
Exercise in Adolescence Enhances Callosal White Matter Refinement in the Female Brain in a Rat Model of Fetal Alcohol Spectrum Disorders

Abstract
:1. Introduction
2. Materials and Methods
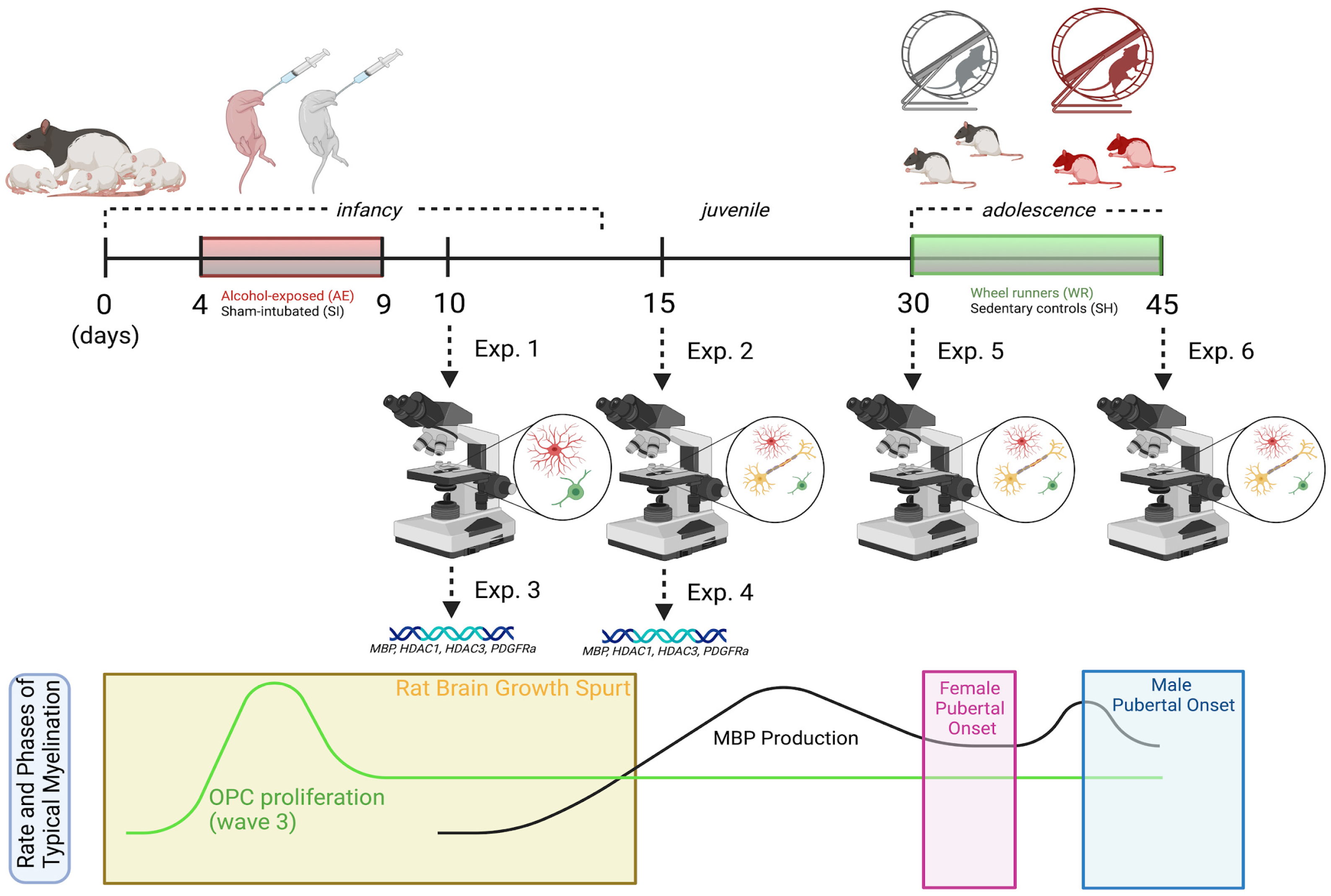
2.1. Rat Model of FASD
2.2. Adolescent Exercise Intervention to Stimulate Myelinogenesis
2.3. Animal Euthanasia for Tissue Collection
2.4. Tissue Preparation for Oligoglia Cell Quantification
2.5. RT-PCR Analysis of Brain Tissue for the Identification of Epigenetic Modifications to Genes Relevant to Developmental Myelination
2.6. Tissue Preparation for Densitometric Analysis of Myelin Basic Protein
2.7. Stereological Estimation of Cell Number and Densitometry
2.8. Statistical Analysis
3. Results
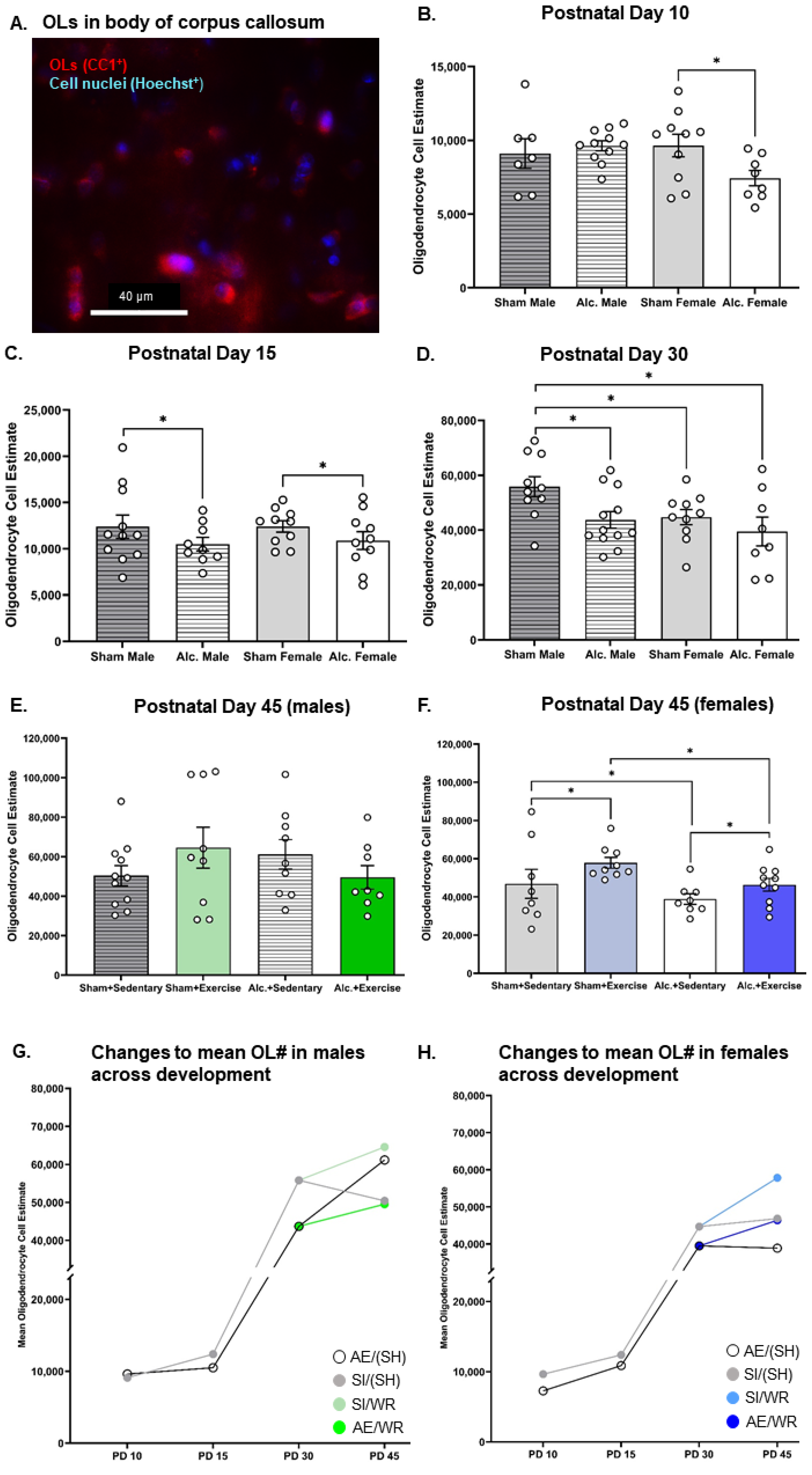
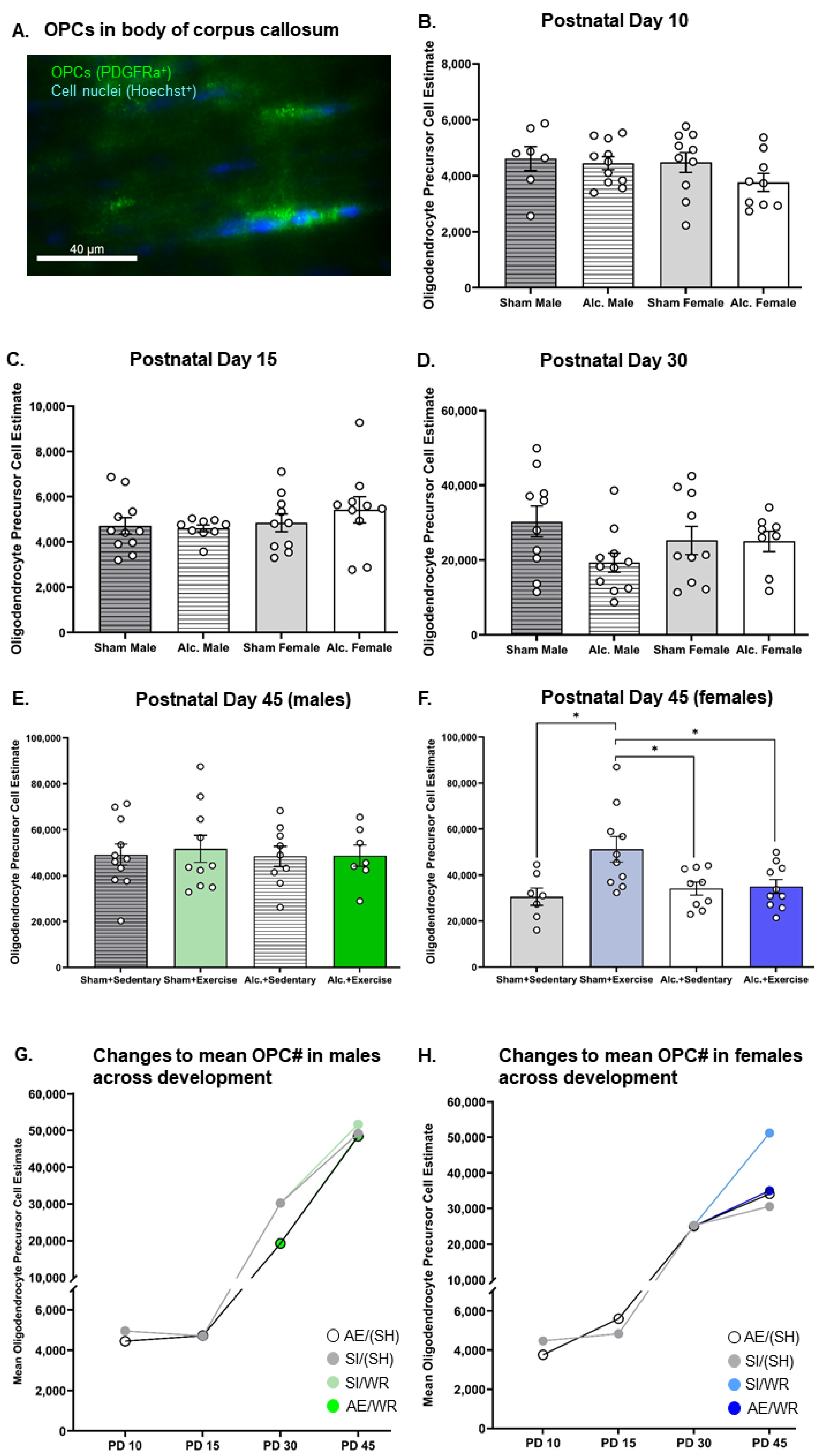
3.1. Estimation of Oligoglia Number in the Body of Corpus Callosum from Infancy to Adolescence
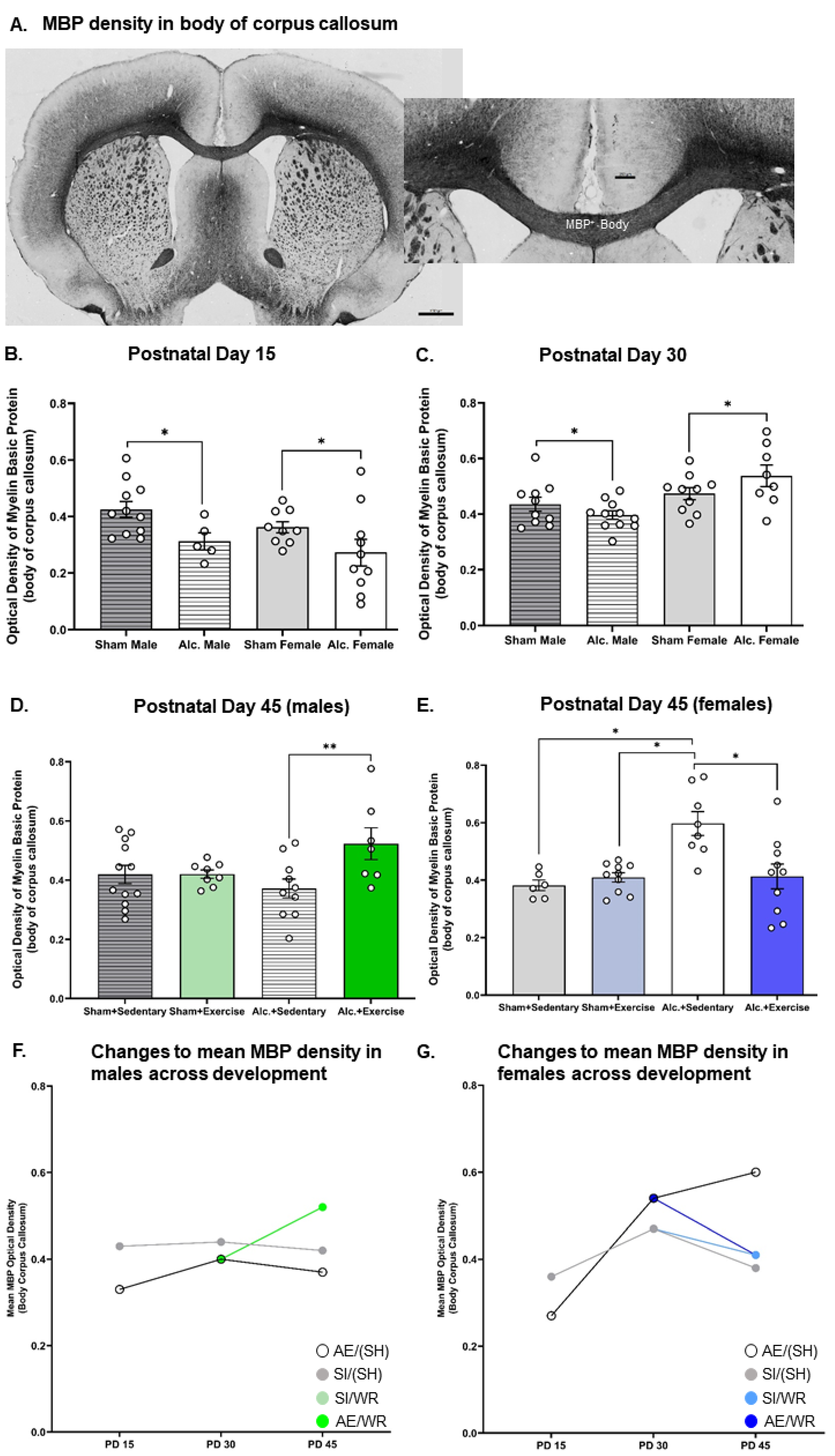
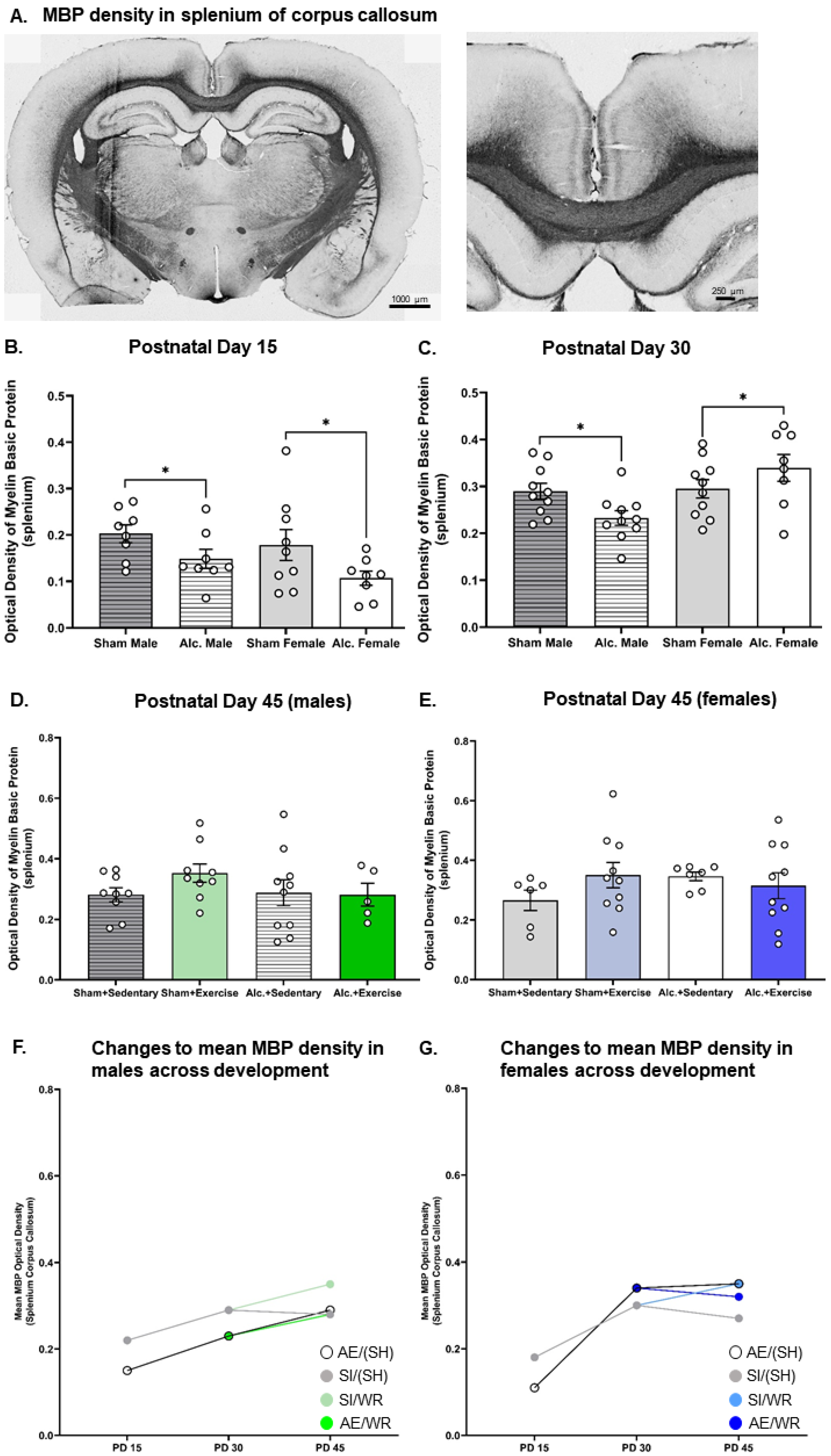
3.2. Densitometric Analysis of MBP Production in the Body and Splenium of Corpus Callosum from Late Infancy to Adolescence
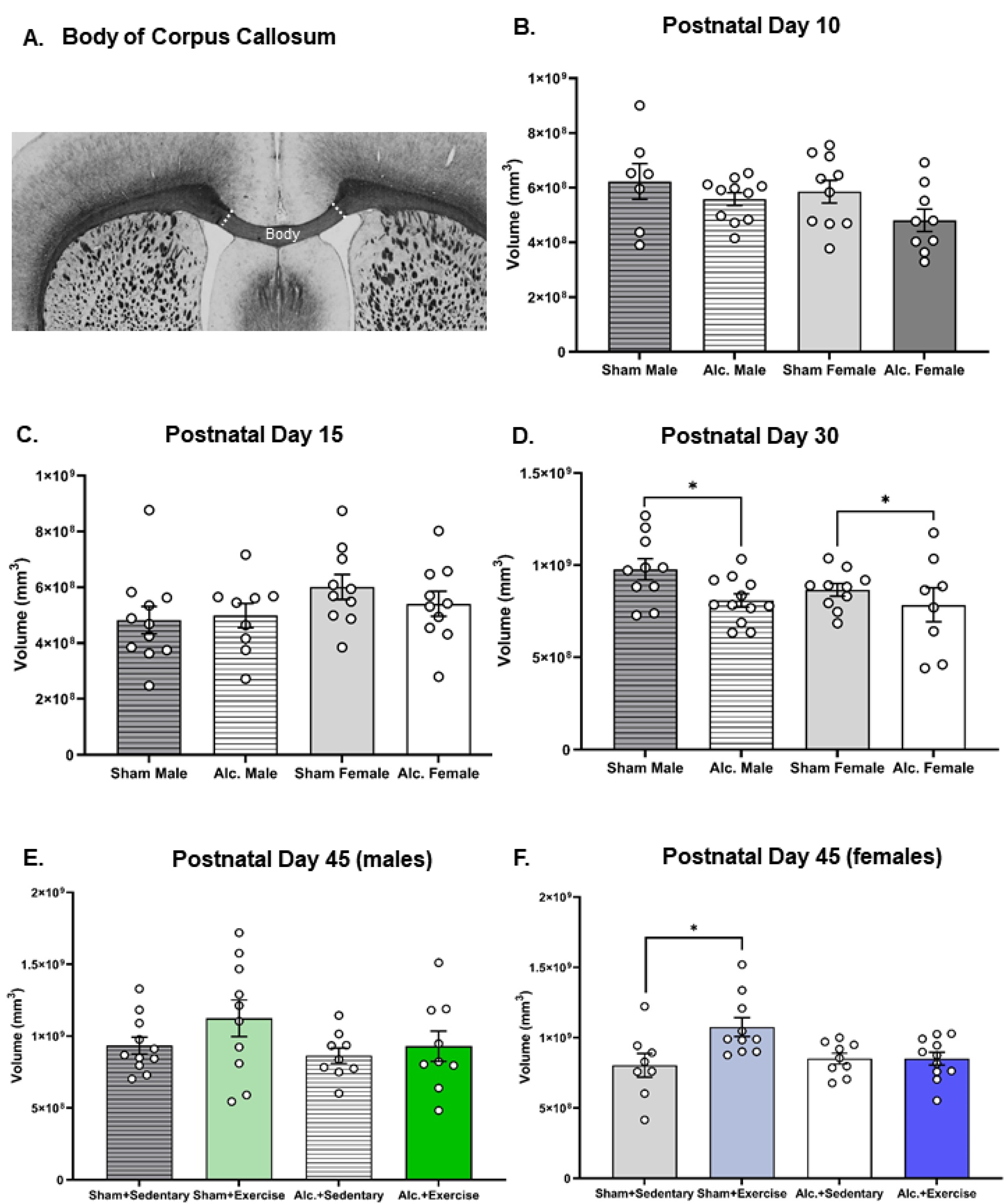
3.3. Volumetric Changes to Body of Corpus Callosum from Infancy to Adolescence
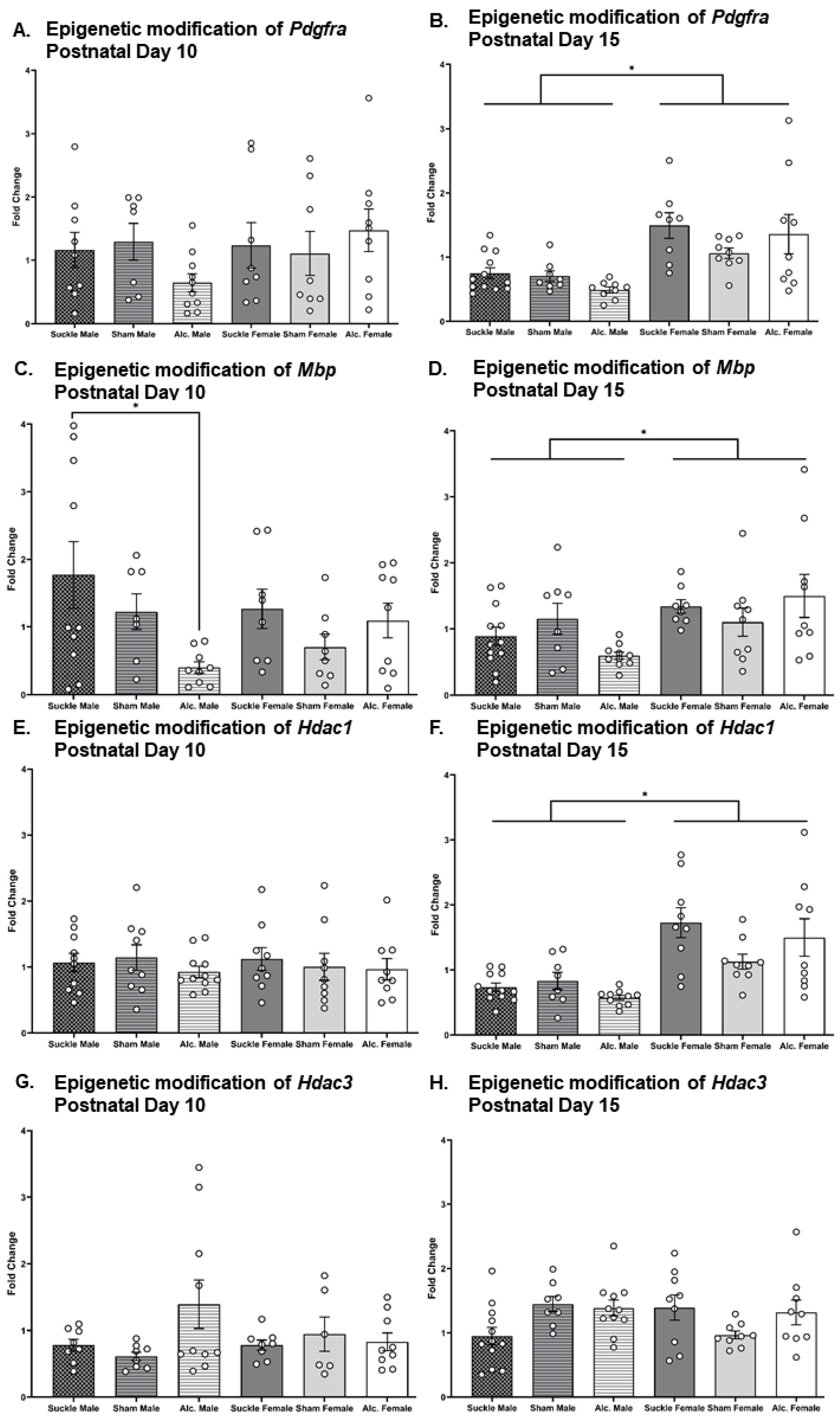
3.4. Analysis of Alcohol and Sex-Related Modifications to the Epigenome of Myelin-Relevant Genes
3.5. Comparing the Trajectories of OPC Proliferation, OL Differentiation, and MBP Production in AE and SI Rats from Each Sex
3.6. Correlation of Ex Vivo Analysis of Corpus Callosum Maturation with Previous In Vivo Neuroimaging Findings
4. Discussion
Summary of Major Findings
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Popova, S.; Lange, S.; Probst, C.; Gmel, G.; Rehm, J. Estimation of National, Regional, and Global Prevalence of Alcohol Use during Pregnancy and Fetal Alcohol Syndrome: A Systematic Review and Meta-Analysis. Lancet Glob. Health 2017, 5, e290–e299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denny, C.H.; Acero, C.S.; Naimi, T.S.; Kim, S.Y. Consumption of Alcohol Beverages and Binge Drinking Among Pregnant Women Aged 18–44 Years—United States, 2015–2017. Morb. Mortal. Wkly. Rep. 2019, 68, 365–368. [Google Scholar] [CrossRef] [Green Version]
- Hoyme, H.E.; Kalberg, W.O.; Elliott, A.J.; Blankenship, J.; Buckley, D.; Marais, A.-S.; Nursing, B.C.; Manning, M.A.; Robinson, L.K.; Adam, M.P.; et al. Updated Clinical Guidelines for Diagnosing Fetal Alcohol Spectrum Disorders. Pediatrics 2016, 138, e20154256. [Google Scholar] [CrossRef] [Green Version]
- May, P.A.; Blankenship, J.; Marais, A.-S.; Gossage, J.P.; Kalberg, W.O.; Joubert, B.; Cloete, M.; Barnard, R.; De Vries, M.; Hasken, J.; et al. Maternal Alcohol Consumption Producing Fetal Alcohol Spectrum Disorders (FASD): Quantity, Frequency, and Timing of Drinking. Drug Alcohol Depend. 2013, 133, 502–512. [Google Scholar] [CrossRef] [Green Version]
- Bonthius, D.J.; West, J.R. Permanent Neuronal Deficits in Rats Exposed to Alcohol during the Brain Growth Spurt. Teratology 1991, 44, 147–163. [Google Scholar] [CrossRef]
- Bonthius, D.J.; Goodlett, C.R.; West, J.R. Blood Alcohol Concentration and Severity of Microencephaly in Neonatal Rats Depend on the Pattern of Alcohol Administration. Alcohol 1988, 5, 209–214. [Google Scholar] [CrossRef]
- Goodlett, C.R.; Kelly, S.J.; West, J.R. Early Postnatal Alcohol Exposure That Produces High Blood Alcohol Levels Impairs Development of Spatial Navigation Learning. Psychobiology 1987, 15, 64–74. [Google Scholar] [CrossRef]
- Bodnar, T.; Weinberg, J. Prenatal Alcohol Exposure: Impact on Neuroendocrine-Neuroimmune Networks. In Neural-Immune Interactions in Brain Function and Alcohol Related Disorders; Springer Science+Business Media: Berlin/Heidelberg, Germany, 2013; pp. 307–357. ISBN 9781461447290. [Google Scholar]
- Peferoen, L.; Kipp, M.; van der Valk, P.; van Noort, J.M.; Amor, S. Oligodendrocyte-Microglia Cross-Talk in the Central Nervous System. Immunology 2013, 141, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Domingues, H.S.; Portugal, C.C.; Socodato, R.; Relvas, J.B. Oligodendrocyte, Astrocyte, and Microglia Crosstalk in Myelin Development, Damage, and Repair. Front. Cell Dev. Biol. 2016, 4, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantacorps, L.; Alfonso-Loeches, S.; Moscoso-Castro, M.; Cuitavi, J.; Gracia-Rubio, I.; López-Arnau, R.; Escubedo, E.; Guerri, C.; Valverde, O. Maternal Alcohol Binge Drinking Induces Persistent Neuroinflammation Associated with Myelin Damage and Behavioural Dysfunctions in Offspring Mice. Neuropharmacology 2017, 123, 368–384. [Google Scholar] [CrossRef] [Green Version]
- Milbocker, K.A.; Campbell, T.S.; Collins, N.; Kim, S.H.; Smith, I.F.; Roth, T.L.; Klintsova, A.Y. Glia-Driven Brain Circuit Refinement Is Altered by Early-Life Adversity: Behavioral Outcomes. Front. Behav. Neurosci. 2021, 15, 786234. [Google Scholar] [CrossRef]
- Treit, S.; Lebel, C.; Baugh, L.; Rasmussen, C.; Andrew, G.; Beaulieu, C. Longitudinal MRI Reveals Altered Trajectory of Brain Development during Childhood and Adolescence in Fetal Alcohol Spectrum Disorders. J. Neurosci. 2013, 33, 10098–10109. [Google Scholar] [CrossRef] [Green Version]
- Inkelis, S.M.; Moore, E.M.; Bischoff-Grethe, A.; Riley, E.P. Neurodevelopment in Adolescents and Adults with Fetal Alcohol Spectrum Disorders (FASD): A Magnetic Resonance Region of Interest Analysis. Brain Res. 2020, 1732, 146654. [Google Scholar] [CrossRef] [PubMed]
- Sowell, E.R.; Johnson, A.; Kan, E.; Lu, L.H.; Van Horn, J.D.; Toga, A.W.; O’Connor, M.J.; Bookheimer, S.Y. Mapping White Matter Integrity and Neurobehavioral Correlates in Children with Fetal Alcohol Spectrum Disorders. J. Neurosci. 2008, 28, 1313–1319. [Google Scholar] [CrossRef] [Green Version]
- Gautam, P.; Nuñez, S.C.; Narr, K.L.; Kan, E.C.; Sowell, E.R. Effects of Prenatal Alcohol Exposure on the Development of White Matter Volume and Change in Executive Function. NeuroImage Clin. 2014, 5, 19–27. [Google Scholar] [CrossRef] [Green Version]
- Archibald, S.L.; Fennema-Notestine, C.; Gamst, A.; Riley, E.P.; Mattson, S.N.; Jernigan, T.L. Brain Dysmorphology in Individuals with Severe Prenatal Alcohol Exposure. Dev. Med. Child Neurol. 2001, 43, 148–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.; Xue, R.; Zhang, J.; Ren, T.; Richards, L.J.; Yarowsky, P.; Miller, M.I.; Mori, S. Anatomical Characterization of Human Fetal Brain Development with Diffusion Tensor Magnetic Resonance Imaging. J. Neurosci. 2009, 29, 4263–4273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wozniak, J.R.; Riley, E.P.; Charness, M.C. Diagnosis, Epidemiology, Assessment, Pathophysiology, and Management of Fetal Alcohol Spectrum Disorders. Lancet Neurol. 2019, 18, 760–770. [Google Scholar] [CrossRef]
- Rakic, P.; Yakovlev, P.I. Development of the Corpus Callosum and Cavum Septi in Man. J. Comp. Neurol. 1968, 132, 45–72. [Google Scholar] [CrossRef]
- Kier, E.L.; Truwit, C.L. The Normal and Abnormal Genu of the Corpus Callosum: An Evolutionary, Embryologic, Anatomic, and MR Analysis. Am. J. Neuroradiol. 1996, 17, 1631–1645. [Google Scholar]
- Tanaka-Arakawa, M.M.; Matsui, M.; Tanaka, C.; Uematsu, A.; Uda, S.; Miura, K.; Sakai, T.; Noguchi, K. Developmental Changes in the Corpus Callosum from Infancy to Early Adulthood: A Structural Magnetic Resonance Imaging Study. PLoS ONE 2015, 10, e0118760. [Google Scholar] [CrossRef] [Green Version]
- Fields, R.D. White Matter in Learning, Cognition and Psychiatric Disorders. Trends Neurosci. 2008, 31, 361–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xin, W.; Chan, J.R. Myelin Plasticity: Sculpting Circuits in Learning and Memory. Nat. Rev. Neurosci. 2020, 21, 682–694. [Google Scholar] [CrossRef]
- Downes, N.; Mullins, P. The Development of Myelin in the Brain of the Juvenile Rat. Toxicol. Pathol. 2014, 42, 913–922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drew, P.D.; Kane, C.J.M. Fetal Alcohol Spectrum Disorders and Neuroimmune Changes. Int. Rev. Neurobiol. 2014, 118, 41–80. [Google Scholar] [CrossRef] [Green Version]
- Jones, K.L.; Smith, D.W.; Hanson, J.W. The Fetal Alcohol Syndrome: Clinical Delineation. Ann. N. Y. Acad. Sci. 1976, 273, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Riley, E.P.; Mattson, S.N.; Sowell, E.R.; Jernigan, T.L.; Sobel, D.F.; Jones, K.L. Abnormalities of the Corpus Callosum in Children Prenatally Exposed to Alcohol. Alcohol. Clin. Exp. Res. 1995, 19, 1198–1202. [Google Scholar] [CrossRef] [Green Version]
- Bergles, D.E.; Richardson, W.D. Oligodendrocyte Development and Plasticity. Cold Spring Harb. Perspect. Biol. 2016, 8, a020453. [Google Scholar] [CrossRef]
- Semple, B.D.; Blomgren, K.; Gimlin, K.; Ferriero, D.M.; Noble-Haeusslein, L.J. Brain Development in Rodents and Humans: Identifying Benchmarks of Maturation and Vulnerability to Injury across Species. Prog. Neurobiol. 2013, 106, 1–16. [Google Scholar] [CrossRef] [Green Version]
- van Tilborg, E.; de Theije, C.G.M.; van Hal, M.; Wagenaar, N.; de Vries, L.S.; Benders, M.J.; Rowitch, D.H.; Nijboer, C.H. Origin and Dynamics of Oligodendrocytes in the Developing Brain: Implications for Perinatal White Matter Injury. Glia 2018, 66, 221–238. [Google Scholar] [CrossRef]
- Biffen, S.C.; Warton, C.M.R.; Lindinger, N.M.; Randall, S.R.; Lewis, C.E.; Molteno, C.D.; Jacobson, J.L.; Jacobson, S.W.; Meintjes, E.M. Reductions in Corpus Callosum Volume Partially Mediate Effects of Prenatal Alcohol Exposure on IQ. Front. Neuroanat. 2018, 11, 132. [Google Scholar] [CrossRef] [Green Version]
- Roebuck-Spencer, T.M.; Mattson, S.N.; Marion, S.D.; Brown, W.S.; Riley, E.P. Bimanual Coordination in Alcohol-Exposed Children: Role of the Corpus Callosum. J. Int. Neuropsychol. Soc. 2004, 10, 536–548. [Google Scholar] [CrossRef] [PubMed]
- Dodge, N.C.; Jacobson, J.L.; Molteno, C.D.; Meintjes, E.M.; Bangalore, S.; Diwadkar, V.; Hoyme, E.H.; Robinson, L.K.; Khaole, N.; Avison, M.J.; et al. Prenatal Alcohol Exposure and Interhemispheric Transfer of Tactile Information: Detroit and Cape Town Findings. Alcohol. Clin. Exp. Res. 2009, 33, 1628–1637. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, J.R.; Muetzel, R.L.; Mueller, B.A.; McGee, C.L.; Freerks, M.A.; Ward, E.E.; Nelson, M.L.; Chang, P.-N.; Lim, K.O. Microstructural Corpus Callosum Anomalies in Children with Prenatal Alcohol Exposure: An Extension of Previous Diffusion Tensor Imaging Findings. Alcohol Clin. Exp. Res. 2009, 33, 1825–1835. [Google Scholar] [CrossRef]
- Fields, R.D.; Bukalo, O. Myelin Makes Memories. Nat. Neurosci. 2020, 23, 469–470. [Google Scholar] [CrossRef]
- Dobbing, J.; Sands, J. Quantitative Growth and Development of Human Brain. Arch. Dis. Child. 1973, 48, 757–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalitz, P.; Cock, M.; Harding, R.; Rees, S. Injurious Effects of Acute Ethanol Exposure during Late Gestation on Developing White Matter in Fetal Sheep. Int. J. Dev. Neurosci. 2008, 26, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Creeley, C.E.; Dikranian, K.T.; Johnson, S.A.; Farber, N.B.; Olney, J.W. Alcohol-Induced Apoptosis of Oligodendrocytes in the Fetal Macaque Brain. Acta Neuropathol. Commun. 2013, 1, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Newville, J.; Valenzuela, C.F.; Li, L.; Jantzie, L.L.; Cunningham, L.A. Acute Oligodendrocyte Loss with Persistent White Matter Injury in a Third Trimester Equivalent Mouse Model of Fetal Alcohol Spectrum Disorder. Glia 2017, 65, 1317–1332. [Google Scholar] [CrossRef] [Green Version]
- Zoeller, R.T.; Butnariu, O.V.; Fletcher, D.L.; Riley, E.P. Limited Postnatal Ethanol Exposure Permanently Alters the Expression of MRNAS Encoding Myelin Basic Protein and Myelin-Associated Glyco- Protein in Cerebellum. Alcohol. Clin. Exp. Res. 1994, 18, 909–916. [Google Scholar] [CrossRef]
- Niedzwiedz-Massey, V.M.; Douglas, J.C.; Rafferty, T.; Wight, P.A.; Kane, C.J.M.; Drew, P.D. Ethanol Modulation of Hippocampal Neuroinflammation, Myelination, and Neurodevelopment in a Postnatal Mouse Model of Fetal Alcohol Spectrum Disorders. Neurotoxicol. Teratol. 2021, 87, 107015. [Google Scholar] [CrossRef] [PubMed]
- Newville, J.; Howard, T.A.; Chavez, G.J.; Valenzuela, C.F.; Cunningham, L.A. Persistent Myelin Abnormalities in a Third Trimester-equivalent Mouse Model of Fetal Alcohol Spectrum Disorder. Alcohol. Clin. Exp. Res. 2021, 46, 77–86. [Google Scholar] [CrossRef]
- Tiane, A.; Schepers, M.; Rombaut, B.; Hupperts, R.; Prickaerts, J.; Hellings, N.; Van Den Hove, D.; Vanmierlo, T. From OPC to Oligodendrocyte: An Epigenetic Journey. Cells 2019, 8, 1236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berry, K.; Wang, J.; Richard Lu, Q. Epigenetic Regulation of Oligodendrocyte Myelination in Developmental Disorders and Neurodegenerative Diseases. F1000Research 2020, 9, F1000 Faculty Rev-105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, F.; Chen, Y.; Hoang, T.; Montgomery, R.L.; Zhao, X.H.; Bu, H.; Hu, T.; Taketo, M.M.; Van Es, J.H.; Clevers, H.; et al. HDAC1 and HDAC2 Regulate Oligodendrocyte Differentiation by Disrupting the B-Catenin-TCF Interaction. Nat. Neurosci. 2009, 12, 829–838. [Google Scholar] [CrossRef] [Green Version]
- Haycock, P.C. Fetal Alcohol Spectrum Disorders: The Epigenetic Perspective. Biol. Reprod. 2009, 81, 607–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaddock-Heyman, L.; Erickson, K.I.; Kienzler, C.; Drollette, E.S.; Raine, L.B.; Kao, S.C.; Bensken, J.; Weisshappel, R.; Castelli, D.M.; Hillman, C.H.; et al. Physical Activity Increases White Matter Microstructure in Children. Front. Neurosci. 2018, 12, 950. [Google Scholar] [CrossRef] [Green Version]
- Ruotsalainen, I.; Tammelin, T.H.; Karvanen, J.; Parviainen, T. Physical Activity, Aerobic Fitness, and Brain White Matter: Their Role for Executive Functions in Adolescence. Dev. Cogn. Neurosci. 2020, 42, 100765. [Google Scholar] [CrossRef]
- Tomlinson, L.; Leiton, C.V.; Colognato, H. Behavioral Experiences as Drivers of Oligodendrocyte Lineage Dynamics and Myelin Plasticity. Neuropharmacology 2016, 110, 548–562. [Google Scholar] [CrossRef]
- Tomlinson, L.; Huang, P.H.; Colognato, H. Prefrontal Cortex NG2 Glia Undergo a Developmental Switch in Their Responsiveness to Exercise. Dev. Neurobiol. 2018, 78, 687–700. [Google Scholar] [CrossRef]
- Gursky, Z.H.; Johansson, J.R.; Klintsova, A.Y. Postnatal Alcohol Exposure and Adolescent Exercise Have Opposite Effects on Cerebellar Microglia in Rat. Int. J. Dev. Neurosci. 2020, 80, 558–571. [Google Scholar] [CrossRef] [PubMed]
- Vivar, C.; Potter, M.C.; van Praag, H. All about Running: Synaptic Plasticity, Growth Factors and Adult Hippocampal Neurogenesis. Curr. Top. Behav. Neurosci. 2013, 15, 189–210. [Google Scholar] [PubMed] [Green Version]
- Voss, M.W.; Vivar, C.; Kramer, A.F.; van Praag, H. Bridging Animal and Human Models of Exercise-Induced Brain Plasticity. Trends Cogn. Sci. 2013, 17, 525–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobbing, J.; Sands, J. Comparative Aspects of the Brain Growth Spurt. Early Hum. Dev. 1979, 311, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Milbocker, K.A.; LeBlanc, G.L.; Brengel, E.K.; Hekmatyar, K.S.; Kulkarni, P.; Ferris, C.F.; Klintsova, A.Y. Reduced and Delayed Myelination and Volume of Corpus Callosum in an Animal Model of Fetal Alcohol Spectrum Disorders Partially Benefit from Voluntary Exercise. Sci. Rep. 2022, 12, 10653. [Google Scholar] [CrossRef]
- Kuhn, S.; Gritti, L.; Crooks, D.; Dombrowski, Y. Oligodendrocytes in Development, Myelin Generation and Beyond. Cells 2019, 11, 1424. [Google Scholar] [CrossRef] [Green Version]
- Goldman, S.A.; Kuypers, N.J. How to Make an Oligodendrocyte. Development 2015, 142, 3983–3995. [Google Scholar] [CrossRef] [Green Version]
- Sil, S.; Periyasamy, P.; Thangaraj, A.; Chivero, E.T.; Buch, S. PDGF/PDGFR Axis in the Neural Systems. Mol. Asp. Med. 2018, 62, 63–74. [Google Scholar] [CrossRef]
- Milbocker, K.A.; Klintsova, A.Y. Examination of Cortically Projecting Cholinergic Neurons Following Exercise and Environmental Intervention in a Rodent Model of Fetal Alcohol Spectrum Disorders. Birth Defects Res. 2021, 113, 299–313. [Google Scholar] [CrossRef]
- Hamilton, A.M.; Forkert, N.D.; Yang, R.; Wu, Y.; Rogers, J.A.; Yong, V.W.; Dunn, J.F. Central Nervous System Targeted Autoimmunity Causes Regional Atrophy: A 9.4T MRI Study of the EAE Mouse Model of Multiple Sclerosis. Sci. Rep. 2019, 9, 8488. [Google Scholar] [CrossRef] [Green Version]
- Slomianka, L.; West, M.J. Estimators of the Precision of Stereological Estimates: An Example Based on the CA1 Pyramidal Cell Layer of Rats. Neuroscience 2005, 136, 757–767. [Google Scholar] [CrossRef]
- Purohit, D.C.; Mandyam, A.D.; Terranova, M.J.; Mandyam, C.D. Voluntary Wheel Running during Adolescence Distinctly Alters Running Output in Adulthood in Male and Female Rats. Behav. Brain Res. 2020, 377, 112235. [Google Scholar] [CrossRef] [PubMed]
- Lam, V.Y.Y.; Raineki, C.; Ellis, L.; Yu, W.; Weinberg, J. Interactive Effects of Prenatal Alcohol Exposure and Chronic Stress in Adulthood on Anxiety-like Behavior and Central Stress-Related Receptor MRNA Expression: Sex- and Time-Dependent Effects. Psychoneuroendocrinology 2018, 97, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Hackett, M.; McQuillan, J.; El-Assaad, F.; Aitken, J.; Levina, A.; Cohen, D.; Siegele, R.; Carter, E.; Grau, G.; Hunt, N.; et al. Chemical Alterations to Murine Brain Tissue Induced by Formalin Fixation: Implications for Biospectroscopic Imaging and Mapping Studies of Disease Pathogenesis. Analyst 2011, 136, 2941–2952. [Google Scholar] [CrossRef]
- Yakovlev, P.; Lecours, A. The Myelogenetic Cycles of Regional Maturation of the Brain. In Regional Development of the Brain in Early Life; Minkowski, A., Ed.; Blackwell: Oxford, UK, 1967; pp. 3–70. [Google Scholar]
- Xiong, X.; Zhu, L.N.; Dong, X.X.; Wang, W.; Yan, J.; Chen, A.G. Aerobic Exercise Intervention Alters Executive Function and White Matter Integrity in Deaf Children: A Randomized Controlled Study. Neural Plast. 2018, 2018, 3735208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talukdar, T.; Nikolaidis, A.; Zwilling, C.E.; Paul, E.J.; Hillman, C.H.; Cohen, N.J.; Kramer, A.F.; Barbey, A.K. Aerobic Fitness Explains Individual Differences in the Functional Brain Connectome of Healthy Young Adults. Cereb. Cortex 2018, 28, 3600–3609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, E.G.; Orthmann-Murphy, J.L.; Langseth, A.J.; Bergles, D.E. Myelin Remodeling through Experience-Dependent Oligodendrogenesis in the Adult Somatosensory Cortex. Nat. Neurosci. 2018, 21, 696–706. [Google Scholar] [CrossRef]
- McKenzie, I.A.; Ohayon, D.; Li, H.; De Faria, J.P.; Emery, B.; Tohyama, K.; Richardson, W.D. Motor Skill Learning Requires Active Central Myelination. Science 2014, 346, 318–322. [Google Scholar] [CrossRef]
- Zheng, J.; Sun, X.; Ma, C.; Li, B.M.; Luo, F. Voluntary Wheel Running Promotes Myelination in the Motor Cortex through Wnt Signaling in Mice. Mol. Brain 2019, 12, 85. [Google Scholar] [CrossRef] [Green Version]
- Schreiber, W.B.; St Cyr, S.A.; Jablonski, S.A.; Hunt, P.S.; Klintsova, A.Y.; Stanton, M.E. Effects of Exercise and Environmental Complexity on Deficits in Trace and Contextual Fear Conditioning Produced by Neonatal Alcohol Exposure in Rats. Dev. Psychobiol. 2013, 55, 483–495. [Google Scholar] [CrossRef]
- Hamilton, G.F.; Criss, K.J.; Klintsova, A.Y. Voluntary Exercise Partially Reverses Neonatal Alcohol-Induced Deficits in MPFC Layer II/III Dendritic Morphology of Male Adolescent Rats. Synapse 2015, 69, 405–415. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, G.F.; Jablonski, S.A.; Schiffino, F.L.; St Cyr, S.A.; Stanton, M.E.; Klintsova, A.Y. Exercise and Environment as an Intervention for Neonatal Alcohol Effects on Hippocampal Adult Neurogenesis and Learning. Neuroscience 2014, 265, 274–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boschen, K.E.; McKeown, S.E.; Roth, T.L.; Klintsova, A.Y. Impact of Exercise and a Complex Environment on Hippocampal Dendritic Morphology, Bdnf Gene Expression, and DNA Methylation in Male Rat Pups Neonatally Exposed to Alcohol. Dev. Neurobiol. 2017, 77, 708–725. [Google Scholar] [CrossRef] [PubMed]
- Darbinian, N.; Darbinyan, A.; Merabova, N.; Bajwa, A.; Tatevosian, G.; Martirosyan, D.; Zhao, H.; Selzer, M.E.; Goetzl, L. Ethanol-Mediated Alterations in Oligodendrocyte Differentiation in the Developing Brain. Neurobiol. Dis. 2021, 148, 105181. [Google Scholar] [CrossRef]
- Juraska, J.M.; Drzewiecki, C.M. Cortical Reorganization during Adolescence: What the Rat Can Tell Us about the Cellular Basis. Dev. Cogn. Neurosci. 2020, 45, 100857. [Google Scholar] [CrossRef]
- Shen, S.; Li, J.; Casaccia-Bonnefil, P. Histone Modifications Affect Timing of Oligodendrocyte Progenitor Differentiation in the Developing Rat Brain. J. Cell Biol. 2005, 169, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Marin-Husstege, M.; Muggironi, M.; Liu, A.; Casaccia-Bonnefil, P. Histone Deacetylase Activity Is Necessary for Oligodendrocyte Lineage Progression. J. Neurosci. 2002, 22, 10333–10345. [Google Scholar] [CrossRef] [Green Version]
- Darling, J.S.; Daniel, J.M. Pubertal Hormones Mediate Sex Differences in Levels of Myelin Basic Protein in the Orbitofrontal Cortex of Adult Rats. Neuroscience 2019, 406, 487–495. [Google Scholar] [CrossRef]
- Terasaki, L.S.; Schwarz, J.M. Effects of Moderate Prenatal Alcohol Exposure during Early Gestation in Rats on Inflammation across the Maternal-Fetal-Immune Interface and Later-Life Immune Function in the Offspring. J. Neuroimmune Pharmacol. 2016, 11, 680–692. [Google Scholar] [CrossRef] [Green Version]
- Walker, W.H.; Bumgarner, J.R.; Nelson, R.J.; Courtney DeVries, A. Transcardial Perfusion Is Not Required to Accurately Measure Cytokines within the Brain. J. Neurosci. Methods 2020, 334, 108601. [Google Scholar] [CrossRef]
- Ahluwalia, B.; Smith, D.; Adeyiga, O.; Akbasak, B.; Rajguru, S. Ethanol Decreases Progesterone Synthesis in Human Placental Cells: Mechanism of Ethanol Effect. Alcohol 1992, 9, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Caldeira, J.C.; Wu, Y.; Mameli, M.; Purdy, R.H.; Li, P.K.; Akwa, Y.; Savage, D.D.; Engen, J.R.; Valenzuela, C.F. Fetal Alcohol Exposure Alters Neurosteroid Levels in the Developing Rat Brain. J. Neurochem. 2004, 90, 1530–1539. [Google Scholar] [CrossRef] [PubMed]
- Ghoumari, A.M.; Baulieu, E.E.; Schumacher, M. Progesterone Increases Oligodendroglial Cell Proliferation in Rat Cerebellar Slice Cultures. Neuroscience 2005, 135, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Ghoumari, A.M.; Ibanez, C.; El-Etr, M.; Leclerc, P.; Eychenne, B.; O’Malley, B.W.; Baulieu, E.E.; Schumacher, M. Progesterone and Its Metabolites Increase Myelin Basic Protein Expression in Organotypic Slice Cultures of Rat Cerebellum. J. Neurochem. 2003, 86, 848–859. [Google Scholar] [CrossRef] [PubMed]
- Hussain, R.; El-Etr, M.; Gaci, O.; Rakotomamonjy, J.; Macklin, W.B.; Kumar, N.; Sitruk-Ware, R.; Schumacher, M.; Ghoumari, A.M. Progesterone and Nestorone Facilitate Axon Remyelination: A Role for Progesterone Receptors. Neuroendocrinology 2011, 152, 3820–3831. [Google Scholar] [CrossRef] [Green Version]
- Cerghet, M.; Skoff, R.P.; Swamydas, M.; Bessert, D. Sexual Dimorphism in the White Matter of Rodents. J. Neurol. Sci. 2009, 286, 76–80. [Google Scholar] [CrossRef] [Green Version]
- González-Orozco, J.C.; Camacho-Arroyo, I. Progesterone Actions during Central Nervous System Development. Front. Neurosci. 2019, 13, 503. [Google Scholar] [CrossRef] [Green Version]
- Chavarria, M.C.; Sánchez, F.J.; Chou, Y.-Y.; Thompson, P.M.; Luders, E. Puberty in the Corpus Callosum. Neuroscience 2014, 265, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Markham, J.A.; Morris, J.R.; Juraska, J.M. Neuron Number Decreases in the Rat Ventral, but Not Dorsal, Medial Prefrontal Cortex between Adolescence and Adulthood. Neuroscience 2007, 144, 961–968. [Google Scholar] [CrossRef]
- Tang, Z.; Cheng, S.; Sun, Y.; Zhang, Y.; Xiang, X.; Ouyang, Z.; Zhu, X.; Wang, B.; Hei, M. Early TLR4 Inhibition Reduces Hippocampal Injury at Puberty in a Rat Model of Neonatal Hypoxic-Ischemic Brain Damage via Regulation of Neuroimmunity and Synaptic Plasticity. Exp. Neurol. 2019, 321, 113039. [Google Scholar] [CrossRef]
- Rao, H.; Betancourt, L.; Giannetta, J.M.; Brodsky, N.L.; Korczykowski, M.; Avants, B.B.; Gee, J.C.; Wang, J.; Hurt, H.; Detre, J.A.; et al. Early Parental Care Is Important for Hippocampal Maturation: Evidence from Brain Morphology in Humans. Neuroimage 2010, 49, 1144–1150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honeycutt, J.A.; Demaestri, C.; Peterzell, S.; Silveri, M.M.; Cai, X.; Kulkarni, P.; Cunningham, M.G.; Ferris, C.F.; Brenhouse, H.C. Altered Corticolimbic Connectivity Reveals Sex-Specific Adolescent Outcomes in a Rat Model of Early Life Adversity. eLife 2020, 9, e52651. [Google Scholar] [CrossRef]
- Strzelewicz, A.R.; Ordoñes Sanchez, E.; Rondón-Ortiz, A.N.; Raneri, A.; Famularo, S.T.; Bangasser, D.A.; Kentner, A.C. Access to a High Resource Environment Protects against Accelerated Maturation Following Early Life Stress: A Translational Animal Model of High, Medium and Low Security Settings. Horm. Behav. 2019, 111, 26–59. [Google Scholar] [CrossRef]
- Kar, P.; Reynolds, J.E.; Grohs, M.N.; Ben Gibbard, W.; McMorris, C.; Tortorelli, C.; Lebel, C. White Matter Alterations in Young Children with Prenatal Alcohol Exposure. Dev. Neurobiol. 2021, 81, 400–410. [Google Scholar] [CrossRef] [PubMed]
- Herting, M.M.; Kim, R.; Uban, K.A.; Kan, E.; Binley, A.; Sowell, E.R. Longitudinal Changes in Pubertal Maturation and White Matter Microstructure. Psychoneuroendocrinology 2017, 81, 70–79. [Google Scholar] [CrossRef]
- Smith, I.F.; Gursky, Z.H.; Klintsova, A.Y. Representation of Prefrontal Axonal Efferents in the Thalamic Nucleus Reuniens in a Rodent Model of Fetal Alcohol Exposure during Third Trimester. Front. Behav. Neurosci. 2022, 16, 993601. [Google Scholar] [CrossRef] [PubMed]
- Treit, S.; Chen, Z.; Rasmussen, C.; Beaulieu, C. White Matter Correlates of Cognitive Inhibition during Development: A Diffusion Tensor Imaging Study. Neuroscience 2014, 276, 87–97. [Google Scholar] [CrossRef]
- Blaauw, J.; Meiners, L.C. The Splenium of the Corpus Callosum: Embryology, Anatomy, Function and Imaging with Pathophysiological Hypothesis. Neuroradiology 2020, 62, 563–585. [Google Scholar] [CrossRef] [Green Version]
- Pritchard Orr, A.B.; Keiver, K.; Bertram, C.P.; Clarren, S. FAST Club: The Impact of a Physical Activity Intervention on Executive Function in Children with Fetal Alcohol Spectrum Disorder. Adapt. Phys. Act. Q. 2018, 35, 403–423. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Independent Samples t-Tests | ||||||||
|---|---|---|---|---|---|---|---|---|
| Group | Comparison | Gene Target | Time Point 1 Mean (SD) | Time Point 2 Mean (SD) | T Statistic | p-Value | Cohen’s d | % Change |
| M SC vs. M SC | PD 10 to PD 15 | Mbp expression | 1.77 (1.56) | 0.89 (0.47) | 1.863 | 0.077 | 0.798 | −49.72 |
| Pdgfra expression | 1.63 (1.66) | 0.75 (0.30) | 1.8 | 0.087 | 0.771 | −53.99 | ||
| Hdac1 expression | 1.07 (0.44) | 0.74 (0.22) | 2.286 | 0.033 | 0.979 | −30.84 | ||
| Hdac3 expression | 1.75 (2.05) | 0.95 (0.48) | 1.312 | 0.204 | 0.562 | −45.71 | ||
| M SI vs. M SI | PD 10 to PD 15 | Mbp expression | 1.22 (0.70) | 1.15 (0.66) | 0.2 | 0.845 | 0.103 | −5.74 |
| Pdgfra expression | 1.29 (0.77) | 0.71 (0.23) | 2.079 | 0.058 | 1.076 | −44.96 | ||
| Hdac1 expression | 1.14 (0.58) | 0.83 (0.37) | 1.303 | 0.212 | 0.633 | −27.19 | ||
| Hdac3 expression | 0.99 (1.15) | 1.45 (0.33) | −1.086 | 0.295 | −0.528 | 46.46 | ||
| M AE vs. M AE | PD 10 to PD 15 | Mbp expression | 0.52 (0.45) | 0.71 (0.40) | −0.987 | 0.336 | −0.431 | 36.54 |
| Pdgfra expression | 0.65 (0.45) | 0.60 (0.39) | 0.272 | 0.789 | 0.119 | −7.69 | ||
| Hdac1 expression | 0.93 (0.29) | 0.68 (0.37) | 1.755 | 0.095 | 0.748 | −26.88 | ||
| Hdac3 expression | 1.85 (1.86) | 1.39 (0.42) | 0.801 | 0.432 | 0.342 | −24.86 | ||
| F SC vs. F SC | PD 10 to PD 15 | Mbp expression | 1.27 (0.83) | 1.52 (0.60) | −0.73 | 0.477 | −0.355 | 19.69 |
| Pdgfra expression | 1.24 (1.01) | 1.85 (1.20) | −1.127 | 0.277 | −0.548 | 49.19 | ||
| Hdac1 expression | 1.12 (0.52) | 1.73 (0.69) | −2.096 | 0.052 | −0.988 | 54.46 | ||
| Hdac3 expression | 1.06 (0.87) | 1.40 (0.59) | −0.941 | 0.361 | −0.443 | 32.08 | ||
| F SI vs. F SI | PD 10 to PD 15 | Mbp expression | 0.71 (0.53) | 1.10 (0.64) | −1.391 | 0.184 | −0.676 | 54.92 |
| Pdgfra expression | 1.11 (0.98) | 1.06 (0.25) | 0.14 | 0.89 | 0.068 | −4.5 | ||
| Hdac1 expression | 1.00 (0.62) | 1.13 (0.34) | −0.54 | 0.596 | −0.255 | 13 | ||
| Hdac3 expression | 1.38 (1.30) | 0.97 (0.18) | 0.963 | 0.352 | 0.485 | −29.71 | ||
| F AE vs. F AE | PD 10 to PD 15 | Mbp expression | 1.09 (0.77) | 1.50 (0.98) | −0.979 | 0.342 | −0.461 | 37.61 |
| Pdgfra expression | 1.48 (1.01) | 1.36 (0.92) | 0.258 | 0.8 | 0.122 | −8.11 | ||
| Hdac1 expression | 0.97 (0.48) | 1.50 (0.86) | −1.616 | 0.126 | −0.762 | 54.64 | ||
| Hdac3 expression | 0.83 (0.40) | 1.32 (0.58) | −2.067 | 0.055 | −0.974 | 59.04 | ||
| Independent Samples t-Tests | ||||||||
|---|---|---|---|---|---|---|---|---|
| Group | Comparison | Histological Parameter | Time Point 1 Mean (SD) | Time Point 2 Mean (SD) | T Statistic | p-Value | Cohen’s d | % Change |
| M SI vs. M SI (SH) | PD 10 to PD 15 | OPC number | 4960.83 (736.40) | 4712.72 (1201.26) | 0.457 | 0.654 | 0.232 | n/a |
| OL number | 9109.29 (2634.41) | 12,394.73 (4152.54) | −1.858 | 0.082 | −0.898 | n/a | ||
| MBP density body of corpus callosum | n/a | n/a | n/a | n/a | n/a | n/a | ||
| MBP density splenium of corpus callosum | n/a | n/a | n/a | n/a | n/a | n/a | ||
| PD 15 to PD 30 | OPC number | 4712.72 (1201.26) | 30,261.80 (13,127.84) | −6.442 | <0.001 | −2.815 | 542.13 | |
| OL number | 12,394.73 (4152.54) | 55,843.40 (11,610.49) | −11.201 | <0.001 | −5.088 | 350.54 | ||
| MBP density body of corpus callosum | 0.43 (0.09) | 0.44 (0.08) | −0.261 | 0.797 | −0.114 | n/a | ||
| MBP density splenium of corpus callosum | 0.22 (0.05) | 0.29 (0.05) | −3.4 | 0.004 | −1.62 | n/a | ||
| PD 30 to PD 45 | OPC number | 30,261.80 (13,127.84) | 49,201.27 (15,212.47) | 4.13 | 0.002 | 1.25 | 62.58 | |
| OL number | 55,843.40 (11,610.49) | 50,416.18 | −1.1 | 0.315 | −0.319 | n/a | ||
| MBP density body of corpus callosum | 0.44 (0.08) | 0.42 (0.11) | −0.667 | 0.52 | −0.193 | n/a | ||
| MBP density splenium of corpus callosum | 0.29 (0.05) | 0.28 (0.07) | −0.37 | 0.73 | −0.12 | n/a | ||
| M SI vs. M SI (WR) | PD 30 to PD 45 | OPC number | 30,261.80 (13,127.84) | 51,705.60 (18,540.18) | 3.66 | 0.005 | 1.16 | 70.86 |
| OL number | 55,843.40 (11,610.49) | 64,613.67 (31,081.10) | 0.85 | 0.42 | 0.28 | n/a | ||
| MBP density body of corpus callosum | 0.44 (0.08) | 0.42 (0.04) | −1.4 | 0.2 | −0.5 | n/a | ||
| MBP density splenium of corpus callosum | 0.29 (0.05) | 0.35 (0.09) | 2.1 | 0.07 | 0.7 | n/a | ||
| M AE vs. M AE (SH) | PD 10 to PD 15 | OPC number | 4453.00 (771.65) | 4727.88 (242.38) | −1.109 | 0.286 | −0.449 | 31.82 |
| OL number | 9627.91 (1124.10) | 10,500.11 (2173.88) | −1.159 | 0.262 | −0.521 | n/a | ||
| MBP density body of corpus callosum | n/a | n/a | n/a | n/a | n/a | n/a | ||
| MBP density splenium of corpus callosum | n/a | n/a | n/a | n/a | n/a | n/a | ||
| PD 15 to PD 30 | OPC number | 4727.88 (242.38) | 19,304.27 (8424.22) | −4.85 | <0.001 | −2.26 | 308.31 | |
| OL number | 10,500.11 (2173.88) | 43,716.5 (10,411.86) | −9.36 | <0.001 | −4.13 | 316.34 | ||
| MBP density body of corpus callosum | 0.33 (0.06) | 0.40 (0.05) | −2.6 | 0.02 | −1.21 | 21.21 | ||
| MBP density splenium of corpus callosum | 0.15 (0.06) | 0.23 (0.05) | −3.4 | 0.004 | −1.6 | 53.33 | ||
| PD 30 to PD 45 | OPC number | 19,304.27 (8424.22) | 48,456.56 (13,015.88) | 6.72 | <0.001 | 2.24 | 151.01 | |
| OL number | 43,716.5 (10,411.86) | 61,180.56 (22,375.24) | 2.34 | 0.047 | 0.78 | 39.95 | ||
| MBP density body of corpus callosum | 0.40 (0.05) | 0.37 (0.11) | −0.877 | 0.4 | −0.28 | n/a | ||
| MBP density splenium of corpus callosum | 0.23 (0.05) | 0.29 (0.14) | 1.35 | 0.21 | 0.43 | n/a | ||
| M AE vs. M AE (WR) | PD 30 to PD 45 | OPC number | 19,304.27 (8424.22) | 48,697 (12,252.38) | 6.35 | <0.001 | 2.4 | 152.26 |
| OL number | 43,716.5 (10,411.86) | 49,567 (16,860.13) | 0.98 | 0.34 | 0.35 | n/a | ||
| MBP density body of corpus callosum | 0.40 (0.05) | 0.52 (0.15) | 2.3 | 0.06 | 0.87 | n/a | ||
| MBP density splenium of corpus callosum | 0.23 (0.05) | 0.28 (0.08) | 1.36 | 0.24 | 0.61 | n/a | ||
| F SI vs. F SI (SH) | PD 10 to PD 15 | OPC number | 4481.30 (1147.03) | 4844.90 (1244.13) | −0.679 | 0.505 | −0.304 | n/a |
| OL number | 9653.100 (2390.77) | 12,409.10 (1943.46) | −2.829 | 0.011 | −1.265 | 28.55 | ||
| MBP density body of corpus callosum | n/a | n/a | n/a | n/a | n/a | n/a | ||
| MBP density splenium of corpus callosum | n/a | n/a | n/a | n/a | n/a | n/a | ||
| PD 15 to PD 30 | OPC number | 4844.90 (1244.13) | 25,258.90 (11,801.67) | −5.44 | <0.001 | −2.43 | 421.35 | |
| OL number | 12,409.10 (1943.46) | 44,692 (8861.01) | −11.25 | <0.001 | −5.03 | 260.16 | ||
| MBP density body of corpus callosum | 0.36 (0.06) | 0.47 (0.07) | −3.77 | 0.002 | −1.73 | 30.55 | ||
| MBP density splenium of corpus callosum | 0.18 (0.10) | 0.30 (0.06) | −3.1 | 0.007 | −1.4 | 66.67 | ||
| PD 30 to PD 45 | OPC number | 25,258.90 (11,801.67) | 30,564.29 (9962.98) | 1.41 | 0.21 | 0.53 | n/a | |
| OL number | 44,692 (8861.01) | 46,855.88 (21,523.32) | 0.28 | 0.78 | 0.1 | n/a | ||
| MBP density body of corpus callosum | 0.47 (0.07) | 0.38 (0.05) | −4.76 | 0.005 | −1.95 | n/a | ||
| MBP density splenium of corpus callosum | 0.30 (0.06) | 0.27 (0.08) | −1 | 0.37 | −0.41 | −19.15 | ||
| F SI vs. F SI (WR) | PD 30 to PD 45 | OPC number | 25,258.90 (11,801.67) | 51,238.60 (17,597.21) | 4.68 | 0.001 | 1.48 | 102.85 |
| OL number | 44,692 (8861.01) | 57,836.44 (8534.98) | 4.62 | 0.002 | 1.54 | 29.41 | ||
| MBP density body of corpus callosum | 0.47 (0.07) | 0.41 (0.05) | −3.76 | 0.004 | −119 | −12.77 | ||
| MBP density splenium of corpus callosum | 0.30 (0.06) | 0.35 (0.13) | 1.2 | 0.26 | 0.38 | |||
| F AE vs. F AE (SH) | PD 10 to PD 15 | OPC number | 3769.18 (954.78) | 5615.14 (462.72) | −4.68 | <0.001 | −2.358 | 48.98 |
| OL number | 7282.78 (1448.05) | 10,884.40 (3066.90) | −3.209 | 0.005 | −1.474 | 49.45 | ||
| MBP density body of corpus callosum | n/a | n/a | n/a | n/a | n/a | n/a | ||
| MBP density splenium of corpus callosum | n/a | n/a | n/a | n/a | n/a | n/a | ||
| PD 15 to PD 30 | OPC number | 5615.14 (462.72) | 25,017 (7707.62) | −7.1 | <0.001 | −3.42 | 345.52 | |
| OL number | 10,884.40 (3066.90) | 39,506.75 (14,860.75) | −5.36 | <0.001 | −2.83 | 263 | ||
| MBP density body of corpus callosum | 0.27 (0.15) | 0.54 (0.11) | −4.2 | <0.001 | −2 | 100 | ||
| MBP density splenium of corpus callosum | 0.11 (0.04) | 0.34 (0.08) | −7.22 | <0.001 | −3.61 | 209.1 | ||
| PD 30 to PD 45 | OPC number | 25017 (7707.62) | 34,108.56 (8416.51) | 3.24 | 0.01 | 1.08 | 36.34 | |
| OL number | 39,506.75 (14,860.75) | 38,872.88 (7914.29) | −0.23 | 0.83 | −0.08 | n/a | ||
| MBP density body of corpus callosum | 0.54 (0.11) | 0.60 (0.12) | 1.38 | 0.21 | 0.49 | n/a | ||
| MBP density splenium of corpus callosum | 0.34 (0.08) | 0.35 (0.04) | 0.43 | 0.69 | 0.16 | n/a | ||
| F AE vs. F AE (WR) | PD 30 to PD 45 | OPC number | 25,017 (7707.62) | 35,028.90 (9536.40) | 3.32 | 0.009 | 1.05 | 40.02 |
| OL number | 39,506.75 (14,860.75) | 46,393.70 (10,521.17) | 2.07 | 0.07 | 0.66 | n/a | ||
| MBP density body of corpus callosum | 0.54 (0.11) | 0.41 (0.14) | −2.95 | 0.02 | −0.93 | −24.07 | ||
| MBP density splenium of corpus callosum | 0.34 (0.08) | 0.32 (0.14) | −0.57 | 0.58 | −0.18 | n/a | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Milbocker, K.A.; Smith, I.F.; Brengel, E.K.; LeBlanc, G.L.; Roth, T.L.; Klintsova, A.Y. Exercise in Adolescence Enhances Callosal White Matter Refinement in the Female Brain in a Rat Model of Fetal Alcohol Spectrum Disorders. Cells 2023, 12, 975. https://doi.org/10.3390/cells12070975
Milbocker KA, Smith IF, Brengel EK, LeBlanc GL, Roth TL, Klintsova AY. Exercise in Adolescence Enhances Callosal White Matter Refinement in the Female Brain in a Rat Model of Fetal Alcohol Spectrum Disorders. Cells. 2023; 12(7):975. https://doi.org/10.3390/cells12070975
Chicago/Turabian StyleMilbocker, Katrina A., Ian F. Smith, Eric K. Brengel, Gillian L. LeBlanc, Tania L. Roth, and Anna Y. Klintsova. 2023. "Exercise in Adolescence Enhances Callosal White Matter Refinement in the Female Brain in a Rat Model of Fetal Alcohol Spectrum Disorders" Cells 12, no. 7: 975. https://doi.org/10.3390/cells12070975