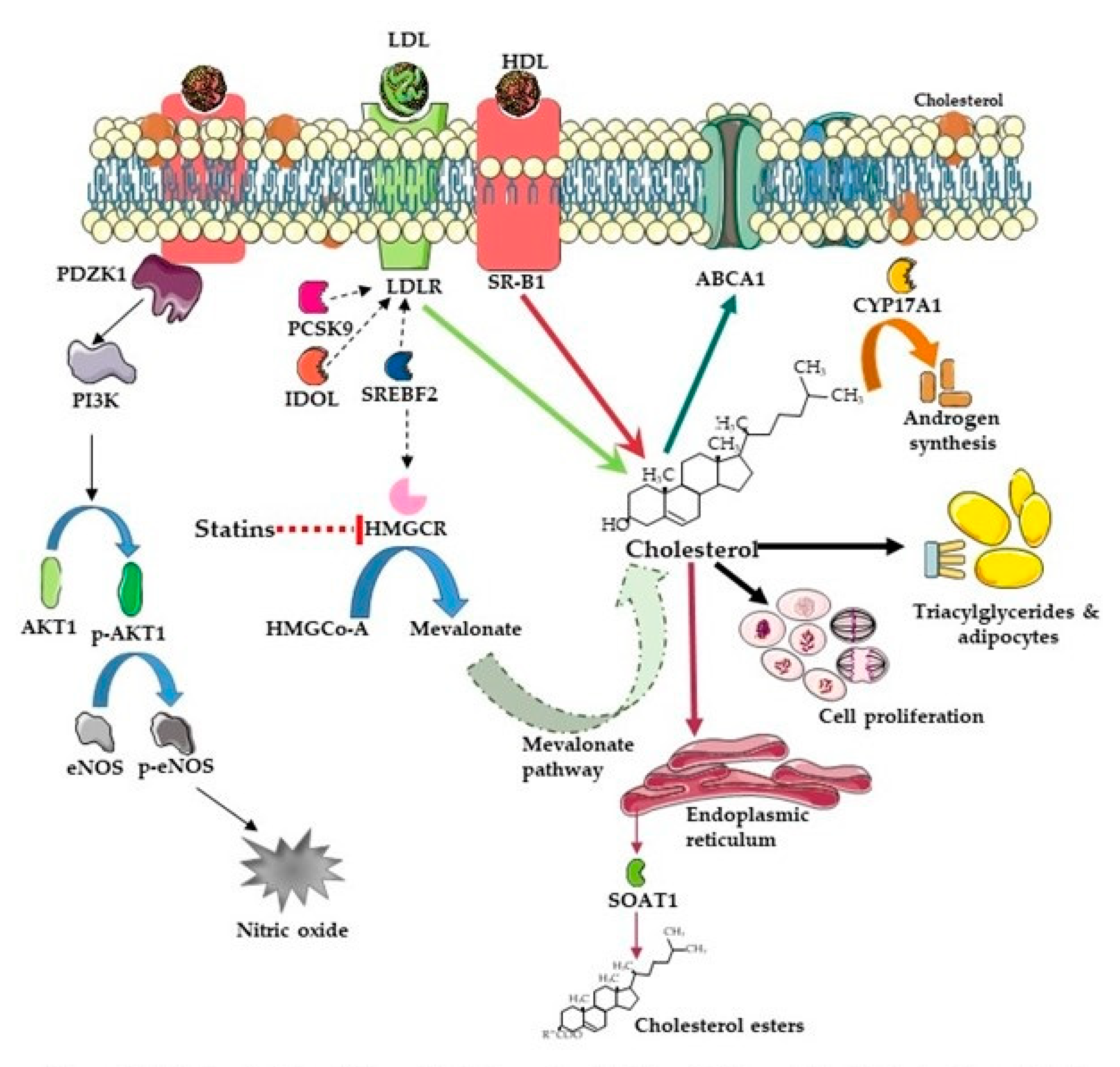{kind=link}
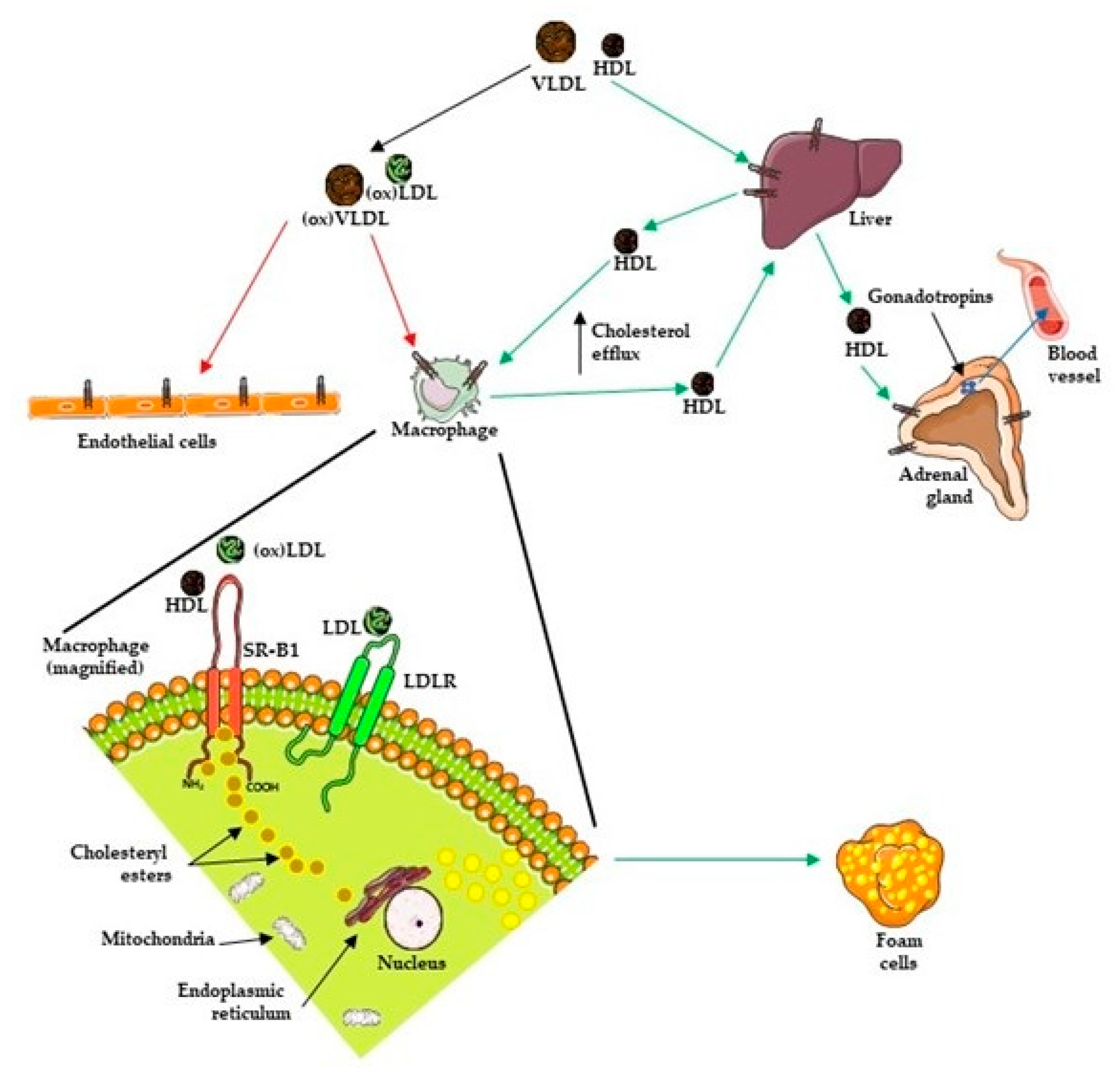{kind=link}
| Target | Mechanism of Action | Drug Class | Examples | Reference |
|---|---|---|---|---|
| HMG-CoA Reductase | Blocks hepatic cholesterol synthesis by inhibiting HMG-CoA reductase from converting HMG-CoA into mevalonic acid, a cholesterol precursor. This results in an increase in the number of LDL receptors, which leads to a reduction in LDL levels. | Statins (HMG-CoA reductase inhibitors) | Atorvastatin, rosuvastatin | [100,101] |
| PCSK9 | A monoclonal antibody that binds specifically to PCSK9, preventing it from degrading LDL receptors, ultimately decreasing LDL levels in blood by increasing the number of LDL receptors available to clear LDL from the body. | PCSK9 inhibitors | Alirocumab, evolocumab | [102,103] |
| CYP17A1 | An androgen synthesis inhibitor that works by selectively inhibiting CYP17A1, which is an enzyme required for androgen synthesis in prostatic tumor tissue, among other tissues. | CYP17A1 inhibitor (androgen biosynthesis inhibitor) | Abiraterone acetate | [104] |
| Reference | Objective | Study Group | Conclusion |
|---|---|---|---|
| Graaf et al. (2004) [129] | To compare the risk of cancer incidence between users of statins and users of other cardiovascular medication |
|
|
| Friis et al. (2005) [130] | To study if Hydroxymethylglutaryl-CoA reductase inhibitors (statins) are linked with potential chemopreventive effects |
|
|
| Shannon et al. (2005) [131] | Case-control study to elucidate the association between statin use and PCa risk |
|
|
| Singal et al. (2005) [132] | To study the association of PCa and statins |
|
|
| Platz et al. (2006) [133] | To investigate the association of statin use with total and advanced PCa; the latter being the most important endpoint to prevent |
|
|
| Flick et al. (2007) [168] | To examine the association between statin use and the risk of PCa |
|
|
| Murtola et al. (2007) [134] | To evaluate the association between serum cholesterol-lowering medication use and PCa risk at the population level |
|
|
| Jacobs et al. (2007) [135] | To examine the association between the use of cholesterol-lowering drugs and PCa incidence by disease stage and grade |
|
|
| Bonovas et al. (2008) [136] | To examine statin use in relation to both total PCa and the more clinically important advanced PCa epidemiologic studies published on the subject in peer-reviewed literature |
|
|
| Breau et al. (2010) [137] | To evaluate the effect of statin medication use on the risk of PCa |
|
|
| Murtola et al. (2010) [138] | To compare the relative risk of advanced PCa between current users and non-users of statins or other cholesterol-lowering medications |
|
|
| Farwell et al. (2011) [139] | To determine if statins can be used as a prevention strategy for high-grade PCas |
|
|
| Tan et al. (2011) [140] | To determine the association between statin use and PCa men who underwent prostate biopsy |
|
|
| Bansal et al. (2012) [141] | To examine the association between statin use and risk of PCa by conducting a detailed meta-analysis of all observational studies published regarding this subject |
|
|
| Marcella et al. (2012) [150] | Population-based case-control investigation to specifically examine the association between statin use and PCa mortality |
|
|
| Nielsen et al. (2012) [151] | To test the hypothesis that statin use begun before a cancer diagnosis is associated with reduced cancer-related mortality |
|
|
| Geybels et al. (2013) [148] | To investigate the associations between statin use begun before PCa (PCa) diagnosis and PCa recurrence/progression and PCa-specific mortality (PCSM) in a prospective, population-based cohort study |
|
|
| Park et al. (2013) [147] | Meta-analysis to evaluate associations between statins and recurrence-free survival (RFS) following treatment of localized PCa, with attention to potential benefits among patients treated primarily with radiotherapy (RT) versus radical prostatectomy |
|
|
| Yu et al. (2014) [153] | To determine whether the use of statins after PCa diagnosis is associated with a decreased risk of cancer-related mortality and all-cause mortality and to assess whether this association is modified by prediagnostic use of statins |
|
|
| Grytli et al. (2014) [152] | To assess the association between β-blockers and PCa-specific mortality in patients with high-risk or metastatic disease and to address potential confounding from the use of statins or acetylsalicylic acid (ASA) |
|
|
| Jespersen et al. (2014) [142] | To examine the association between statin use and risk of PCa in a Denmark-based case-control study |
|
|
| Lustman et al. (2014) [143] | To look for evidence for long-term statin use as an effective chemoprevention for PCa |
|
|
| Harshman et al. (2015) [149] | To evaluate whether statin use prolongs TTP during ADT for hormone-sensitive PCa |
|
|
| Larsen et al. (2017) [154] | To examine whether postdiagnosis statin use was associated with reduced cancer-specific mortality or all-cause mortality among patients with PCa in Denmark |
|
|
| Van Rompay et al. (2019) [144] | To test the hypothesis that cholesterol-lowering drugs affect PC incidence and severity |
|
|
| Allot et al. (2020) [145] | To examine statins and the risk of lethal PCa in the Health Professionals Follow-up Study (HPFS), test associations with molecular subtypes, and integrate gene expression profiling to identify putative mechanisms |
|
|
| Studies Demonstrating No Link Between Incidence of PCa with Use of Statins | |||
| Reference | Objective | Study Group | Conclusion |
| Boudreau et al. (2008) [166] | To evaluate the relation between statin use and PCa risk |
|
|
| Friedman et al. (2008) [155] | To determine the risk of cancer in statin users |
|
|
| Kuoppala et al. (2008) [156] | To review the overall evidence on the association between statin therapy and cancer |
|
|
| Smeeth et al. (2009) [157] | To assess the effect of statins on a range of health outcomes |
|
|
| Coogan et al. (2010) [165] | To update the findings by cancer stage and to evaluate the joint use of statins and NSAIDs |
|
|
| Haukka et al. (2010) [158] | To examine the association between consumption of statins and the risk of cancer, including PCa |
|
|
| Hippisley-Cox et al. (2010) [159] | To quantify the unintended effects of statins according to type, dose, and duration of use |
|
|
| Fowke et al. (2011) [164] | To investigate the association between statin use and the likelihood of having a PSA or DRE test, blood PSA levels, prostate volume, and the severity of lower urinary tract symptoms |
|
|
| Jacobs et al. (2011) [160] | To examine the association between the long-term use of cholesterol-lowering drugs, predominantly statins, and the incidence of ten common cancers including PCa, as well as overall cancer incidence |
|
|
| Chan et al. (2012) [161] | To examine the evidence of an association between statins and PCa risk |
|
|
| Freedland et al. (2013) [162] | To examine the association between statins and cancer and high-grade cancer in REDUCE, where biopsies were largely PSA-independent |
|
|
| Plat et al. (2014) [163] | To investigate whether statin drug use influences the risk of screen detected PCa in a setting in which men had a low baseline serum PSA concentration and were screened annually |
|
|
| Fulcher et al. (2015) [107] | To examine the accuracy of popular risk calculators amongst primary prevention, control arm patients of statin trials in the Cholesterol Treatment Trialists’ Collaboration (CTTC) database |
|
|
| Kantor et al. (2015) [146] | To evaluate the association between statin use and PCa risk in the Southern Community Cohort Study (SCCS) |
|
|
| Study Demonstrating Increased Risk of PCa with Use of Statins | |||
| Reference | Objective | Study Group | Conclusion |
| Chang et al. (2011) [167] | To investigate whether the use of statins was associated with PCa risk |
|
|
| Study | Objectives | Key Findings | Conclusions |
|---|---|---|---|
| Leon et al. (2010) [248] | To assess if cholesterol and its regulation are altered in CRPC cells using a murine PCa xenograft model |
|
|
| Twiddy et al. (2012) [249] | To silence SRB1 and assess the ability of PCa cells to maintain internal supplies of androgens without the presence of cholesterol |
|
|
| Schörghofer et al. (2015) [90] | To study SR-B1 expression in clinical PCa samples |
|
|
| Patel et al. (2018) [251] | To demonstrate that a SPRY2 deficiency leads to treatment resistance and an androgen self-sufficient CRPC To identify important processes that influence treatment response in CRPC |
|
|
| Gordon et al. (2019) [250] | To demonstrate that SR-B1 expression may contribute to malignant transformation by increasing levels of available cholesterol To identify SR-B1 expression in the progression from normal to cancerous prostatic tissue and to CRPC To see the effect of SR-B1 antagonism in PCa cell lines |
|
|
| Traughber et al. (2020) [252] | To examine the effect of HDL on PCa cell growth, proliferation, and tumor progression |
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pandey, M.; Cuddihy, G.; Gordon, J.A.; Cox, M.E.; Wasan, K.M. Inhibition of Scavenger Receptor Class B Type 1 (SR-B1) Expression and Activity as a Potential Novel Target to Disrupt Cholesterol Availability in Castration-Resistant Prostate Cancer. Pharmaceutics 2021, 13, 1509. https://doi.org/10.3390/pharmaceutics13091509
Pandey M, Cuddihy G, Gordon JA, Cox ME, Wasan KM. Inhibition of Scavenger Receptor Class B Type 1 (SR-B1) Expression and Activity as a Potential Novel Target to Disrupt Cholesterol Availability in Castration-Resistant Prostate Cancer. Pharmaceutics. 2021; 13(9):1509. https://doi.org/10.3390/pharmaceutics13091509
Chicago/Turabian StylePandey, Mitali, Grace Cuddihy, Jacob A. Gordon, Michael E. Cox, and Kishor M. Wasan. 2021. "Inhibition of Scavenger Receptor Class B Type 1 (SR-B1) Expression and Activity as a Potential Novel Target to Disrupt Cholesterol Availability in Castration-Resistant Prostate Cancer" Pharmaceutics 13, no. 9: 1509. https://doi.org/10.3390/pharmaceutics13091509