
Advanced Bioinformatics Analysis and Genetic Technologies for Targeting Autophagy in Glioblastoma Multiforme

Abstract
:
1. Introduction
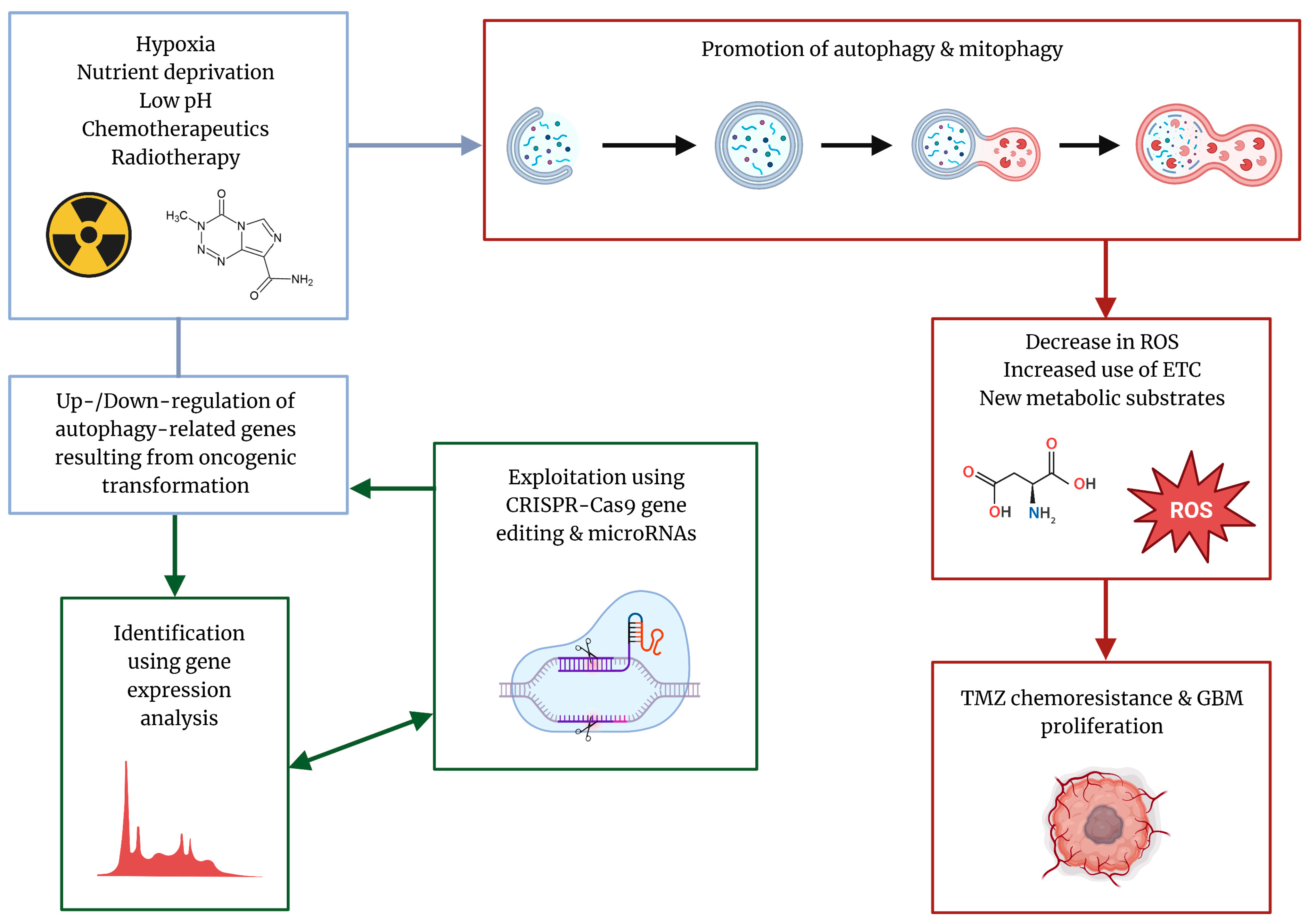
2. Autophagy over the Course of GBM Progression
3. TMZ Can Function as an Autophagy Promoter in GBM
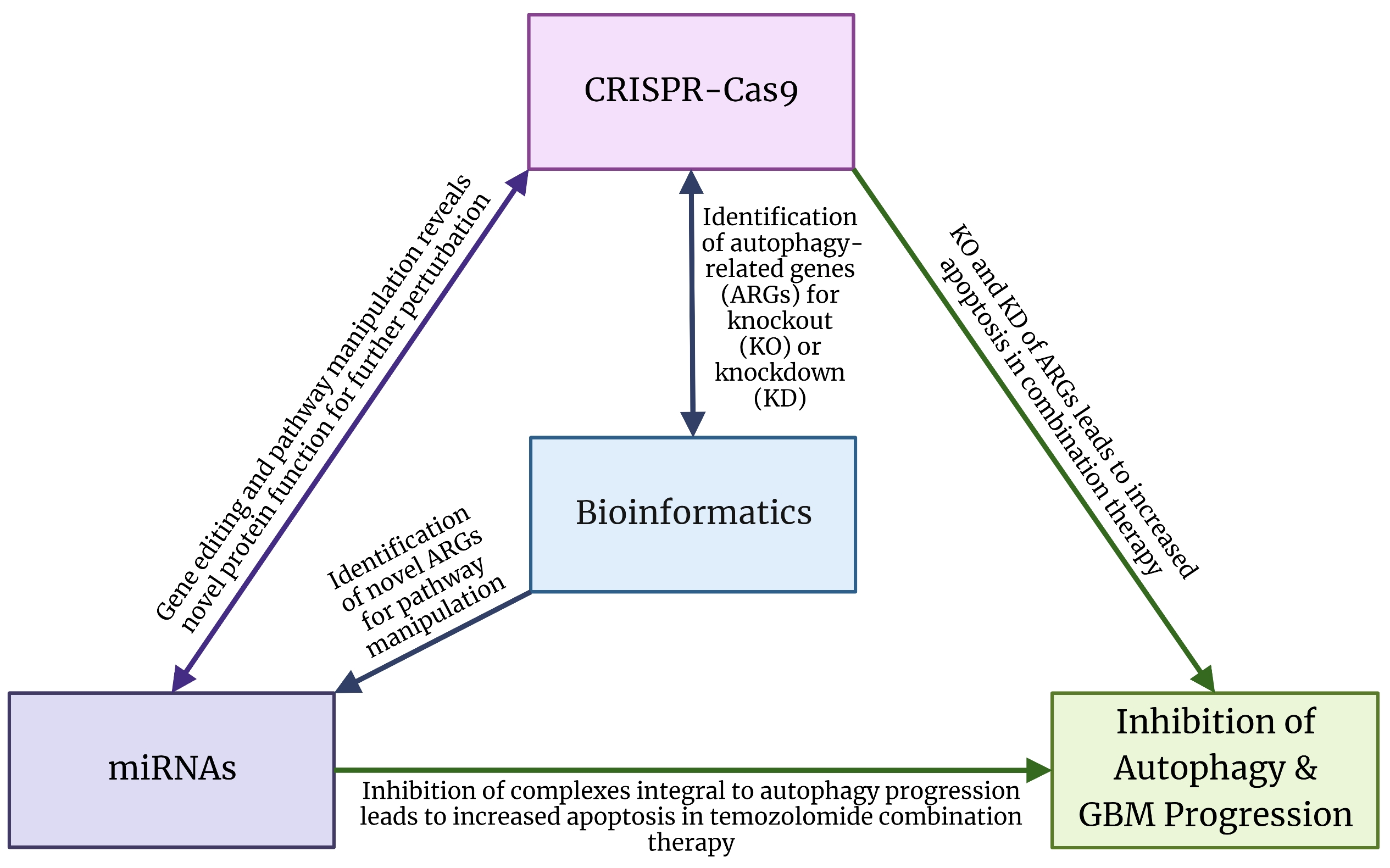
4. Bioinformatics Analysis for Identification of Biomarkers of Autophagy in GBM

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Identified ARGs | Role in Autophagy and GBM Progression | Database(s) Used | AUC Values | References |
|---|---|---|---|---|
| CDK12A | Dysregulates p53 tumor suppressor pathway | TCGA | NA | [55,60,63] |
| BIRC5 | Reduces sensitivity to EGFR tyrosine kinase inhibitors | |||
| ITGB1 | Involved in autophagosome formation | |||
| ITGA3 | Promotes FAK/PI3K/Akt pathway; interacts with ITGB1 to act as cell surface adhesion molecule | TCGA (Identification), CGGA (Verification) | 0.76 (0.5 yr), 0.72 (1 yr), 0.69 (2 yr) | [61,64,65,66,67] |
| NRG1 | Promotes autophagy via ERK1/2 activation | |||
| MAP1LC3A | Biomarker of autophagy progression | |||
| DIRAS3 | Promotes autophagy via EGFR/Akt axis | TCGA (Identification) REMBRANDT and Gravendeel (Verification) | 0.627 (1 yr), 0.733 (3 yr), 0.64 (5 yr) | [62,68,69] |
| LGALS8 | mTOR inhibitor | |||
| STAM | ULK1 stabilization and JNK1 upregulation | |||
| UBC | Inhibits autophagosome formation | TCGA (Identification and verification) | 0.811 (Combined models) | [70,71,72,73,74,75,76] |
| VHL | Inhibits autophagy by decreasing HIF-1α stability | |||
| KCTD7 | Dysfunction correlated with autophagy defects | |||
| FBXL19 | Induces chemotherapy resistance via miR-203 sponging | |||
| RNF7 | Indirectly activates autophagy via RNF7/CARD14/NF-κB axis |
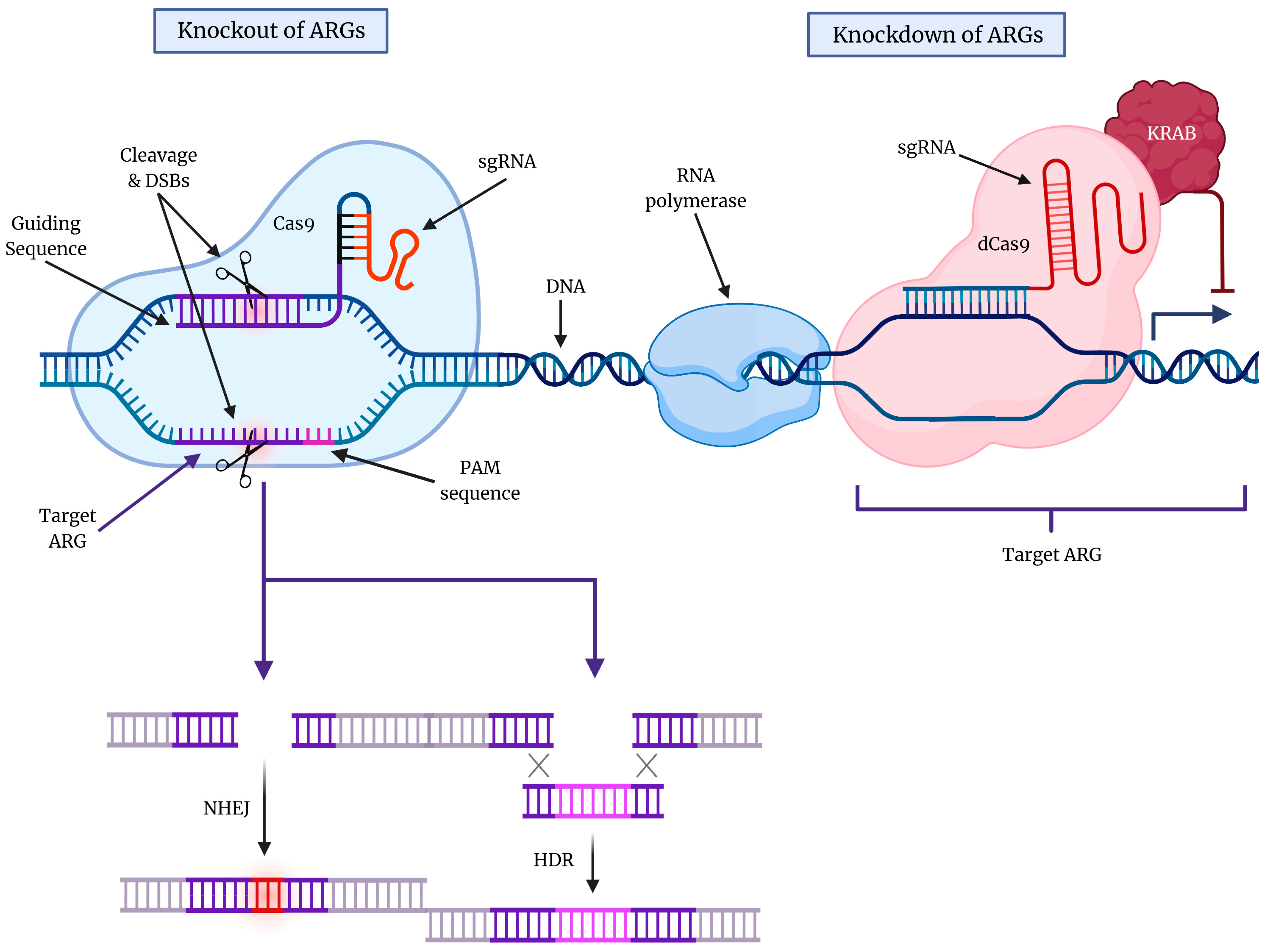
5. Gene Editing Technology for Targeting Cytoprotective Autophagy in GBM
5.1. CRISPR-Cas9 Technology for Targeting Molecular Components of Autophagy
5.2. CRISPR-Cas9 Technology for Targeting GBM Growth and Recurrence
| General Macroautophagy | |||
| KO of Gene(s) | Cell Line/Type | Therapeutic Outcome and Mechanism | References |
| ATG7, ULK1, and ATG5 | HEK293T | Inhibition of autophagy and decreased TNF secretion | [88,89,90] |
| TSC2 | HEK293T and LN18 | Inhibition of autophagy even in purine starvation conditions; increased sensitivity to photodynamic therapy | [95,96] |
| TMEM14B | H4 | Inhibition of late-stage autophagy by preventing formation of mature autophagosomes | [97,98] |
| BIRC6 | H4 | Inhibition of autophagy via upstream ubiquitination of LC3B | [100] |
| STAT3 | GSCs | Inhibition of GBM proliferation and autophagy via association of Bcl-2 and Beclin 1 | [99,102,103,104] |
| Mitophagy | |||
| KO of Gene(s) | Cell Line | Therapeutic Outcome and Mechanism | References |
| GABARAP and 6 other ATG8 genes | HeLa | Inhibition of autophagosome-lysosome fusion due to impaired recruitment of PLEKHM1 | [91] |
| ATG7 and RAB9A | K562 | Prevented initiation of alternative mitophagy, leading to ROS accumulation and DNA damage | [92] |
| HK2, SEC22B, and WIPI2 | HeLa | Demonstrated greatest inhibition of mitophagy in screen | [94] |
| ER-phagy | |||
| KD of Gene(s) | Cell Line | Therapeutic Outcome/Mechanism | References |
| NDUFB4 and NDUFB2 | HCT116 | KD of components required for OXPHOS impaired ER-phagy independent of AMPK signaling | [105] |
| Other Applications | |||
| KO of Gene(s) | Cell Line/Type | Therapeutic Outcome/Mechanism | References |
| MKI67 | GBM12 | Decrease in MGMT expression and restoration of TMZ sensitivity | [101] |
| PKMYT1 | GSCs | Essential for mitosis and GSC proliferation by inhibition of cyclin B-CDK1 activity | [106] |
| DGK | CAR-T cells | Significant regression of U87MG xenografted model via activation of ERK | [107] |
| PD-1, TRAC, and B2M | CAR-T cells | Enhanced survival of GBM xenografts via increased production of proinflammatory cytokines | [108] |
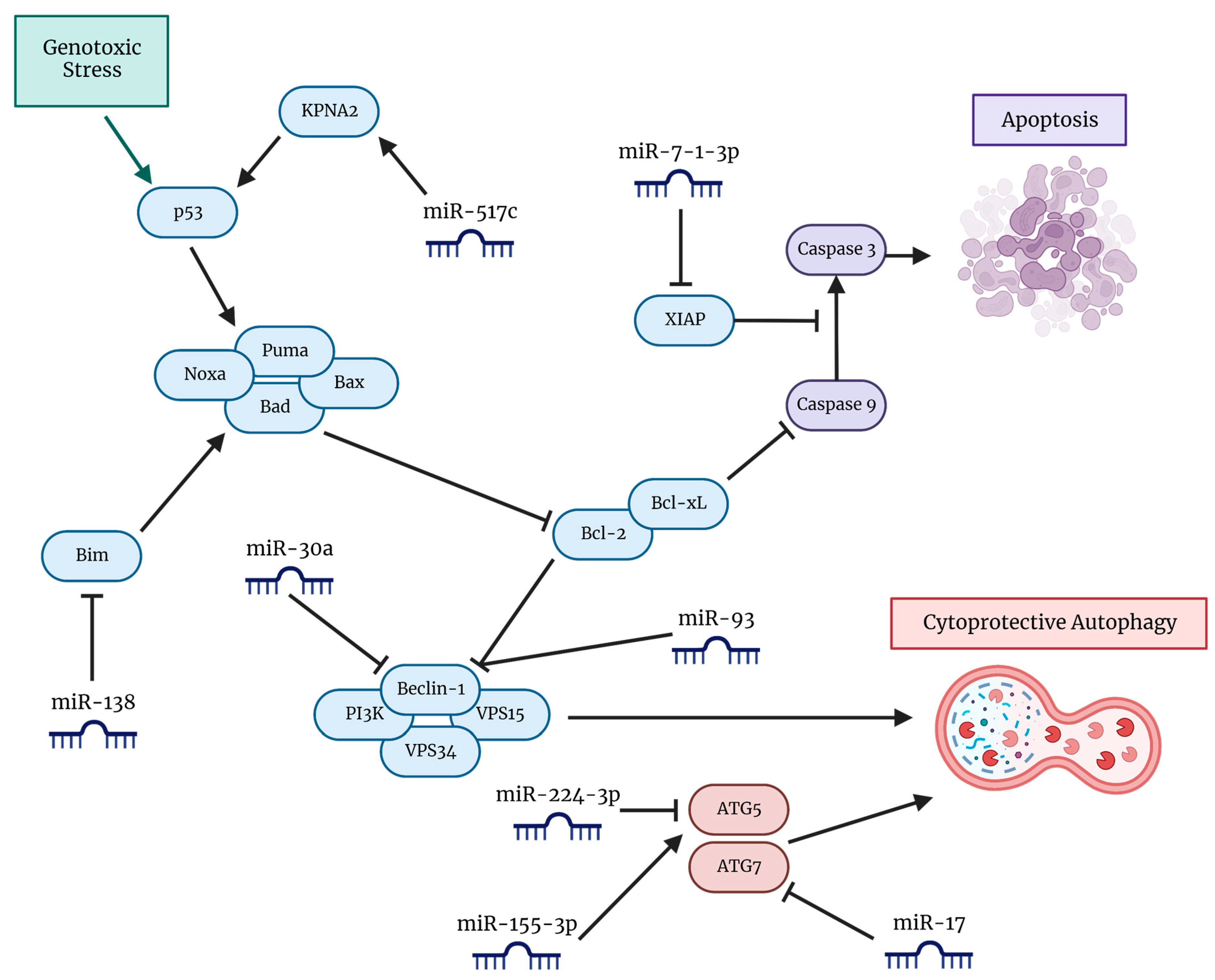
6. miRNAs and Inhibition of Autophagy for Potentiation of TMZ Efficacy in GBM
7. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Zygogianni, A.; Protopapa, M.; Kougioumtzopoulou, A.; Simopoulou, F.; Nikoloudi, S.; Kouloulias, V. From imaging to biology of glioblastoma: New clinical oncology perspectives to the problem of local recurrence. Clin. Transl. Oncol. 2018, 20, 989–1003. [Google Scholar] [CrossRef]
- Abdullah, K.G.; Adamson, C.; Brem, S. The Molecular Pathogenesis of Glioblastoma. In Glioblastoma, 1st ed.; Brem, S., Abdullah, K.G., Eds.; Elsevier, Inc.: Amsterdam, The Netherlands, 2016; pp. 21–31. [Google Scholar]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, R.; Carter, H.; Siu, I.-M.; Gallia, G.L.; et al. An Integrated Genomic Analysis of Human Glioblastoma Multiforme. Science 2008, 321, 1807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. An integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR and NF1. Cancer Cell 2010, 17, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weller, M.; van den Bent, M.; Preusser, M.; Le Rhun, E.; Tonn, J.C.; Minniti, G.; Bendszuz, M.; Balana, C.; Chinot, O.; Dirven, L.; et al. EANO guidelines on the diagnosis and treatment of diffuse gliomas of adulthood. Nat. Rev. Clin. Oncol. 2021, 18, 170–186. [Google Scholar] [CrossRef]
- Koukourakis, G.V.; Kouloulias, V.; Zacharias, G.; Papadimitriou, C.; Pantelakos, P.; Maravelis, G.; Fotineas, A.; Beli, I.; Chaldeopoulos, D.; Kouvaris, J. Temozolomide with Radiation Therapy in High Grade Brain Gliomas: Pharmaceuticals Considerations and Efficacy: A Review Article. Molecules 2009, 14, 1561–1577. [Google Scholar] [CrossRef] [Green Version]
- Hegi, M.E.; Diserens, A.-C.; Gorlia, T.; Harnou, M.-F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT Gene Silencing and Benefit from Temozolomide in Glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [Green Version]
- Malmstrom, A.; Gronberg, B.H.; Marosi, C.; Stupp, R.; Frappaz, D.; Schultz, H.; Abacioglu, U.; Tavelin, B.; Lhermitte, B.; Hegi, M.E.; et al. Temozolomide versus standard 6-week radiotherapy versus hypofractionated radiotherapy in patients older than 60 years with glioblastoma: The Nordic randomised, phase 3 trial. Lancet Oncol. 2012, 13, 916–926. [Google Scholar] [CrossRef]
- Wick, W.; Platten, M.; Meisner, C.; Felsberg, J.; Tabatabai, G.; Simon, M.; Nikkhah, G.; Papsdorf, K.; Steinbach, J.P.; Sable, M.; et al. Temozolomide chemotherapy alone versus radiotherapy alone for malignant astrocytoma in the elderly: The NOA-08 randomised, phase 3 trial. Lancet Oncol. 2012, 13, 707–715. [Google Scholar] [CrossRef] [Green Version]
- Tomar, M.S.; Kumar, A.; Srivastava, C.; Shrivastava, A. Elucidating the mechanisms of Temozolomide resistance in gliomas and the strategies to overcome resistance. Biochem. Biophys. Acta Rev. Cancer 2021, 1876, 188616. [Google Scholar] [CrossRef]
- Kimmelman, A.C.; White, E. Autophagy and Tumor Metabolism. Cell Metab. 2017, 25, 1037–1043. [Google Scholar] [CrossRef] [PubMed]
- Al-Sammarraie, N.; Ray, S.K. Application of CRISPR-Cas9 Technology to Genome Editing in Glioblastoma Multiforme. Cells 2021, 10, 2342. [Google Scholar] [CrossRef] [PubMed]
- Manea, A.J.; Ray, S.K. Regulation of autophagy as a therapeutic option in glioblastoma. Apoptosis 2021, 26, 574–599. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Vicente, M.; Cuervo, A.M. Autophagy and neurodegeneration: When the cleaning crew goes on strike. Lancet Neurol. 2007, 6, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Bejarano, E.; Cuervo, A.M. Chaperone-Mediated Autophagy. Proc. Am. Thorac. Soc. 2010, 7, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Morselli, E.; Galluzzi, L.; Kepp, O.; Marino, G.; Michaud, M.; Vitale, I.; Maiuri, M.C.; Kroemer, G. Oncosuppressive functions of autophagy. Antioxid. Redox Signal. 2011, 14, 2251–2269. [Google Scholar] [CrossRef]
- White, E. Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer 2012, 12, 401–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Galluzzi, L.; Zitvogel, L.; Kroemer, G. Autophagy and cellular immune response. Immunity 2013, 39, 211–227. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Pietrocola, F.; Bravo-San Pedro, J.M.; Amaravadi, R.; Baehrecke, E.H.; Cecconi, F.; Codogno, P.; Debnath, J.; Gewirtz, D.A.; Karantza, V.; et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015, 34, 856–880. [Google Scholar] [CrossRef]
- Cordani, M.; Butera, G.; Pacchiana, R.; Donadelli, M. Molecular interplay between mutant p53 proteins and autophagy in cancer cells. Biochim. Biophys. Acta Rev. Cancer 2017, 1867, 19–28. [Google Scholar] [CrossRef]
- Mathew, R.; Karp, C.M.; Beaudoin, B.; Vuong, N.; Chen, G.; Chen, H.-Y.; Bray, K.; Reddy, A.; Bhanot, G.; Gelinas, C.; et al. Autophagy Suppresses Tumorigenesis Through Elimination of p62. Cell 2009, 137, 1062–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leao Barros, M.B.; Pinheiro, D.R.; Borges, B.N. Mitochondrial DNA Alterations in Glioblastoma (GBM). Int. J. Mol. Sci. 2021, 22, 5855. [Google Scholar] [CrossRef] [PubMed]
- Dugenhardt, K.; Mathew, M.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gelinas, C.; Fan, Y.; et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.J.; Chee, C.E.; Huang, S.; Sinicrope, F.A. The Role of Autophagy in Cancer: Therapeutic Implications. Mol. Cancer Ther. 2011, 10, 1533–1541. [Google Scholar] [CrossRef] [Green Version]
- Gammoh, N.; Fraser, J.; Puente, C.; Syred, H.M.; Kang, H.; Ozawa, T.; Lam, D.; Acosta, J.C.; Finch, A.J.; Holland, E.; et al. Suppression of autophagy impedes glioblastoma development and induces senescence. Autophagy 2016, 12, 1431–1439. [Google Scholar] [CrossRef] [PubMed]
- Padmakrishnan, C.J.; Easwer, H.V.; Vijayakurup, V.; Menon, G.V.; Nair, S.; Gopala, S. High LC3/Beclin Expression Correlates with Poor Survival in Glioma: A Definitive Role for Autophagy as Evidenced by In Vitro Autophagic Flux. Pathol. Oncol. Res. 2017, 25, 137–148. [Google Scholar]
- Simpson, J.E.; Gammoh, N. The impact of autophagy during the development and survival of glioblastoma. Open Biol. 2020, 10, 200184. [Google Scholar] [CrossRef]
- Galavotti, S.; Bartesaghi, S.; Faccenda, D.; Shaked-Rabi, M.; Sanzone, S.; McEvoy, A.; Dinsdale, D.; Condorelli, F.; Brandner, S.; Campanella, M.; et al. The autophagy-associated factors DRAM1 and p62 regulate cell migration and invasion in glioblastoma stem cells. Oncogene 2012, 32, 699–712. [Google Scholar] [CrossRef] [Green Version]
- Hou, J.; Han, Z.-P.; Jing, Y.-Y.; Yang, X.; Zhang, S.-S.; Sun, K.; Hao, C.; Meng, Y.; Yu, F.-H.; Liu, X.-Q.; et al. Autophagy prevents irradiation injury and maintains stemness through decreasing ROS generation in mesenchymal stem cells. Cell Death Dis. 2013, 4, e844. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Prat, L.; Martinez-Vincente, M.; Periguero, E.; Ortet, L.; Rodriguez-Ubreva, J.; Rebollo, E.; Ruiz-bonilla, V.; Gutarra, S.; Ballestar, E.; Serrano, A.L.; et al. Autophagy maintains stemness by preventing senescence. Nature 2016, 529, 37–42. [Google Scholar] [CrossRef]
- Dolma, S.; Selvadurai, H.J.; Lan, X.; Lee, L.; Kushida, M.; Voisin, V.; Whetstone, H.; So, M.; Aviv, T.; Park, N.; et al. Inhibition of Dopamine Receptor D4 Impedes Autophagic Flux, Proliferation, and Survival of Glioblastoma Stem Cells. Cancer Cell 2016, 29, 859–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahase, S.; Rattenni, R.N.; Wesseling, P.; Leenders, W.; Baldotto, C.; Jain, R.; Zagzag, D. Hypoxia-Mediated Mechanisms Associated with Antiangiogenic Treatment Resistance in Glioblastomas. Am. J. Pathol. 2017, 187, 940–953. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Zhang, M.; Cheng, K.; Li, P.; Han, S.; Li, R.; Su, M.; Zeng, W.; Liu, J.; Guo, J.; et al. Resistance of glioma cells to nutrient-deprived microenvironment can be enhanced by CD133-mediated autophagy. Oncotarget 2016, 7, 76238. [Google Scholar] [CrossRef] [Green Version]
- Yang, K.; Niu, L.; Bai, Y.; Le, W. Glioblastoma: Targeting the autophagy in tumorigenesis. Brain Res. Bull. 2019, 153, 334–440. [Google Scholar] [CrossRef]
- Skoda, J.; Borankova, K.; Jansson, P.J.; Huang, M.L.-H.; Veselska, R.; Richardson, D.R. Pharmacological targeting of mitochondria in cancer stem cells: An ancient organelle at the crossroad of novel anti-cancer therapies. Pharmacol. Res. 2019, 139, 298–313. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, T.; Ferraz, L.S. Therapeutic potential of targeting mitochondrial dynamics in cancer. Biochem. Pharmacol. 2020, 182, 114282. [Google Scholar] [CrossRef]
- Capparelli, C.; Guido, C.; Whitaker-Menezes, D.; Bonuccelli, G.; Balliet, R.; Pestell, T.G.; Goldberg, A.F.; Pestell, R.G.; Howell, A.; Sneddon, S.; et al. Autophagy and senescence in cancer-associated fibroblasts metabolically supports tumor growth and metastasis, via glycolysis and ketone production. Cell Cycle 2012, 11, 2285–2302. [Google Scholar] [CrossRef] [Green Version]
- Newlands, E.S.; Blackledge, G.R.P.; Slack, J.A.; Rustin, G.J.; Smith, D.B.; Stuart, N.S.; Quarterman, C.P.; Hoffman, R.; Stevens, M.F.; Bramptom, M.H.; et al. Phase I trial of temozolomide (CCRG 81045: M&B 39831: NSC 362856). Br. J. Cancer 1992, 65, 287–291. [Google Scholar] [PubMed] [Green Version]
- Newlands, E.S.; Stevens, M.F.G.; Wedge, S.R.; Wheelhouse, R.T.; Brock, C. Temozolomide: A review of its discovery, chemical properties, and pre-clinical development and clinical trials. Cancer Treat. Rev. 1997, 23, 35–61. [Google Scholar] [CrossRef] [PubMed]
- Kanzawa, T.; Germano, I.M.; Komata, T.; Ito, H.; Kondo, Y.; Kondo, S. Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Cell Death Differ. 2004, 11, 448–457. [Google Scholar] [CrossRef] [Green Version]
- Yelton, C.J.; Ray, S.K. Multiple Mechanisms of Drug Resistance in Glioblastoma and Novel Therapeutic Opportunities. In Horizons in Cancer Research; Watanabe, H.S., Ed.; Nova Science Publishers, Inc.: Hauppauge, NY, USA, 2019; Volume 71, pp. 111–145. [Google Scholar]
- Thomas, A.; Tanaka, M.; Trepel, J.; Reinhold, W.C.; Rajapakse, V.N.; Pommier, Y. Temozolomide in the Era of Precision Medicine. Cancer Res. 2017, 77, 823–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Natsumeda, M.; Aoki, J.; Miyahara, H.; Yajima, N.; Uzuka, T.; Toyoshima, Y.; Kakita, A.; Takahashi, H.; Fujii, Y. Induction of autophagy in temozolomide treated malignant gliomas. Neuropathology 2011, 31, 486–493. [Google Scholar] [CrossRef] [PubMed]
- Koukourakis, M.I.; Mitraka, A.G.; Giatromanolaki, A. Therapeutic interaction of autophagy with radiation and temozolomide in glioblastoma: Evidence and issues to resolve. Br. J. Cancer 2016, 114, 485–496. [Google Scholar] [CrossRef]
- Rangwala, R.; Leone, R.; Chang, Y.C.; Fecher, L.A.; Schuchter, L.M.; Kramer, A.; Tan, K.-S.; Heithan, D.F.; Rodgers, G.; Gallagher, M.; et al. Phase I trial of hydroxychloroquine with dose-intense temozolomide in patients with advanced solid tumors and melanoma. Autophagy 2014, 10, 1369–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenfeld, M.R.; Ye, X.; Supko, J.G.; Desideri, S.; Grossman, S.A.; Brem, S.; Mikkelson, T.; Wang, D.; Chang, Y.C.; Hu, J.; et al. A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy 2013, 10, 1359–1368. [Google Scholar] [CrossRef]
- Sotelo, J.; Briceno, E.; Lopez-Gonzales, M.A. Adding chloroquine to conventional treatment for glioblastoma multiforme: A randomized, double-blind, placebo-controlled trial. Ann. Intern. Med. 2006, 144, 337–343. [Google Scholar] [CrossRef]
- National Library of Medicine (U.S.). A Phase II Randomized Controlled Trial for the Addition of Chloroquine, an Autophagy Inhibitor, to Concurrent Chemoradiation for Newly Diagnosed Glioblastoma, Identifier NCT02378532. Available online: https://clinicaltrials.gov/ct2/show/NCT02432417 (accessed on 25 February 2023).
- National Library of Medicine (U.S.). TN-TC11G (THC+CBD) Combination with Temozolomide and Radiotherapy in Patients With Newly-diagnosed Glioblastoma (GEINOCANN), Identifier NCT03529448. Available online: https://clinicaltrials.gov/ct2/show/NCT03529448 (accessed on 6 March 2023).
- National Library of Medicine (U.S.). Bortezomib and Temozolomide in Recurrent Glioblastoma with Unmethylated MGMT Promoter (BORTEM-17), Identifier NCT03643549. Available online: https://clinicaltrials.gov/ct2/show/NCT03643549 (accessed on 6 March 2023).
- Katayama, M.; Kawaguchi, T.; Berger, M.S.; Pieper, R.O. DNA damaging agent-induced autophagy produces a cytoprotective adenosine triphosphate surge in malignant glioma cells. Cell Death Differ. 2007, 14, 548–558. [Google Scholar] [CrossRef] [Green Version]
- Knizhnik, A.V.; Roos, W.P.; Nikolova, T.; Quiros, S.; Tomaszowski, K.-H.; Christmann, M.; Kaina, B. Survival and death strategies in glioma cells: Autophagy, senescence and apoptosis triggered by a single type of temozolomide-induced DNA damage. PLoS ONE 2013, 8, e55665. [Google Scholar] [CrossRef] [Green Version]
- Pawlowska, E.; Szczepanska, J.; Szatkowska, M.; Blasiak, J. An Interplay between Senescence, Apoptosis and Autophagy in Glioblastoma Multiforme-Role in Pathogenesis and Therapeutic Perspective. Int. J. Mol. Sci. 2018, 19, 889. [Google Scholar] [CrossRef] [Green Version]
- Vilar, J.B.; Christmann, M.; Tomicic, M.T. Alterations in Molecular Profiles Affecting Glioblastoma Resistance to Radiochemotherapy: Where Does the Good Go? Cancers 2022, 14, 2416. [Google Scholar] [CrossRef]
- Young, A.R.J.; Narita, M.; Ferreira, M.; Kirschner, K.; Sadaie, M.; Darot, J.F.J.; Tavare, S.; Arakawa, S.; Shimizu, S.; Watt, F.M.; et al. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009, 23, 798–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunther, W.; Pawlak, E.; Damasceno, R.; Arnold, H.; Terzis, A.J. Temozolomide induces apoptosis and senescence in glioma cells cultured as multicellular spheroids. Br. J. Cancer 2003, 88, 463–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunel, A.; Hombourger, S.; Barthout, E.; Battu, S.; Kogel, D.; Antonietti, P.; Deluche, E.; Saada, S.; Durand, S.; Lalloue, F.; et al. Autophagy inhibition reinforces stemness together with exit from dormancy of polydisperse glioblastoma stem cells. Aging 2021, 13, 18106–18130. [Google Scholar] [CrossRef]
- Zheng, Y.; Liu, L.; Wang, Y.; Xiao, S.; Mai, R.; Zhu, Z.; Cao, Y. Glioblastoma stem cell (GSC)-derived PD-L1-containing exosomes activates AMPK/ULK1 pathway mediated autophagy to increase temozolomide-resistance in glioblastoma. Cell Biosci. 2021, 11, 63. [Google Scholar] [CrossRef] [PubMed]
- Kondapuram, S.K.; Coumar, M.S. Pan-cancer gene expression analysis: Identification of deregulated autophagy genes and drugs to target them. Gene 2022, 844, 146821. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gao, L.; Guo, X.; Feng, C.; Lian, W.; Deng, K.; Xing, B. Development and validation of a nomogram with an autophagy-related gene signature for predicting survival in patients with glioblastoma. Aging 2019, 11, 12246–12269. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, W.; Xiao, Z.; Guan, G.; Liu, X.; Zhuang, M. A risk signature with four autophagy-related genes for predicting survival of glioblastoma multiforme. J. Cell. Mol. Med. 2020, 24, 3807–3821. [Google Scholar] [CrossRef] [Green Version]
- Zanca, C.; Villa, G.R.; Benitez, J.A.; Thorne, A.H.; Koga, T.; D’Antonio, M.; Ikegami, S.; Ma, J.; Boyer, A.D.; Banisadr, A.; et al. Glioblastoma cellular cross-talk converges on NF-κB to attenuate EGFR inhibitor sensitivity. Genes Dev. 2017, 31, 1212–1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, J.; Zhao, Y.; Hu, D.; Li, H.; Gao, L.; Liu, Z.; Shi, K. Silencing of integrin subunit α3 inhibits the proliferation, invasion, migration and autophagy of esophageal squamous cell carcinoma cells. Oncol. Lett. 2022, 24, 271. [Google Scholar] [CrossRef]
- Huang, Y.; Kong, Y.; Zhang, L.; He, T.; Zhou, X.; Yan, Y.; Zhang, L.; Zhou, D.; Lu, S.; Zhou, J.; et al. High Expression of ITGA3 Promotes Proliferation and Cell Cycle Progression and Indicates Poor Prognosis in Intrahepatic Cholangiocarcinoma. Biomed Res. Int. 2018, 2018, 2352139. [Google Scholar] [CrossRef] [Green Version]
- Lin, W.-W.; Ou, G.-Y.; Lin, J.-Z.; Yi, S.-J.; Yao, W.-C.; Pan, H.-C.; Zhao, W.-J. Neuregulin 1 enhances cell adhesion molecule L1 like expression levels and promotes malignancy in human glioma. Oncol. Lett. 2020, 20, 326–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zois, C.E.; Giatromanolaki, A.; Sivridis, E.; Papaiakovou, M.; Kainulainen, H.; Koukourakis, M.I. "Autophagic flux" in normal mouse tissues: Focus on endogenous LC3A processing. Autophagy 2011, 7, 1371–1378. [Google Scholar] [CrossRef] [Green Version]
- Tian, S.; Jin, S.; Wu, Y.; Liu, T.; Luo, M.; Ou, J.; Xie, W.; Cui, J. High-throughput screening of functional deubiquitinating enzymes in autophagy. Autophagy 2021, 17, 1367–1378. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.-Y.; Li, Y.; Jiang, W.-Q.; Zhou, L.-F. MAPK/JNK signalling: A potential autophagy regulation pathway. Biosci. Rep. 2015, 35, e00199. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.-C.; Wu, H.-Y.; Dang, Y.-W.; Chen, G. Role of alternative splicing signatures in the prognosis of glioblastoma. Cancer Med. 2019, 8, 7623–7636. [Google Scholar] [CrossRef] [Green Version]
- Yan, C.; Huo, H.; Yang, C.; Zhang, T.; Chu, Y.; Liu, Y. Ubiquitin C-Terminal Hydrolase L1 regulates autophagy by inhibiting autophagosome formation through its deubiquitinating enzyme activity. Biochem. Biophys. Res. Commun. 2018, 497, 726–733. [Google Scholar] [CrossRef]
- Feng, J.; Zhang, Y.; She, X.; Sun, Y.; Fan, L.; Ren, X.; Fu, H.; Liu, C.; Li, P.; Zhao, C.; et al. Hypermethylated gene ANKDD1A is a candidate tumor suppressor that interacts with FIH1 and decreases HIF1α stability to inhibit cell autophagy in the glioblastoma multiforme hypoxia microenvironment. Oncogene 2019, 38, 103–119. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wang, H.; Wang, C. Lysosomal dysfunction, autophagic defects, and CLN5 accumulation underlie the pathogenesis of KCTD7-mutated neuronal ceroid lipofuscinoses. Autophagy 2022, 2022, 1–3, Epub ahead of print. [Google Scholar] [CrossRef]
- Mellet, M. Regulation and dysregulation of CARD14 signalling and its physiological consequences in inflammatory skin disease. Cell. Immunol. 2020, 354, 104147. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhao, Y.-F.; Liu, R.-S.; Xiong, Y.-J.; Shen, X.; Wang, Y.; Liang, Z.-Q. Olanzapine induced autophagy through suppression of NF-κB activation in human glioma cells. CNS Neurosci. Ther. 2019, 25, 911–921. [Google Scholar] [CrossRef] [Green Version]
- Shen, B.; Yuan, Y.; Zhang, Y.; Yu, S.; Peng, W.; Huang, X.; Feng, J. Long non-coding RNA FBXL19-AS1 plays oncogenic role in colorectal cancer by sponging miR-203. Biochem. Biophys. Res. Commun. 2017, 488, 67–73. [Google Scholar] [CrossRef]
- Wang, D.; Jiang, Y.; Wang, T.; Wang, Z.; Zou, F. Identification of a novel autophagy-related prognostic signature and small molecule drugs for glioblastoma by bioinformatics. BMC Med. Genom. 2022, 15, 111. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-Y.; Feun, L.G.; Thongkum, A.; Tu, C.-H.; Chen, S.-M.; Wangpaichitr, M.; Wu, C.; Kuo, M.T.; Savaraj, N. Autophagic Mechanism in Anti-Cancer Immunity: Its Pros and Cons for Cancer Therapy. Int. J. Mol. Sci. 2017, 18, 1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouyang, D.-Y.; Xu, L.-H.; He, X.-H.; Zhang, Y.-T.; Zeng, L.-H.; Cai, J.-Y.; Ren, S. Autophagy is differentially induced in prostate cancer LNCaP, DU145 and PC-3 cells via distinct splicing profiles of ATG5. Autophagy 2013, 9, 20–32. [Google Scholar] [CrossRef] [Green Version]
- Torres, P.; Nunez-Ramirez, O.; Romero-Guevara, R.; Bares, G.; Granado-Serrano, A.B.; Ayala, V.; Boada, J.; Fontdevila, L.; Povedano, M.; Sanchis, D.; et al. Cryptic exon splicing function of TARDBP interacts with autophagy in nervous tissue. Autophagy 2018, 14, 1398–1403. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Chen, Q.; Liu, H.; Zeng, L.; Zhou, Y.; Liu, X.; Bai, Y.; Zhang, J.; Pan, Y.; Shao, C. SDC1-dependent TGM2 determines radiosensitivity in glioblastoma by coordinating EPG5-mediated fusion of autophagosomes with lysosomes. Autophagy 2023, 19, 839–857. [Google Scholar] [CrossRef]
- Zhao, Z.; Liu, M.; Long, W.; Yuan, J.; Li, H.; Zhang, C.; Tang, G.; Jiang, W.; Yuan, X.; Qu, M.; et al. Knockdown lncRNA CRNDE enhances temozolomide chemosensitivity by regulating autophagy in glioblastoma. Cancer Cell Int. 2021, 21, 456. [Google Scholar] [CrossRef]
- Adli, M. The CRISPR tool kit for genome editing and beyond. Nat. Commun. 2018, 9, 1911. [Google Scholar] [CrossRef] [Green Version]
- Doudna, J.A.; Charpentier, E. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef]
- Ishino, Y.; Shinagawa, H.; Makino, K.; Amemura, M.; Nakata, A. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J. Bacteriol. 1987, 169, 5429–5433. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Chew, S.J.L.; Shi, Y.; Gong, Z.; Shen, H.-M. CRISPR system for genome engineering: The application for autophagy study. BMB Rep. 2017, 50, 247–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, J.R.; Corn, J.E. A CRISPR view on autophagy. Trends Cell Biol. 2022, 32, 1008–1022. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J., 3rd; Kang, R.; Tang, D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428. [Google Scholar] [CrossRef]
- Horne, D.J.; Graustein, A.D.; Shah, J.A.; Peterson, G.; Savlov, M.; Steele, S.; Narita, M.; Hawn, T.R. Human ULK1 Variation and Susceptibility to Mycobacterium tuberculosis Infection. J. Infect. Dis. 2016, 214, 1260–1267. [Google Scholar] [CrossRef] [Green Version]
- Kim, N.-Y.; Han, B.-I.; Lee, M. Cytoprotective role of autophagy against BH3 mimetic gossypol in ATG5 knockout cells generated by CRISPR-Cas9 endonuclease. Cancer Lett. 2016, 370, 19–26. [Google Scholar] [CrossRef]
- Padman, B.S.; Nguyen, T.N.; Lazarou, M. Autophagosome formation and cargo sequestration in the absence of LC3/GABARAPs. Autophagy 2016, 13, 772–774. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Fang, Y.; Yan, L.; Yuan, N.; Zhang, S.; Xu, L.; Nie, M.; Zhang, X.; Wang, J. Erythroleukemia cells acquire an alternative mitophagy capability. Sci. Rep. 2016, 6, 24641. [Google Scholar] [CrossRef] [Green Version]
- Hoshino, A.; Wang, W.-J.; Wada, S.; McDermott-Roe, C.; Evans, C.S.; Gosis, B.; Morley, M.P.; Rathi, K.S.; Li, J.; Li, K.; et al. The ADP/ATP translocase drives mitophagy independent of nucleotide exchange. Nature 2019, 575, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.-M.; Harper, N.J.; Paulo, J.A.; Li, M.; Xu, Q.; Coughlin, M.; Elledge, S.J.; Harper, J.W. Integrated proteogenetic analysis reveals the landscape of a mitochondrial-autophagosome synapse during PARK2-dependent mitophagy. Sci. Adv. 2019, 5, eaay4624. [Google Scholar] [CrossRef] [Green Version]
- Mimura, K.; Sakamaki, J.-I.; Morishita, H.; Kawazu, M.; Mano, H.; Mizushima, N. Genome-wide CRISPR screening reveals nucleotide synthesis negatively regulates autophagy. J. Biol. Chem. 2021, 296, 100780. [Google Scholar] [CrossRef]
- Fettweis, G.; Valentin, E.D.; L’homme, L.; Lassence, C.; Dequiedt, F.; Fillet, M.; Coupienne, I.; Piette, J. RIP3 antagonizes a TSC2-mediated pro-survival pathway in glioblastoma cell death. Biochem. Biophys. Acta Mol. Cell Res. 2017, 1864, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Moretti, F.; Bergman, P.; Dodgson, S.; Marcellin, D.; Claerr, I.; Goodwin, J.M.; DeJesus, R.; Kang, Z.; Antczak, C.; Begue, D.; et al. TMEM41B is a novel regulator of autophagy and lipid mobilization. EMBO Rep. 2018, 19, e45889. [Google Scholar] [CrossRef]
- Shoemaker, C.; Huang, T.Q.; Weir, N.R.; Polyakov, N.J.; Schultz, S.W.; Denic, V. CRISPR screening using an expanded toolkit of autophagy reporters identifies TMEM41B as a novel autophagy factor. PLoS Biol. 2019, 17, e2007044. [Google Scholar] [CrossRef] [Green Version]
- Morita, K.; Hama, Y.; Izume, T.; Tamura, N.; Ueno, T.; Yamashita, Y.; Sakamaki, Y.; Mimura, K.; Morishita, H.; Shihoya, W.; et al. Genome-wide CRISPR screen identifies TMEM41B as a gene required for autophagosome formation. J. Cell Biol. 2018, 217, 3817–3828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, R.; Bonifacino, J.S. Negative regulation of autophagy by UBA6-BIRC6–mediated ubiquitination of LC3. Elife 2019, 8, e50034. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, D.; Fan, M.; Yang, C.H.; Zbytek, B.; Finkelstein, D.; Roussel, M.F.; Pfeffer, L.M. The critical role that STAT3 plays in glioma-initiating cells: STAT3 addiction in glioma. Oncotarget 2018, 9, 22095–22112. [Google Scholar] [CrossRef] [Green Version]
- Yuan, G.; Yan, S.-F.; Xue, H.; Zhang, P.; Sun, J.-T.; Li, G. Cucurbitacin I Induces Protective Autophagy in Glioblastoma in Vitro and in Vivo. Signal Transduct. 2014, 285, P10607–P10619. [Google Scholar] [CrossRef] [Green Version]
- Yuan, S.; Yan, Q.; Zhao, Z.-Y.; Zhang, J.-L.; Zhang, H.; Hang, Y.; Yuan, Z. STAT3-mediated upregulation of LINC00520 contributed to temozolomide chemoresistance in glioblastoma by interacting with RNA-binding protein LIN28B. Cancer Cell Int. 2022, 22, 248. [Google Scholar] [CrossRef]
- Liang, J.R.; Lingeman, E.; Luong, T.; Ahmed, S.; Muhar, M.; Nguyen, T.; Olzmann, J.A.; Corn, J.E. A Genome-wide ER-phagy Screen Highlights Key Roles of Mitochondrial Metabolism and ER-Resident UFMylation. Cell 2020, 180, 1160–1177. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, M.; Gan, H.; Wang, H.; Lee, J.-H.; Fang, D.; Kitange, G.J.; He, L.; Hu, Z.; Parney, I.F.; et al. A novel enhancer regulates MGMT expression and promotes temozolomide resistance in glioblastoma. Nat. Commun. 2018, 9, 2949. [Google Scholar] [CrossRef] [Green Version]
- Toledo, C.M.; Ding, Y.; Hoellerbauer, P.; Davis, R.J.; Basom, R.; Girard, E.J.; Lee, E.; Corrin, P.; Hart, T.; Bolouri, H.; et al. Genome-wide CRISPR-Cas9 Screens Reveal Loss of Redundancy between PKMYT1 and WEE1 in Glioblastoma Stem-like Cells. Cell Rep. 2015, 13, 2425–2439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, I.-Y.; Kim, Y.-Y.; Yu, H.-S.; Lee, M.; Kim, S.; Lee, J. CRISPR/Cas9-Mediated Knockout of DGK Improves Antitumor Activities of Human T Cells. Cancer Res. 2018, 78, 4692–4703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, B.D.; Yu, X.; Castano, A.P.; Darr, H.; Henderson, D.B.; Bouffard, A.A.; Larson, R.C.; Scarfo, I.; Bailey, S.R.; Gerhard, G.M.; et al. CRISPR-Cas9 disruption of PD-1 enhances activity of universal EGFRvIII CAR T cells in a preclinical model of human glioblastoma. J. Immunother. Cancer 2019, 7, 304. [Google Scholar] [CrossRef]
- Ma, W.; Zhou, Y.; Liu, M.; Qin, Q.; Cui, Y. Long non-coding RNA LINC00470 in serum derived exosome: A critical regulator for proliferation and autophagy in glioma cells. Cancer Cell Int. 2021, 21, 149. [Google Scholar] [CrossRef]
- Oldrini, B.; Curiel-Garcia, A.; Marques, C.; Matia, V.; Uluckan, O.; Grana-Castro, O.; Torres-Ruiz, R.; Rodriguez-Perales, S.; Huse, J.T.; Squatrito, M. Somatic genome editing with the RCAS-TVA-CRISPR-Cas9 system for precision tumor modeling. Nat. Commun. 2018, 9, 1466. [Google Scholar] [CrossRef] [Green Version]
- Cai, X.; Sughrue, M.E. Glioblastoma: New therapeutic strategies to address cellular and genomic complexity. Oncotarget 2017, 9, 9540–9554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, F.L.; Marques-Torrejon, M.-A.; Morrison, G.M.; Pollar, S.M. Experimental models and tools to tackle glioblastoma. Dis. Model Mech. 2019, 12, dmm040386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuckermann, M.; Hovestadt, V.; Knobbe-Thomsen, C.B.; Zapatka, T.; Northcott, P.A.; Schramm, K.; Belic, J.; Jones, D.T.W.; Tschida, B.; Moriarity, B.; et al. Somatic CRISPR/Cas9-mediated tumour suppressor disruption enables versatile brain tumour modelling. Nat. Commun. 2015, 6, 7391. [Google Scholar] [CrossRef] [Green Version]
- Bian, S.; Repic, M.; Guo, Z.; Kavirayani, A.; Burkard, T.; Bagley, J.A.; Krauditsch, C.; Knoblich, J.A. Genetically engineered cerebral organoids model brain tumour formation. Nat. Methods 2019, 15, 631–639. [Google Scholar] [CrossRef]
- Ogawa, J.; Pao, G.M.; Shokhirev, M.N.; Verma, I.M. Glioblastoma Model Using Human Cerebral Organoids. Cell Rep. 2019, 23, 1220–1229. [Google Scholar] [CrossRef] [Green Version]
- Bedoya, D.M.; Dutoit, V.; Migliorini, D. Allogeneic CAR T Cells: An Alternative to Overcome Challenges of CAR T Cell Therapy in Glioblastoma. Front. Immunol. 2021, 12, 640082. [Google Scholar] [CrossRef] [PubMed]
- Arnesen, V.S.; Navarro, A.G.; Chekenya, M. Challenges and Prospects for Designer T and NK Cells in Glioblastoma Immunotherapy. Cancers 2021, 13, 4986. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-RNA–Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocinol. 2018, 9, 402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calin, G.A.; Dumitru, C.D.; Shimizu, M.; Bichi, R.; Zupo, S.; Noch, E.; Aldler, H.; Rattan, S.; Keating, M.; Rai, K.; et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 15524–15529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sufianov, A.; Begliarzade, S.; Ilyasova, T.; Liang, Y.; Beylerli, O. MicroRNAs as prognostic markers and therapeutic targets in gliomas. Noncoding RNA Res. 2022, 7, 171–177. [Google Scholar] [CrossRef]
- Lu, Y.; Xiao, L.; Liu, Y.; Wang, H.; Li, H.; Zhou, Q.; Pan, J.; Lei, B.; Huang, A.; Qi, S. MIR517C inhibits autophagy and the epithelial-to-mesenchymal (-like) transition phenotype in human glioblastoma through KPNA2-dependent disruption of TP53 nuclear translocation. Autophagy 2015, 11, 2213–2232. [Google Scholar] [CrossRef] [Green Version]
- Comincini, S.; Allavena, G.; Palumbo, S.; Morini, M.; Durando, F.; Angeletti, F.; Pirtoli, L.; Miracco, C. microRNA-17 regulates the expression of ATG7 and modulates the autophagy process, improving the sensitivity to temozolomide and low-dose ionizing radiation treatments in human glioblastoma cells. Cancer Biol. Ther. 2013, 14, 574–586. [Google Scholar] [CrossRef]
- Xu, J.; Huang, H.; Peng, R.; Ding, X.; Jiang, B.; Yuan, X.; Xi, J. MicroRNA-30a increases the chemosensitivity of U251 glioblastoma cells to temozolomide by directly targeting beclin 1 and inhibiting autophagy. Exp. Ther. Med. 2018, 15, 4798–4804. [Google Scholar]
- Chakrabarti, M.; Ray, S.K. Anti-tumor activities of luteolin and silibinin in glioblastoma cells: Overexpression of miR-7-1-3p augmented luteolin and silibinin to inhibit autophagy and induce apoptosis in glioblastoma in vivo. Apoptosis 2016, 21, 312–328. [Google Scholar] [CrossRef]
- Luo, S.; Garcia-Arencibia, M.; Zhao, R.; Puri, C.; Toh, P.P.C.; Sadiq, O.; Rubinsztein, D.C. Bim Inhibits Autophagy by Recruiting Beclin 1 to Microtubules. Mol Cell. 2012, 47, 359–370. [Google Scholar] [CrossRef] [Green Version]
- Stojcheva, N.; Schechtmann, G.; Sass, S.; Roth, P.; Florea, A.-M.; Stefanski, A.; Stuhler, K.; Wolter, M.; Muller, N.S.; Theis, F.J.; et al. MicroRNA-138 promotes acquired alkylator resistance in glioblastoma by targeting the Bcl-2-interacting mediator BIM. Oncotarget 2016, 7, 12937–12950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.; Qi, P.; Zheng, T.; Li, F.; He, X. The HIF-1α/miR-224-3p/ATG5 axis affects cell mobility and chemosensitivity by regulating hypoxia-induced protective autophagy in glioblastoma and astrocytoma. Oncol. Rep. 2019, 41, 1759–1768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, H.; Yuan, g.; Guo, X.; Liu, Q.; Zhang, J.; Gao, X.; Guo, X.; Xu, S.; Li, T.; Shao, Q.; et al. A novel tumor-promoting mechanism of IL6 and the therapeutic efficacy of tocilizumab: Hypoxia-induced IL6 is a potent autophagy initiator in glioblastoma via the p-STAT3-MIR155-3p-CREBRF pathway. Autophagy 2016, 12, 1129–1152. [Google Scholar] [CrossRef] [Green Version]
- Huang, T.; Wan, X.; Alvares, A.A.; James, C.D.; Song, X.; Yang, Y.; Sastry, N.; Nakano, I.; Sulman, E.P.; Hu, B.; et al. MIR93 (microRNA -93) regulates tumorigenicity and therapy response of glioblastoma by targeting autophagy. Autophagy 2019, 15, 1100–1111. [Google Scholar] [CrossRef]
- Han, Y.; Zhou, L.; Wu, T.; Huang, Y.; Cheng, Z.; Li, X.; Sun, T.; Zhou, Y.; Du, Z. Downregulation of lncRNA-MALAT1 Affects Proliferation and the Expression of Stemness Markers in Glioma Stem Cell Line SHG139S. Cell. Mol. Neurobiol. 2015, 36, 1097–1107. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Luo, W.; Wang, J.; Pent, T.; Sun, G.; Shi, J.; Li, Z.; Zhang, B. Malat1 activates autophagy and promotes cell proliferation by sponging miR-101 and upregulating STMN1, RAB5A and ATG4D expression in glioma. Biochem. Biophys. Res. Commun. 2017, 492, 480–486. [Google Scholar] [CrossRef] [PubMed]
- Baspinar, Y.; Elmaci, I.; Ozpinar, A.; Altinoz, M.A. Long non-coding RNA MALAT1 as a key target in pathogenesis of glioblastoma. Janus faces or Achilles’ heal? Gene 2020, 739, 144518. [Google Scholar] [CrossRef]
- Yang, M.C.; Loh, J.K.; Li, Y.Y.; Huang, W.S.; Chou, C.H.; Cheng, J.; Wang, Y.; Lieu, A.S.; Howng, S.L.; Hong, Y.R.; et al. Bcl2L12 with a BH3-like domain in regulating apoptosis and TMZ-induced autophagy: A prospective combination of ABT-737 and TMZ for treating glioma. Int. J. Oncol. 2015, 46, 1304–1316. [Google Scholar] [CrossRef] [Green Version]
- Calis, S.; Dogan, B.; Durdagi, S.; Celebi, A.; Yapicier, O.; Kilic, T.; Turanli, E.T.; Avsar, T. A novel BH3 mimetic Bcl-2 inhibitor promotes autophagic cell death and reduces in vivo Glioblastoma tumor growth. Cell Death Discov. 2022, 8, 433. [Google Scholar] [CrossRef]
- Xiang, H.; Liu, R.; Zhang, X.; An, R.; Zhou, M.; Tan, C.; Li, Q.; Su, M.; Guo, C.; Zhou, L.; et al. Discovery of Small-Molecule Autophagy Inhibitors by Disrupting the Protein−Protein Interactions Involving Autophagy-Related 5. J. Med. Chem. 2023, 66, 2457–2476. [Google Scholar] [CrossRef] [PubMed]



| Drug Combined with TMZ | Role in Autophagy | Phase | Dosage | OS and PFS Clinical Outcomes | Adverse Effects | References |
|---|---|---|---|---|---|---|
| HCQ | Late-stage inhibitor | I | 200–1200 mg/day | Partial response in melanoma | Fatigue, anorexia, nausea, constipation, and diarrhea | [46] |
| I/II | 600 mg/day (MTD) | MS of 15.6 months in GBM | Grade 3 and 4 neutropenia and thrombocytopenia at 800 mg/day HCQ | [47] | ||
| CQ | Late-stage inhibitor | I | 150 mg/day | MS of 24 months in GBM | Seizures due to neoplasm; no other adverse effects | [48] |
| II | 400 mg/day | Not yet recruiting | NA | [49] | ||
| TN-TC11G (THC-CBD) | Promoter via TRB3 pathway | IB | 5–40 mg/3 times/day | Not yet recruiting | NA | [50] |
| Bortezomib | Inhibitor | IB/II | 1.3 mg/m2/3 times/week | Currently recruiting | NA | [51] |
| miRNA | Mechanism of Action | Results | References |
|---|---|---|---|
| miR-517c | miR-517c/KPNA2/cytoplasmic p53 axis; inhibition of autophagy in wild type-p53 U87MG cell line | Combination with TMZ reduced migration and infiltration, increased expression of epithelial markers and inhibition of EMT | [122] |
| miR-17 | ATG7 inhibitor | Increased TMZ sensitivity and cell death | [123] |
| miR-30a | Beclin 1 inhibitor | TMZ treatment downregulated miR-30a in a dose-dependent manner | [124] |
| miR-7-1-3p | Targets BIRC4 to promote apoptosis | Combination with SIL and LUT led to autophagy inhibition in rapamycin-treated GBM cell lines | [125] |
| miR-138 | miR-138/Bim axis; upstream promotion of Beclin 1 | Upregulation led to promotion of autophagy and TMZ resistance | [126,127] |
| miR-224-3p | Downregulation of ATG5 | Inhibition of autophagy, decreased cell mobility, increased sensitivity to TMZ | [128] |
| miR-155-3p | Hypoxia-induced IL-6/pSTAT3/miR-155-3p/CREB3/ATG5 axis | KD of miRNA led to inhibition of IL-6-induced cytoprotective autophagy; synergism with TMZ therapy | [129] |
| miR-93 | Beclin 1, ATG5, and ATG4B inhibitor | Overexpression led to autophagy inhibition and GSC sensitization to TMZ therapy | [130] |
| MALAT1 (lncRNA) | miR-101 and miR-203 inhibitor | Maintained stemness and induced protective autophagy; decreased TMZ sensitivity due to an increase in EMT | [131,132,133] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manea, A.J.; Ray, S.K. Advanced Bioinformatics Analysis and Genetic Technologies for Targeting Autophagy in Glioblastoma Multiforme. Cells 2023, 12, 897. https://doi.org/10.3390/cells12060897
Manea AJ, Ray SK. Advanced Bioinformatics Analysis and Genetic Technologies for Targeting Autophagy in Glioblastoma Multiforme. Cells. 2023; 12(6):897. https://doi.org/10.3390/cells12060897
Chicago/Turabian StyleManea, Amanda J., and Swapan K. Ray. 2023. "Advanced Bioinformatics Analysis and Genetic Technologies for Targeting Autophagy in Glioblastoma Multiforme" Cells 12, no. 6: 897. https://doi.org/10.3390/cells12060897