
Concurrent Activation of Both Survival-Promoting and Death-Inducing Signaling by Chloroquine in Glioblastoma Stem Cells: Implications for Potential Risks and Benefits of Using Chloroquine as Radiosensitizer
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Cell-Based Assays
2.2. Cell Treatments and Cell-Based Assays
2.3. Animal Experiments
2.4. Protein Analyses
2.5. Gene Expression and Bioinformatics
2.6. Statistical Analysis
3. Results
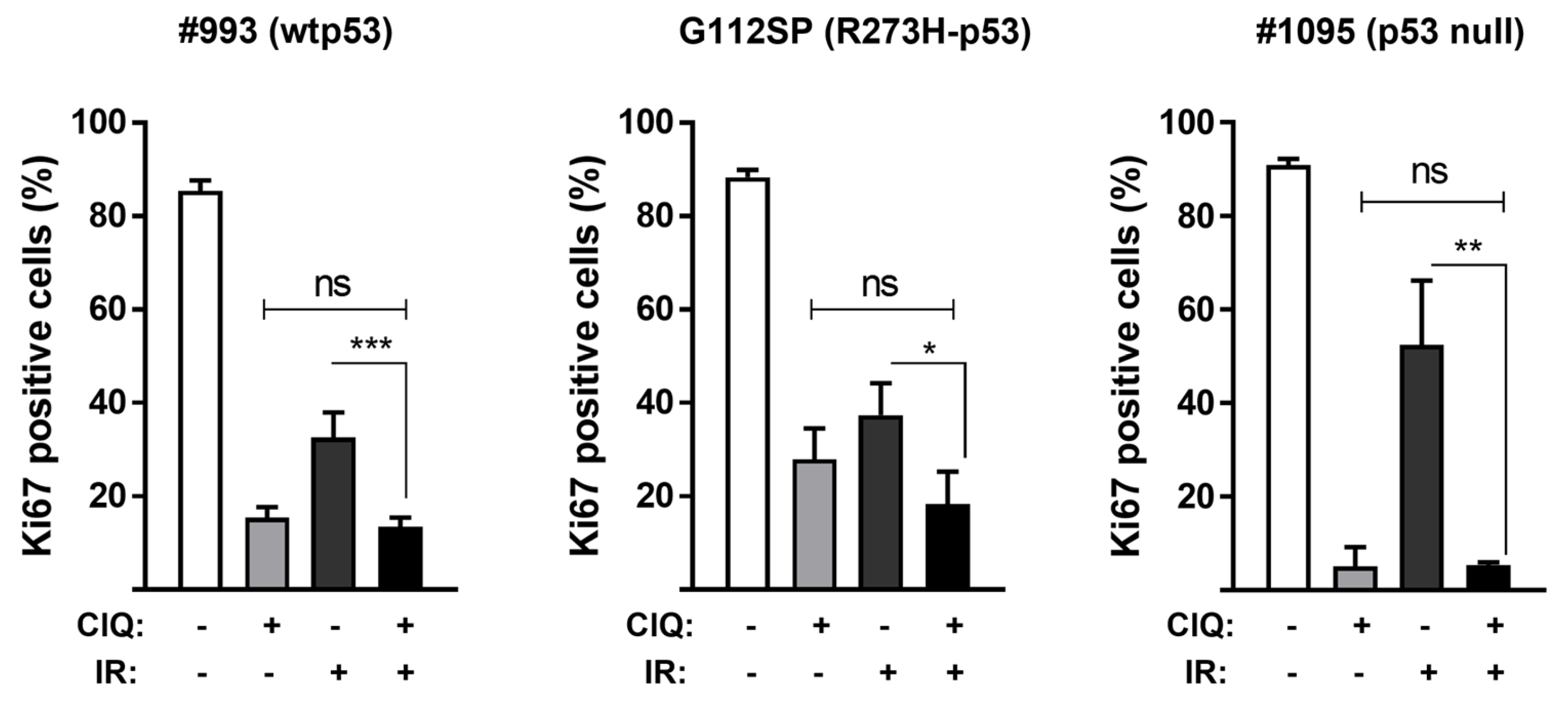
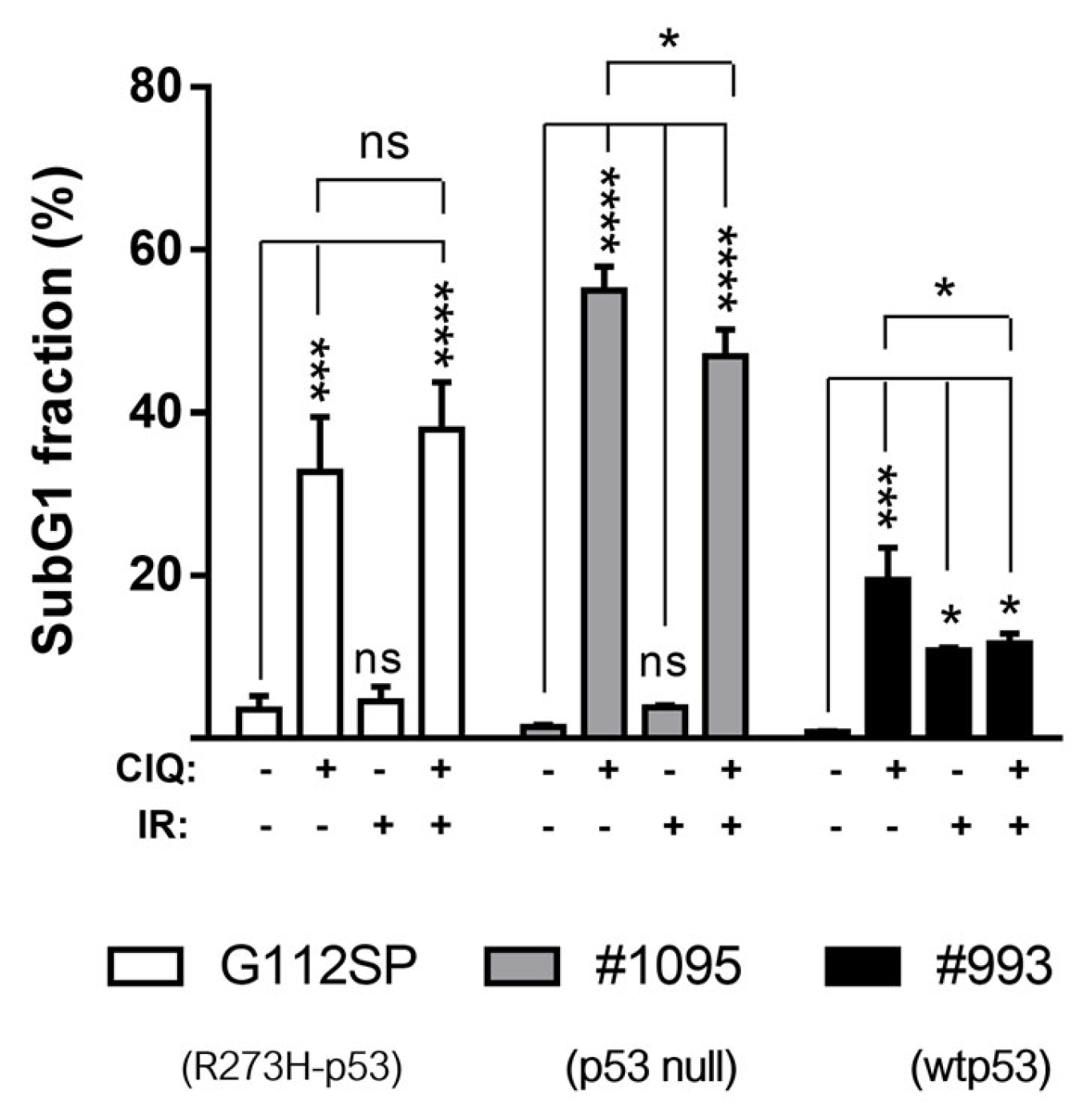
3.1. Impact of ClQ Alone or in Combination with IR on GSCs Proliferation and Viability In Vitro
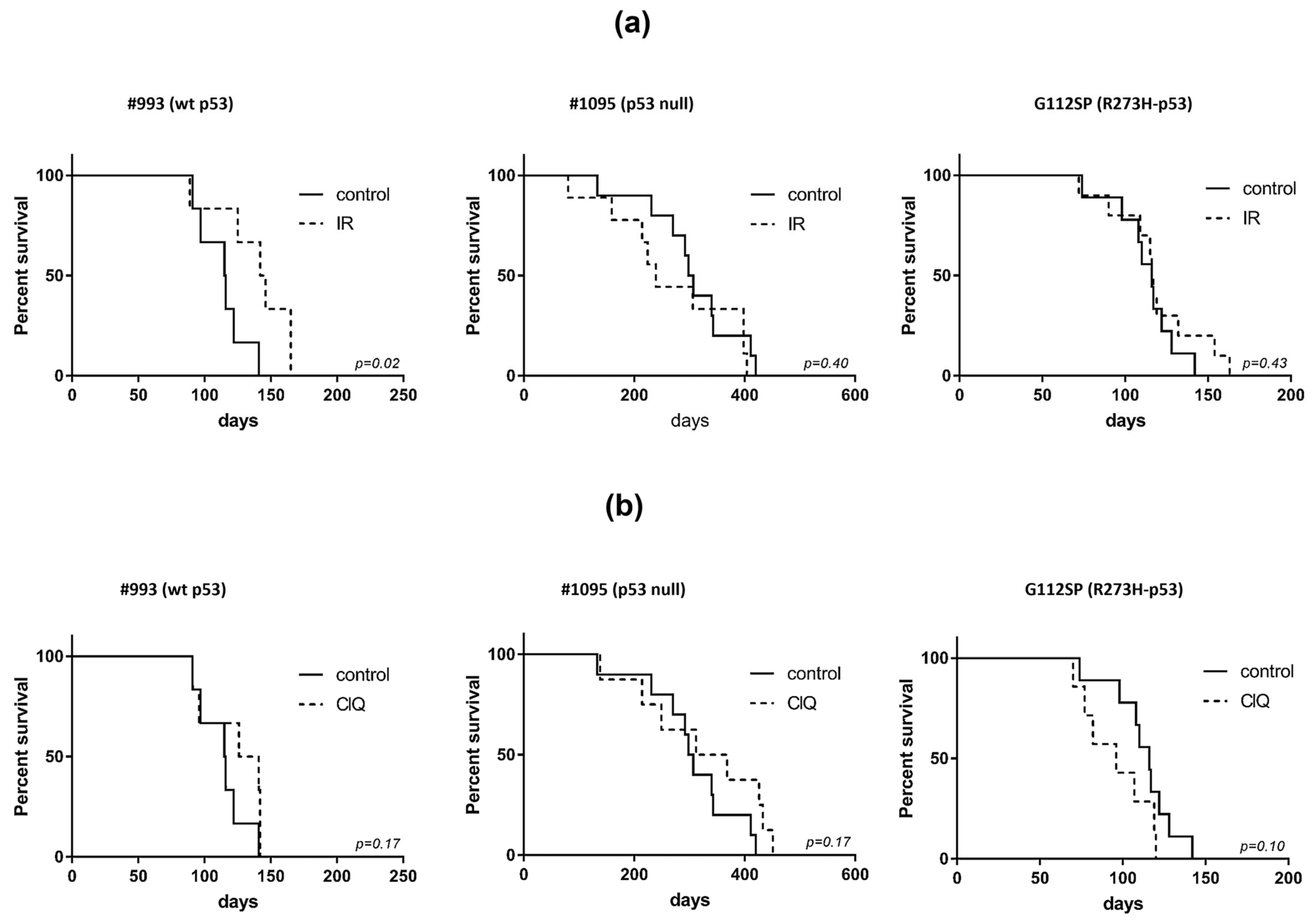
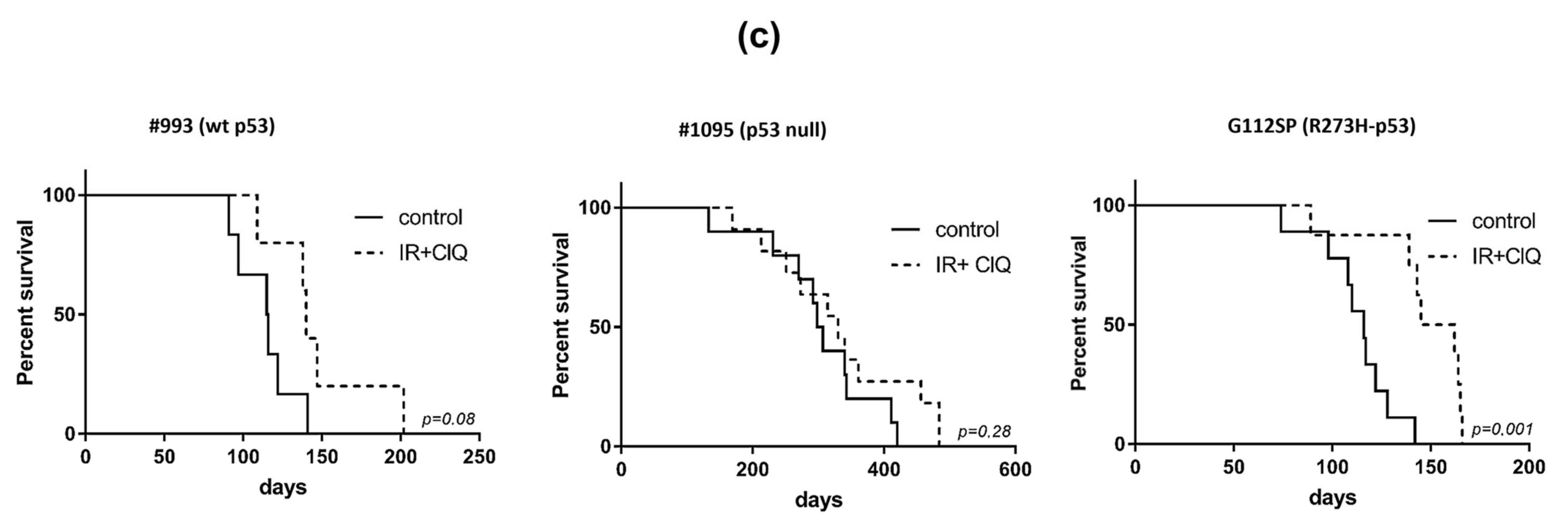
3.2. In Vivo Effects of ClQ Alone or in Combination with IR on the Tumor-Propagating Capacity of GSCs
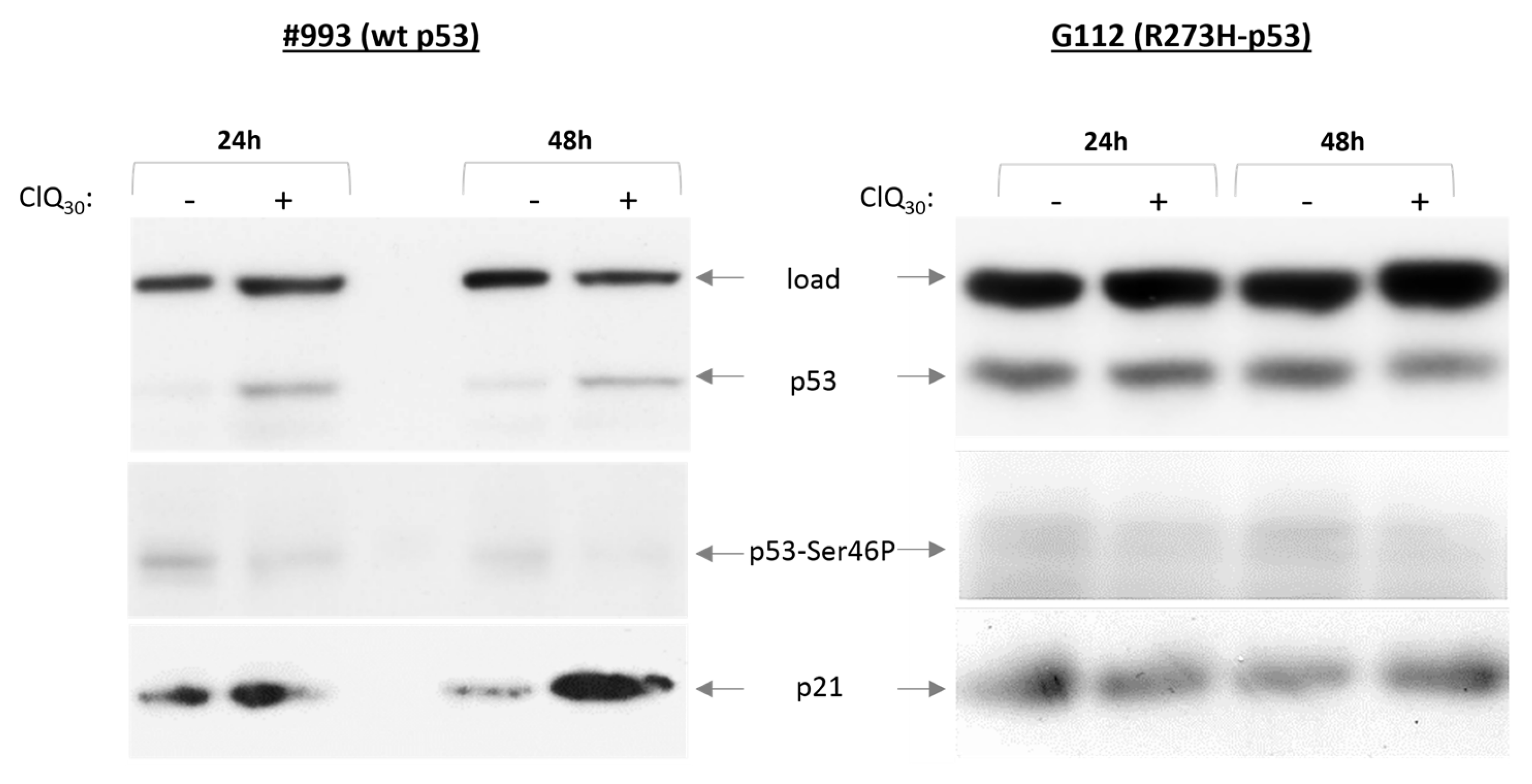
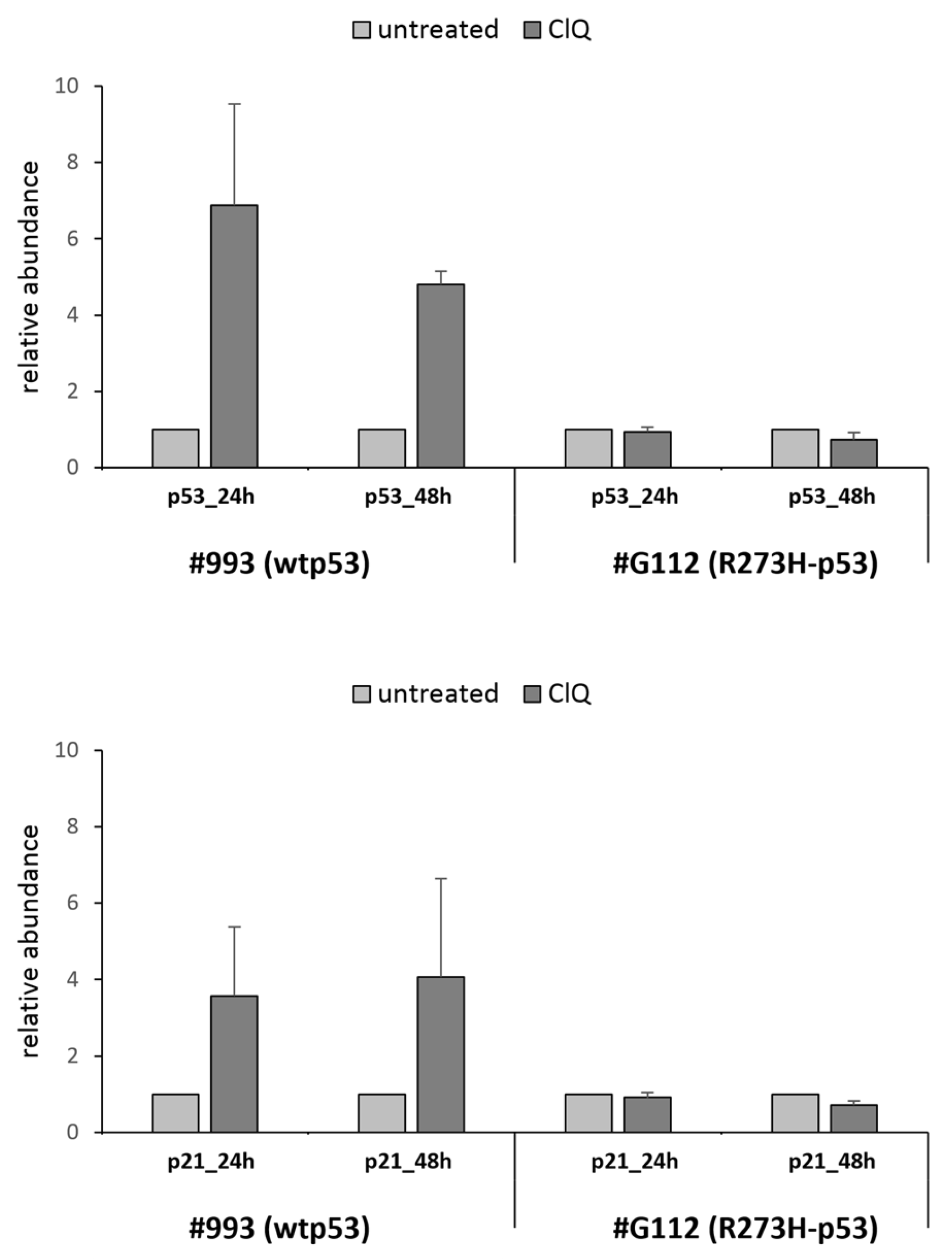
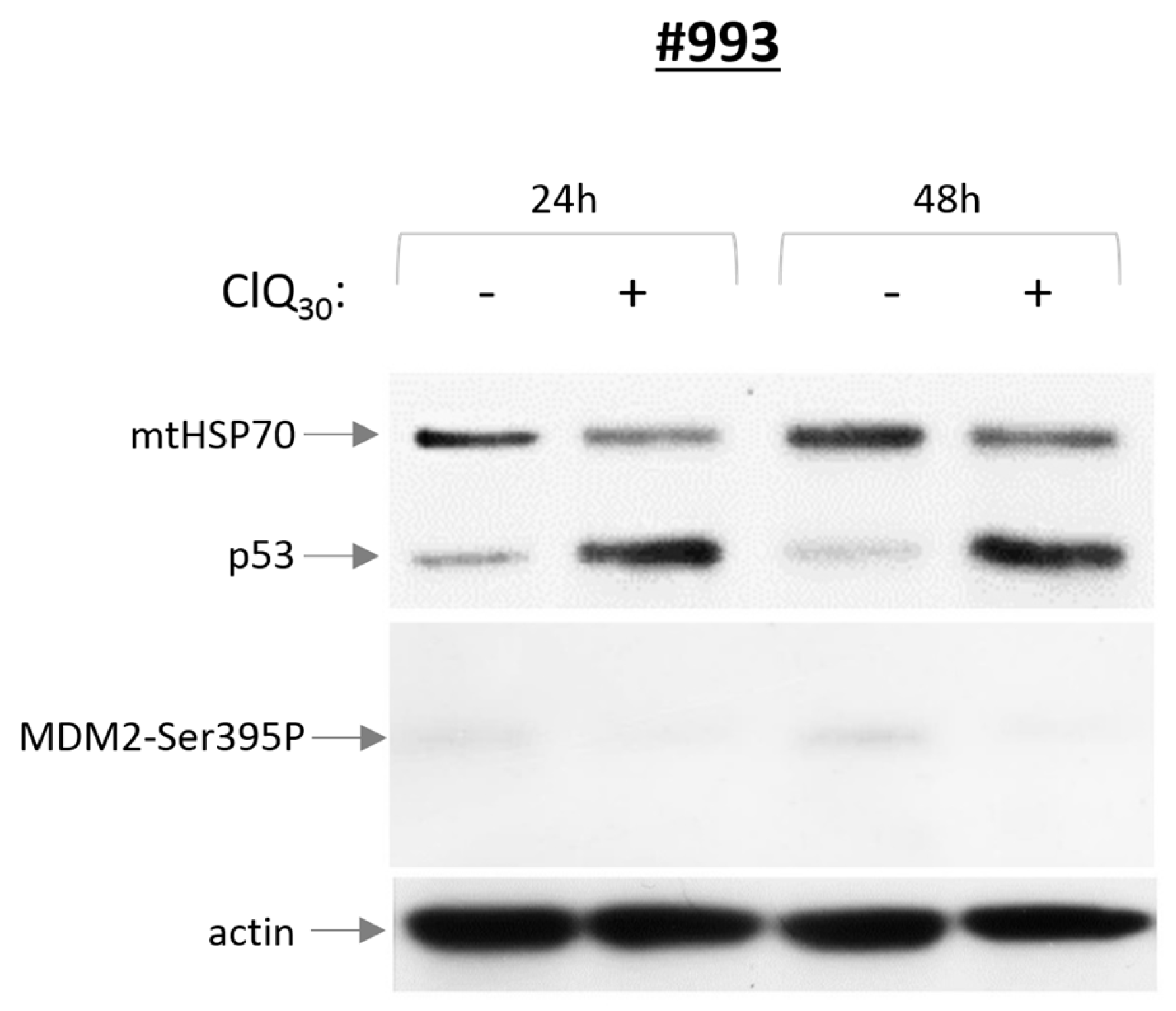
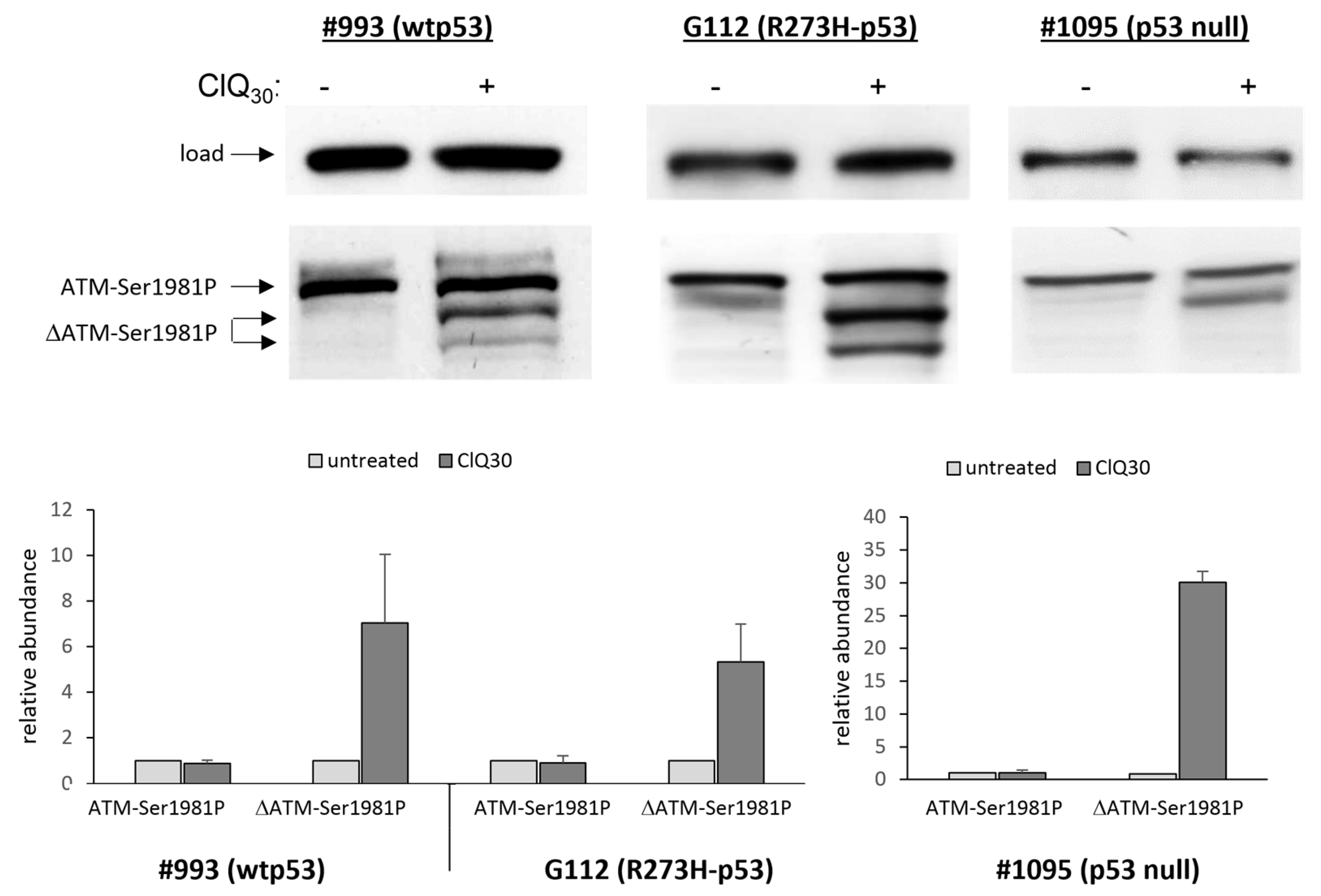
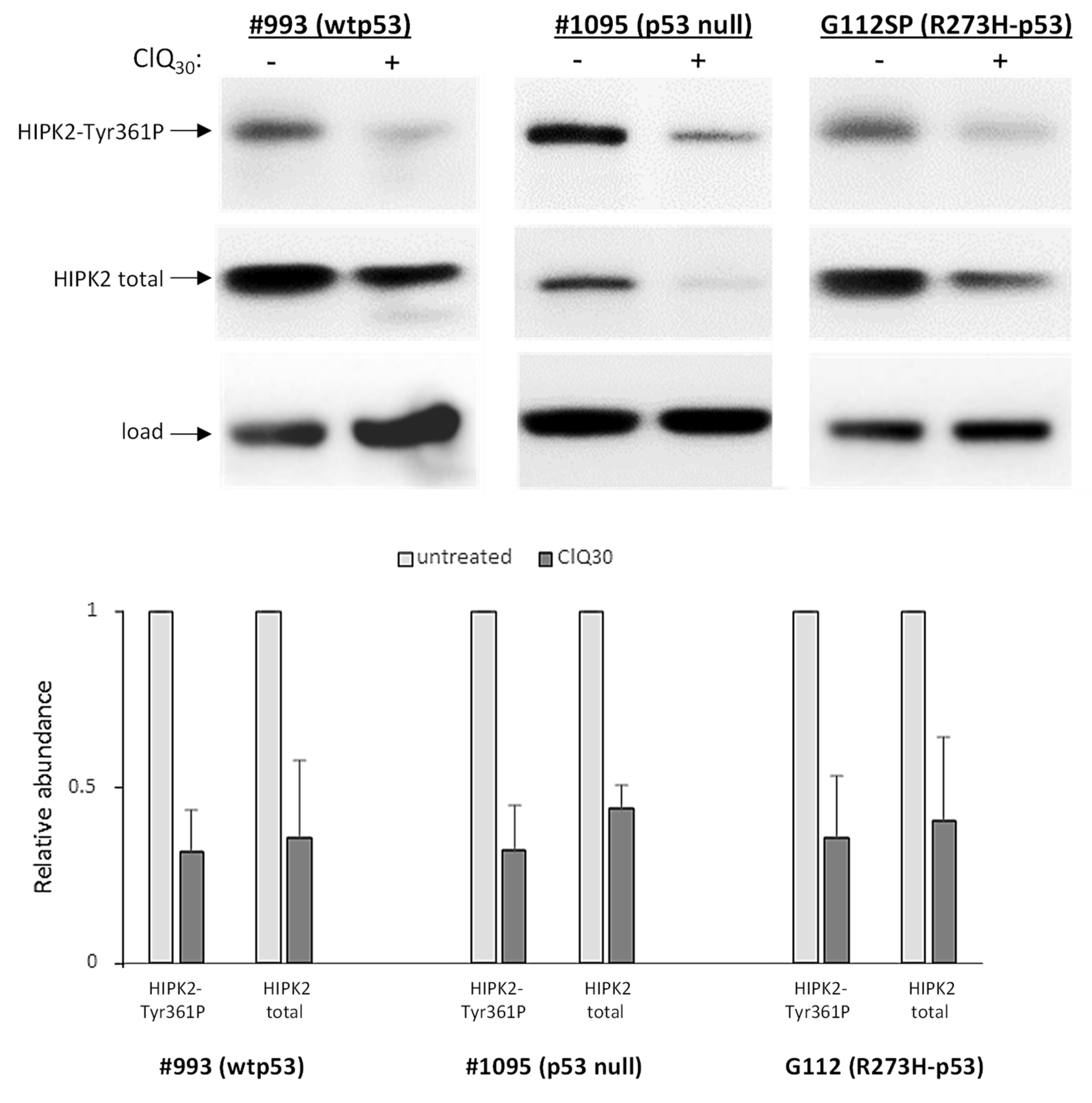
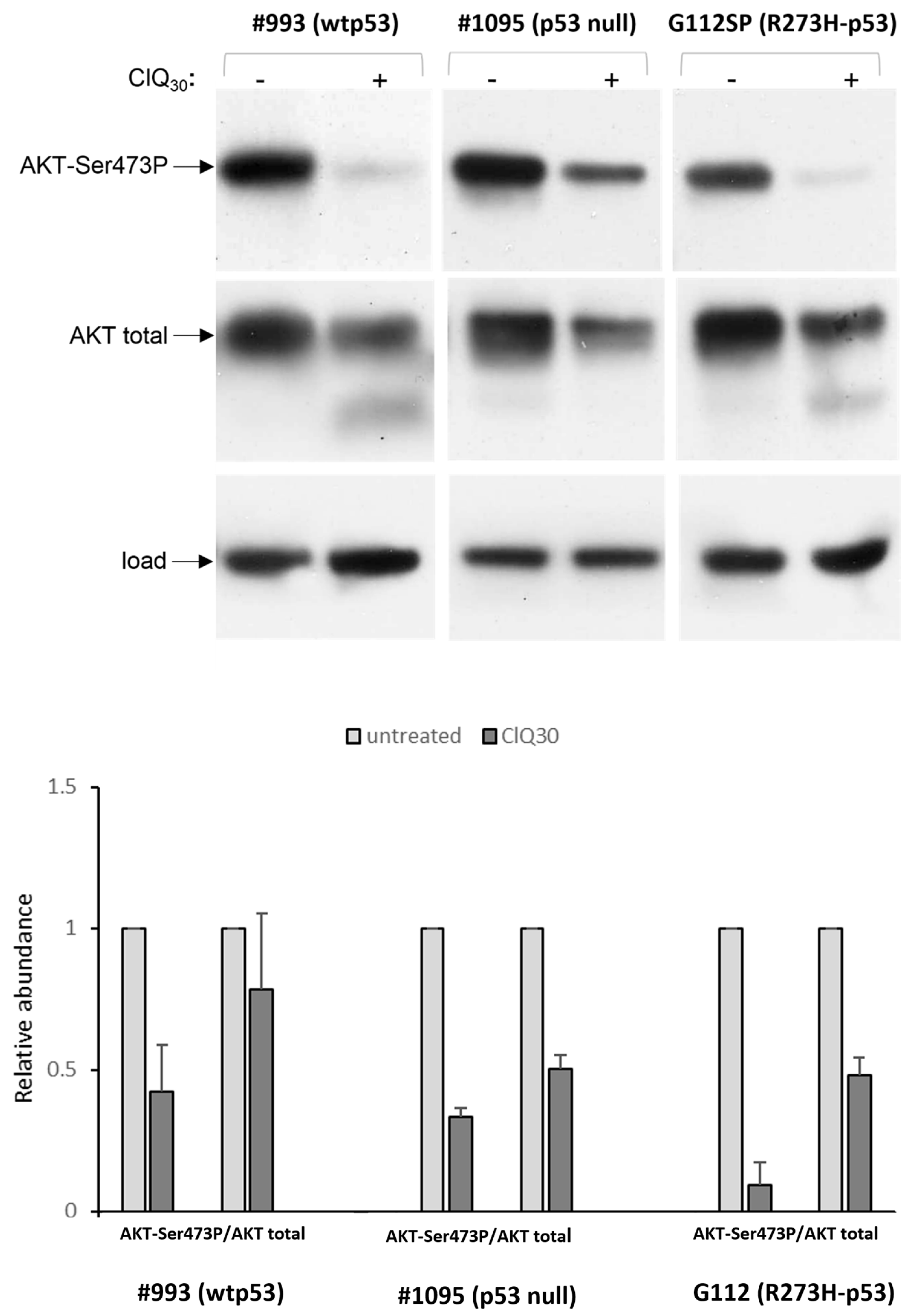
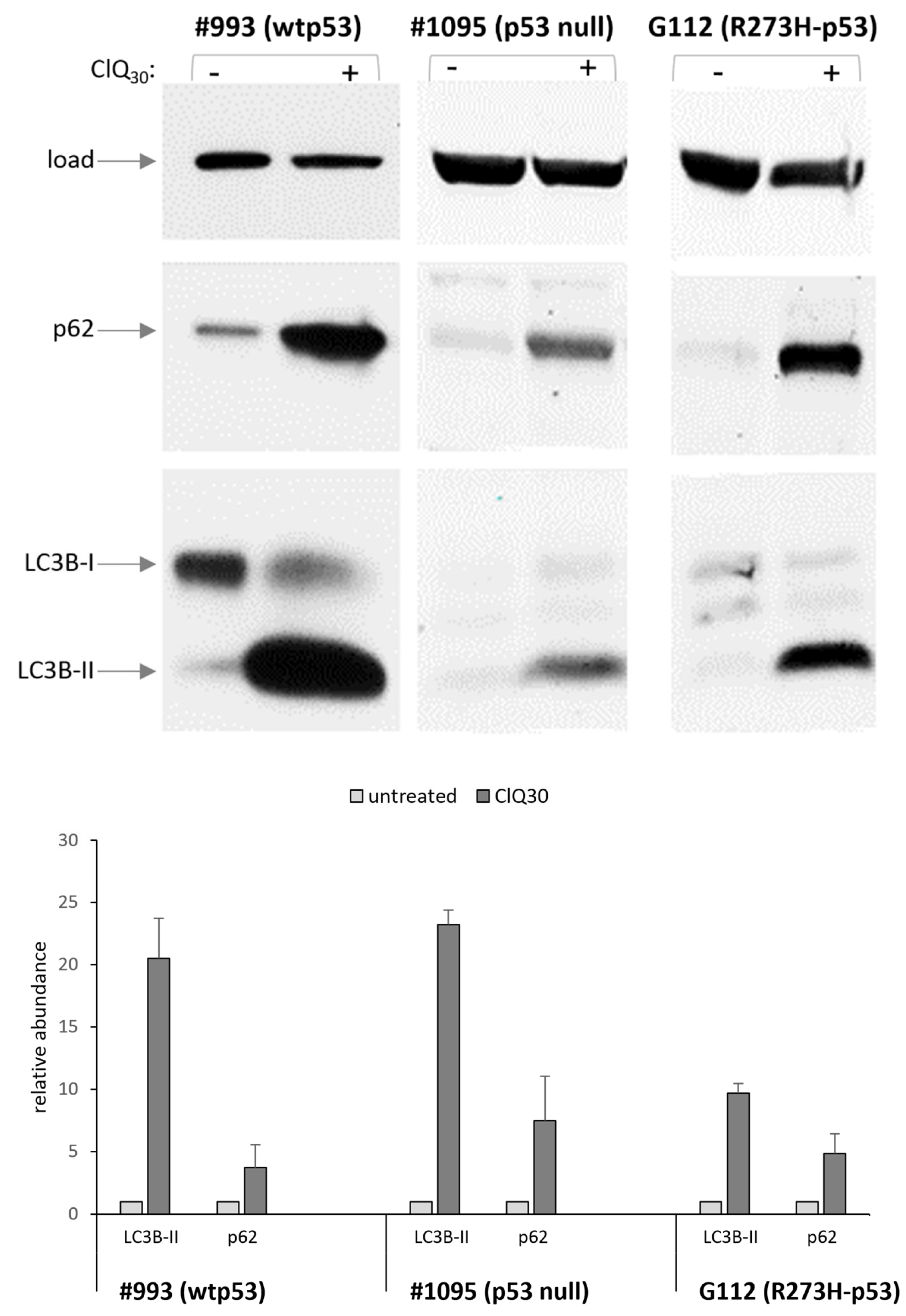
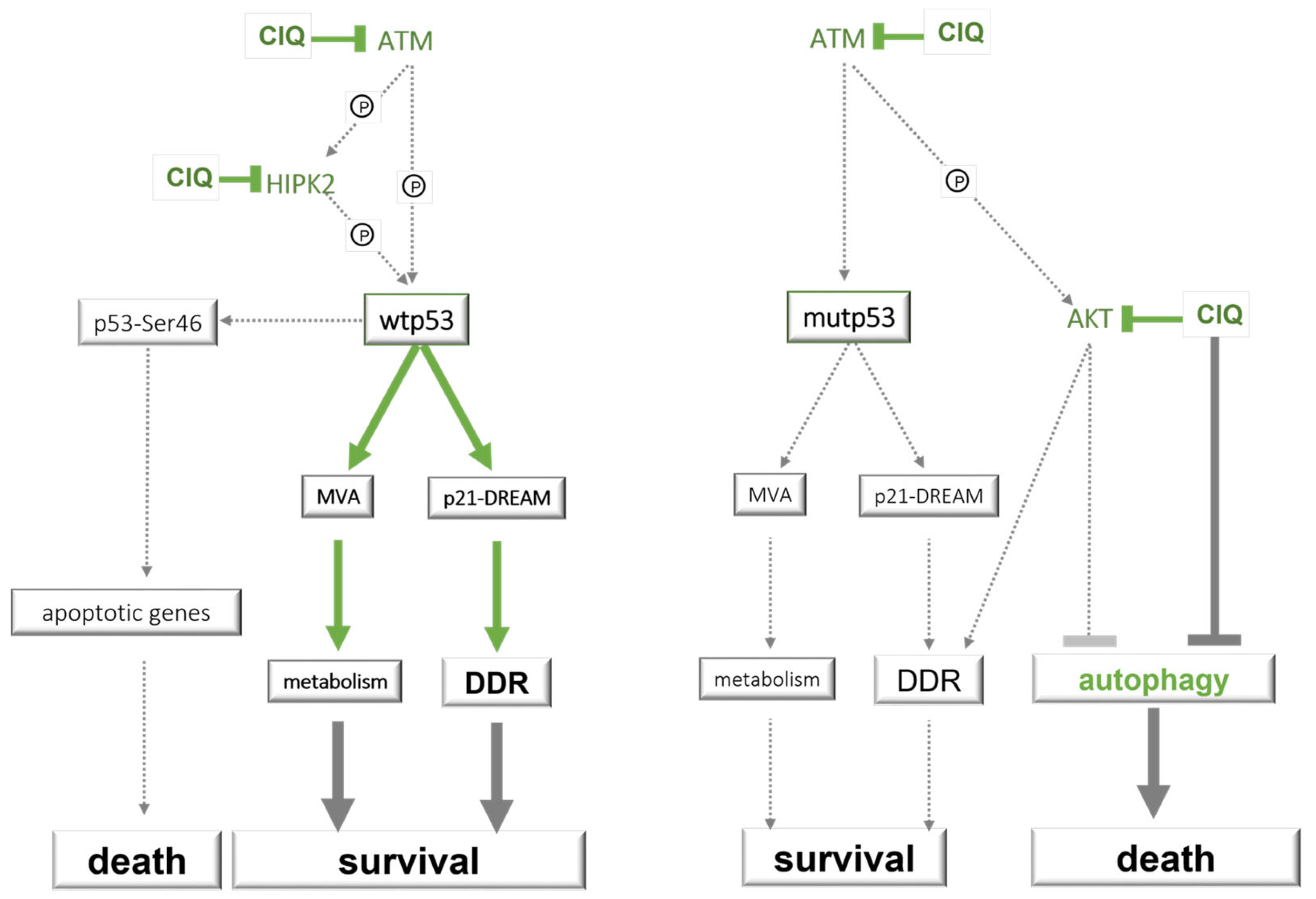
3.3. ClQ Elicits Distinct Molecular Outcomes in GSCs Differing for the Status of p53
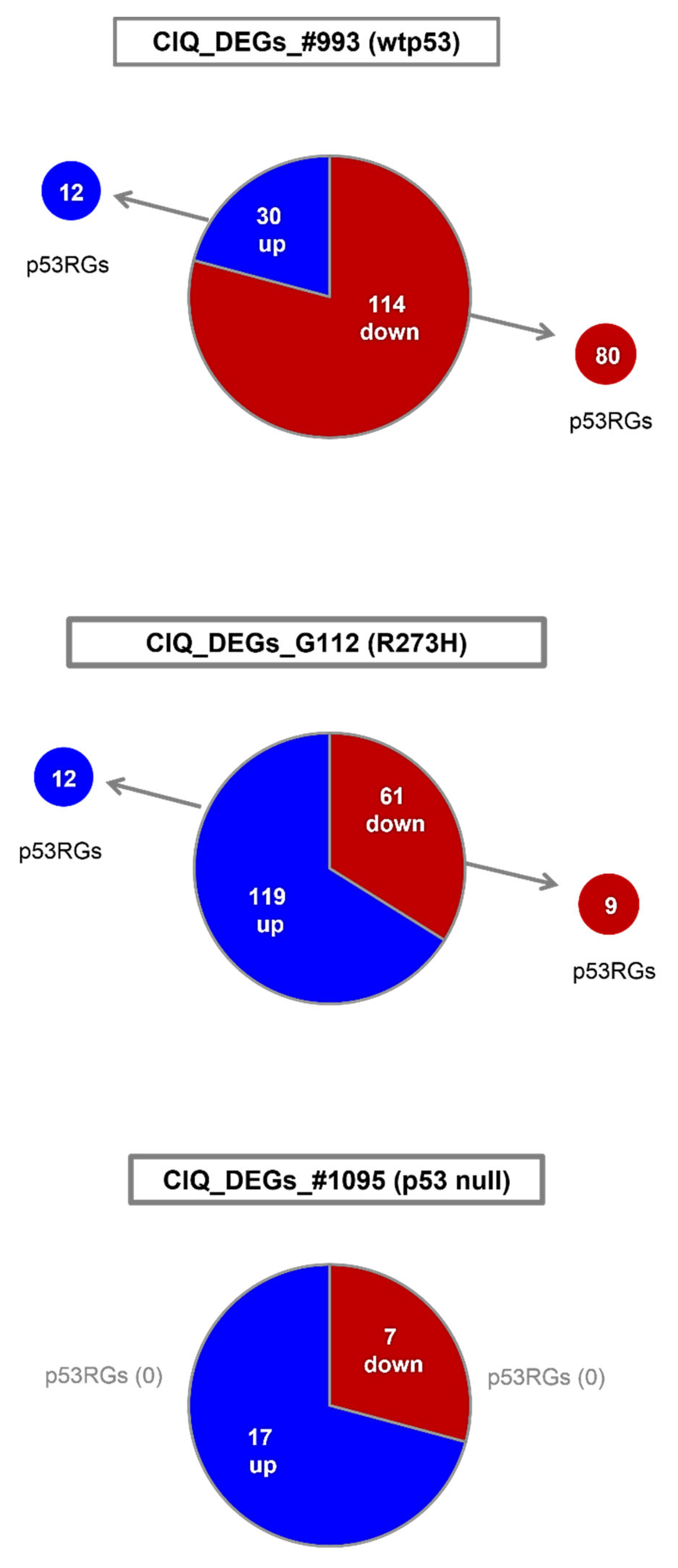
3.4. ClQ Induces Transcriptional Repression via the p53-p21-DREAM Pathway Concurrently with Transcriptional Activation of the Mevalonate Pathway
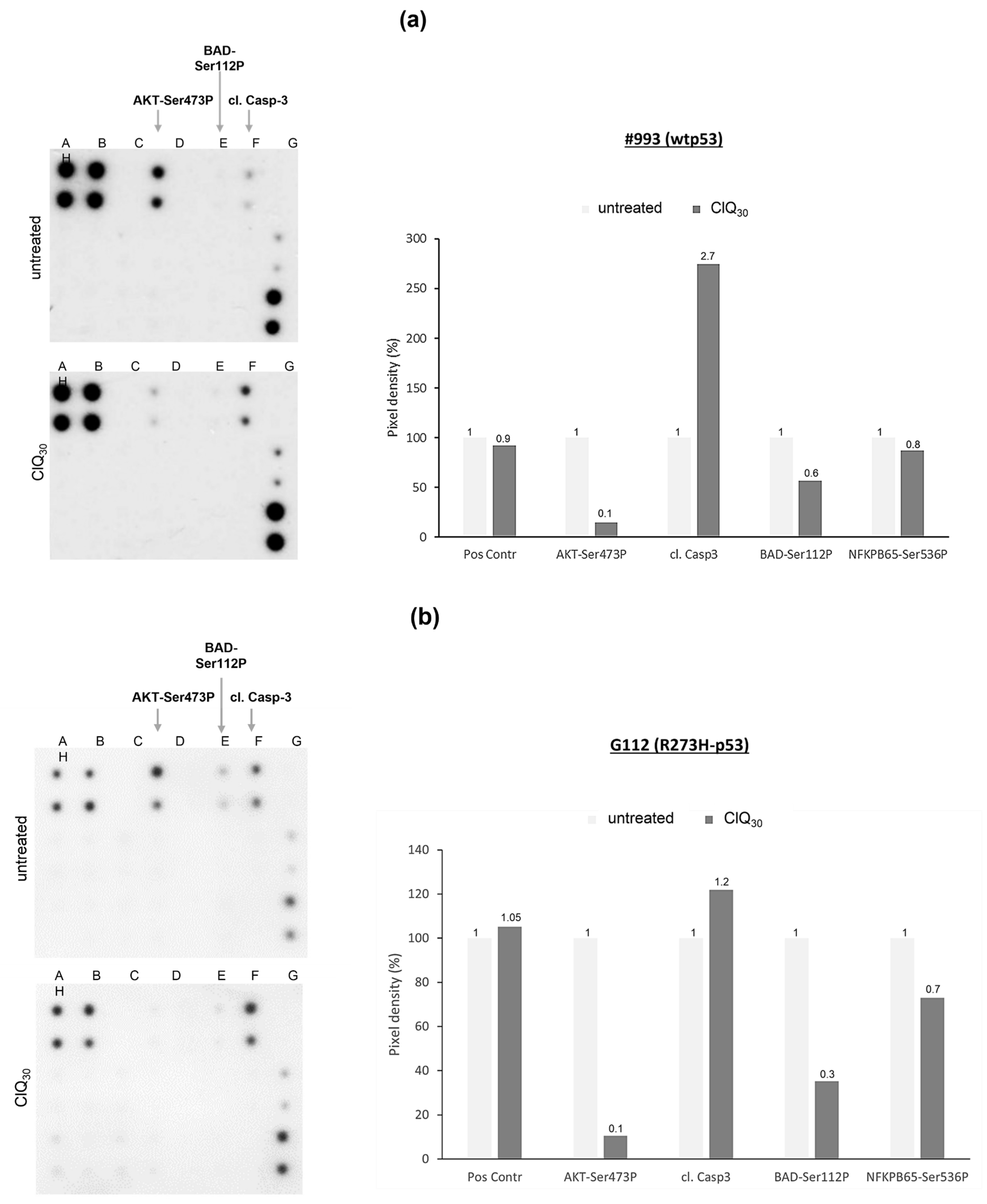
3.5. Impacts of ClQ on the Apoptotic Signaling
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wen, P.Y.; Weller, M.; Lee, E.Q.; Alexander, B.M.; Barnholtz-Sloan, J.S.; Barthel, F.P.; Batchelor, T.T.; Bindra, R.S.; Chang, S.M.; Chiocca, E.A.; et al. Glioblastoma in adults: A Society for Neuro-Oncology (SNO) and European Society of Neuro-Oncology (EANO) consensus review on current management and future directions. Neuro.-Oncol. 2020, 22, 1073–1113. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; van den Bent, M.; Preusser, M.; Le Rhun, E.; Tonn, J.C.; Minniti, G.; Bendszus, M.; Balana, C.; Chinot, O.; Dirven, L.; et al. EANO guidelines on the diagnosis and treatment of diffuse gliomas of adulthood. Nat. Rev. Clin. Oncol. 2021, 18, 170–186. [Google Scholar] [CrossRef] [PubMed]
- Delgado-López, P.; Corrales-García, E. Survival in glioblastoma: A review on the impact of treatment modalities. Clin. Transl. Oncol. 2016, 18, 1062–1071. [Google Scholar] [CrossRef] [PubMed]
- Oliver, L.; Lalier, L.; Salaud, C.; Heymann, D.; Cartron, P.F.; Vallette, F.M. Drug resistance in glioblastoma: Are persisters the key to therapy? Cancer Drug Resist. 2020, 3, 287. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.B.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; De Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [Green Version]
- Shergalis, A.; Bankhead, A., 3rd; Luesakul, U.; Muangsin, N.; Neamati, N. Current Challenges and Opportunities in Treating Glioblastoma. Pharmacol. Rev. 2018, 70, 412–445. [Google Scholar] [CrossRef] [Green Version]
- Pearson, J.R.D.; Regad, T. Targeting cellular pathways in glioblastoma multiforme. Signal Transduct. Target. Ther. 2017, 2, 17040. [Google Scholar] [CrossRef] [Green Version]
- Gimple, R.C.; Bhargava, S.; Dixit, D.; Rich, J.N. Glioblastoma stem cells: Lessons from the tumor hierarchy in a lethal cancer. Genes Dev. 2019, 33, 591–609. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Osuka, S.; Van Meir, E.G. Overcoming therapeutic resistance in glioblastoma: The way forward. J. Clin. Investig. 2017, 127, 415–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascolo, S. Time to use a dose of Chloroquine as an adjuvant to anti-cancer chemotherapies. Eur. J. Pharmacol. 2016, 771, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Weyerhäuser, P.; Kantelhardt, S.R.; Kim, E.L. Re-purposing Chloroquine for Glioblastoma: Potential Merits and Confounding Variables. Front. Oncol. 2018, 8, 335. [Google Scholar] [CrossRef]
- Bilger, A.; Bittner, M.I.; Grosu, A.L.; Wiedenmann, N.; Meyer, P.T.; Firat, E.; Milanovic, D. FET-PET-based reirradiation and chloroquine in patients with recurrent glioblastoma: First tolerability and feasibility results. Strahlenther. Onkol. 2014, 190, 957–961. [Google Scholar] [CrossRef] [PubMed]
- Sotelo, J.; Briceño, E.; López-González, M.A. Adding chloroquine to conventional treatment for glioblastoma multiforme: A randomized, double-blind, placebo-controlled trial. Ann. Intern. Med. 2006, 144, 337–343. [Google Scholar] [CrossRef]
- Briceño, E.; Reyes, S.; Sotelo, J. Therapy of glioblastoma multiforme improved by the antimutagenic chloroquine. Neurosurg. Focus. 2003, 14, 1–6. [Google Scholar] [CrossRef]
- Compter, I.; Eekers, D.B.P.; Hoeben, A.; Rouschop, K.M.A.; Reymen, B.; Ackermans, L.; Beckervordersantforth, J.; Bauer, N.J.C.; Anten, M.M.; Wesseling, P.; et al. Chloroquine combined with concurrent radiotherapy and temozolomide for newly diagnosed glioblastoma: A phase IB trial. Autophagy 2021, 17, 2604–2612. [Google Scholar] [CrossRef]
- Sotelo, J. Chloroquine for Treatment of Glioblastoma Multiforme (NCT00224978); U.S. National Library of Medicine: Bethesda, MD, USA, 2009. Available online: https://ClinicalTrials.gov/ (accessed on 1 March 2023).
- DeNittis, A. IDO2 Genetic Status Informs the Neoadjuvant Efficacy of Chloroquine (CQ) in Brain Metastasis Radiotherapy (NCT01727531); U.S. National Library of Medicine: Bethesda, MD, USA, 2015. Available online: https://ClinicalTrials.gov/ (accessed on 1 March 2023).
- Rodríguez, O.G.A. Study of Whole-Brain Irradiation with Chloroquine for Brain Metastases (CLQ) (NCT01894633); U.S. National Library of Medicine: Bethesda, MD, USA, 2017. Available online: https://ClinicalTrials.gov/ (accessed on 1 March 2023).
- Wilmink, J.W. Metformin and Chloroquine in IDH1/2-Mutated Solid Tumors (MACIST) (NCT02496741); U.S. National Library of Medicine: Bethesda, MD, USA, 2020. Available online: https://ClinicalTrials.gov/ (accessed on 1 March 2023).
- De Ruysscher, D. The Addition of Chloroquine to Chemoradiation for Glioblastoma (CHLOROBRAIN) (NCT02378532); U.S. National Library of Medicine: Bethesda, MD, USA, 2020. Available online: https://ClinicalTrials.gov/ (accessed on 1 March 2023).
- Azab, M.A. Chloroquine for Glioblastoma (NCT04772846); U.S. National Library of Medicine: Bethesda, MD, USA, 2021. Available online: https://ClinicalTrials.gov/ (accessed on 1 March 2023).
- Kramm, C. International Cooperative Phase III Trial of the HIT-HGG Study Group (HIT-HGG-2013) (HIT-HGG-2013) (NCT03243461); U.S. National Library of Medicine: Bethesda, MD, USA, 2022. Available online: https://ClinicalTrials.gov/ (accessed on 1 March 2023).
- Lambin, P. The Addition of Chloroquine to Chemoradiation for Glioblastoma (NCT02432417); U.S. National Library of Medicine: Bethesda, MD, USA, 2022. Available online: https://ClinicalTrials.gov/ (accessed on 1 March 2023).
- Dominello, M.; Partial Brain, R.T. Temozolomide, Chloroquine, and TTF Therapy for the Treatment of Newly Diagnosed Glio-Blastoma (NCT04397679); U.S. National Library of Medicine: Bethesda, MD, USA, 2022. Available online: https://ClinicalTrials.gov/ (accessed on 1 March 2023).
- Maycotte, P.; Aryal, S.; Cummings, C.T.; Thorburn, J.; Morgan, M.J.; Thorburn, A. Chloroquine sensitizes breast cancer cells to chemotherapy independent of autophagy. Autophagy 2012, 8, 200–212. [Google Scholar] [CrossRef] [Green Version]
- Eng, C.H.; Wang, Z.; Tkach, D.; Toral-Barza, L.; Ugwonali, S.; Liu, S.; Fitzgerald, S.L.; George, E.; Frias, E.; Cochran, N.; et al. Macroautophagy is dispensable for growth of KRAS mutant tumors and chloroquine efficacy. Proc. Natl. Acad. Sci. USA 2016, 113, 182–187. [Google Scholar] [CrossRef] [Green Version]
- King, M.; Ganley, I.; Flemington, V. Inhibition of cholesterol metabolism underlies synergy between mTOR pathway inhibition and chloroquine in bladder cancer cells. Oncogene 2016, 35, 4518–4528. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.L.; Wüstenberg, R.; Rübsam, A.; Schmitz-Salue, C.; Warnecke, G.; Bücker, E.-M.; Pettkus, N.; Speidel, D.; Rohde, V.; Schulz-Schaeffer, W.; et al. Chloroquine activates the p53 pathway and induces apoptosis in human glioma cells. Neuro-Oncology 2010, 12, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Burikhanov, R.; Hebbar, N.; Noothi, S.K.; Shukla, N.; Sledziona, J.; Araujo, N.; Kudrimoti, M.; Wang, Q.J.; Watt, D.S.; Welch, D.R.; et al. Chloroquine-Inducible Par-4 Secretion Is Essential for Tumor Cell Apoptosis and Inhibition of Metastasis. Cell Rep. 2017, 18, 508–519. [Google Scholar] [CrossRef] [PubMed]
- Park, E.J.; Min, K.-J.; Choi, K.S.; Kubatka, P.; Kruzliak, P.; Kim, D.E.; Kwon, T.K. Chloroquine enhances TRAIL-mediated apoptosis through up-regulation of DR5 by stabilization of mRNA and protein in cancer cells. Sci. Rep. 2016, 6, 22921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, J.R.; Kim, W.Y.; Cho, Y.J.; Ryu, J.Y.; Choi, J.J.; Jeong, S.Y.; Kim, M.S.; Kim, J.H.; Paik, E.S.; Lee, Y.Y.; et al. Chloroquine reverses chemoresistance via upregulation of p21WAF1/CIP1 and autophagy inhibition in ovarian cancer. Cell Death Dis. 2020, 11, 1034. [Google Scholar] [CrossRef] [PubMed]
- Eloranta, K.; Cairo, S.; Liljeström, E.; Soini, T.; Kyrönlahti, A.; Judde, J.-G.; Wilson, D.B.; Heikinheimo, M.; Pihlajoki, M. Chloroquine Triggers Cell Death and Inhibits PARPs in Cell Models of Aggressive Hepatoblastoma. Front. Oncol. 2020, 10, 1138. [Google Scholar] [CrossRef]
- McLendon, R.; Friedman, A.; Bigneret, D.; Van Meir, E.G.; Brat, D.J.; Mastrogianakis, G.M.; Olson, J.J.; Mikkelsen, T.; Lehman, N.; Aldape, K.; et al. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar]
- Sabapathy, K.; Lane, D.P. Therapeutic targeting of p53: All mutants are equal, but some mutants are more equal than others. Nat. Rev. Clin. Oncol. 2018, 15, 13–30. [Google Scholar] [CrossRef]
- Pfister, N.T.; Prives, C. Transcriptional Regulation by Wild-Type and Cancer-Related Mutant Forms of p53. Cold Spring Harb. Perspect. Med. 2017, 7, a026054. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, F.; Collavin, L.; Del Sal, G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 2019, 26, 199–212. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Cao, J.; Topatana, W.; Juengpanich, S.; Li, S.; Bin Zhang, B.; Shen, J.; Cai, L.; Cai, X.; Chen, M. Targeting mutant p53 for cancer therapy: Direct and indirect strategies. J. Hematol. Oncol. 2021, 14, 157. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrantes-Freer, A.; Kim, E.; Bielanska, J.; Giese, A.; Mortensen, L.S.; Schulz-Schaeffer, W.J.; Stadelmann, C.; Brück, W.; Pardo, L.A. Human Glioma–Initiating Cells Show a Distinct Immature Phenotype Resembling but Not Identical to NG2 Glia. J. Neuropathol. Exp. Neurol. 2013, 72, 307–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrantes-Freer, A.; Renovanz, M.; Eich, M.; Braukmann, A.; Sprang, B.; Spirin, P.; Pardo, L.A.; Giese, A.; Kim, E.L. CD133 Expression Is Not Synonymous to Immunoreactivity for AC133 and Fluctuates throughout the Cell Cycle in Glioma Stem-like Cells. PLoS ONE 2015, 10, e0130519. [Google Scholar] [CrossRef] [Green Version]
- Kalasauskas, D.; Sorokin, M.; Sprang, B.; Elmasri, A.; Viehweg, S.; Salinas, G.; Opitz, L.; Rave-Fraenk, M.; Schulz-Schaeffer, W.; Kantelhardt, S.R.; et al. Diversity of Clinically Relevant Outcomes Resulting from Hypofractionated Radiation in Human Glioma Stem Cells Mirrors Distinct Patterns of Transcriptomic Changes. Cancers 2020, 12, 570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fauß, J.; Sprang, B.; Leukel, P.; Sommer, C.; Nikolova, T.; Ringel, F.; Kim, E.L. ALDH1A3 Segregated Expression and Nucleus-Associated Proteasomal Degradation Are Common Traits of Glioblastoma Stem Cells. Biomedicines 2021, 10, 7. [Google Scholar] [CrossRef]
- Hanisch, D.; Krumm, A.; Diehl, T.; Stork, C.M.; Dejung, M.; Butter, F.; Kim, E.; Brenner, W.; Fritz, G.; Hofmann, T.G.; et al. Class I HDAC overexpression promotes temozolomide resistance in glioma cells by regulating RAD18 ex-pression. Cell Death Dis. 2022, 13, 293. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.L.; Yoshizato, K.; Kluwe, L.; Meissner, H.; Warnecke, G.; Zapf, S.; Westphal, M.; Deppert, W.; Giese, A. Comparative assessment of the functional p53 status in glioma cells. Anticancer Res. 2005, 25, 213–224. [Google Scholar]
- Hu, Y.; Smyth, G.K. ELDA: Extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J. Immunol. Methods 2009, 347, 70–78. [Google Scholar] [CrossRef]
- Kim, E.L.; Sorokin, M.; Kantelhardt, S.R.; Kalasauskas, D.; Sprang, B.; Fauss, J.; Ringel, F.; Garazha, A.; Albert, E.; Gaifullin, N.; et al. Intratumoral Heterogeneity and Longitudinal Changes in Gene Expression Predict Differential Drug Sensitivity in Newly Diagnosed and Recurrent Glioblastoma. Cancers 2020, 12, 520. [Google Scholar] [CrossRef] [Green Version]
- Gautier, L.; Cope, L.; Bolstad, B.M.; Irizarry, R.A. Affy—Analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 2004, 20, 307–315. [Google Scholar] [CrossRef] [Green Version]
- Wettenhall, J.M.; Smyth, G.K. limmaGUI: A graphical user interface for linear modeling of microarray data. Bioinformatics 2004, 20, 3705–3706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gentleman, R.C.; Carey, V.J.; Bates, D.M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004, 5, R80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smyth, G.K. Linear Models and Empirical Bayes Methods for Assessing Differential Expression in Microarray Experiments. Stat. Appl. Genet. Mol. Biol. 2004, 3, 3. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Golden, E.B.; Cho, H.-Y.; Jahanian, A.; Hofman, F.M.; Louie, S.G.; Schönthal, A.H.; Chen, T.C. Chloroquine enhances temozolomide cytotoxicity in malignant gliomas by blocking autophagy. Neurosurg. Focus. 2014, 37, E12. [Google Scholar] [CrossRef] [Green Version]
- Ye, H.; Chen, M.; Cao, F.; Huang, H.; Zhan, R.; Zheng, X. Chloroquine, an autophagy inhibitor, potentiates the radiosensitivity of glioma initiating cells by inhibiting autophagy and activating apoptosis. BMC Neurol. 2016, 16, 178. [Google Scholar] [CrossRef] [Green Version]
- Vessoni, A.T.; Quinet, A.; de Andrade-Lima, L.C.; Martins, D.J.; Garcia, C.C.M.; Rocha, C.R.R.; Vieira, D.B.; Menck, C.F.M. Chloroquine-induced glioma cells death is associated with mitochondrial membrane potential loss, but not oxidative stress. Free. Radic. Biol. Med. 2016, 90, 91–100. [Google Scholar] [CrossRef]
- Geng, Y.; Kohli, L.; Klocke, B.J.; Roth, K.A. Chloroquine-induced autophagic vacuole accumulation and cell death in glioma cells is p53 independent. Neuro-Oncology 2010, 12, 473–481. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.; Luo, X.; Nie, P.; Wu, B.; Xu, W.; Shi, X.; Chang, H.; Li, B.; Yu, X.; Zou, Z. CQ synergistically sensitizes human colorectal cancer cells to SN-38/CPT-11 through lysosomal and mitochondrial apoptotic pathway via p53-ROS cross-talk. Free. Radic. Biol. Med. 2017, 104, 280–297. [Google Scholar] [CrossRef]
- Lee, S.W.; Kim, H.-K.; Lee, N.-H.; Yi, H.-Y.; Kim, H.-S.; Hong, S.H.; Hong, Y.-K.; Joe, Y.A. The synergistic effect of combination temozolomide and chloroquine treatment is dependent on autophagy formation and p53 status in glioma cells. Cancer Lett. 2015, 360, 195–204. [Google Scholar] [CrossRef]
- Bakkenist, C.J.; Kastan, M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003, 421, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Shiloh, Y.; Ziv, Y. The ATM protein kinase: Regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol. 2013, 14, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Maya, R.; Balass, M.; Kim, S.-T.; Shkedy, D.; Leal, J.-F.M.; Shifman, O.; Moas, M.; Buschmann, T.; Ronai, Z.; Shiloh, Y.; et al. ATM-dependent phosphorylation of Mdm2 on serine 395: Role in p53 activation by DNA damage. Genes Dev. 2001, 15, 1067–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Q.; Chen, L.; Li, Z.; Lane, W.S.; Chen, J. ATM activates p53 by regulating MDM2 oligomerization and E3 processivity. EMBO J. 2009, 28, 3857–3867. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Pabla, N.; Wang, C.-Y.; Wang, W.; Schoenlein, P.V.; Dong, Z. Caspase-mediated cleavage of ATM during cisplatin-induced tubular cell apoptosis: Inactivation of its kinase activity toward p53. Am. J. Physiol.-Ren. Physiol. 2006, 291, F1300–F1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liebl, M.C.; Hofmann, T.G. Cell Fate Regulation upon DNA Damage: p53 Serine 46 Kinases Pave the Cell Death Road. BioEssays 2019, 41, e1900127. [Google Scholar] [CrossRef] [Green Version]
- Engeland, K. Cell cycle arrest through indirect transcriptional repression by p53: I have a DREAM. Cell Death Differ. 2018, 25, 114–132. [Google Scholar] [CrossRef] [Green Version]
- Engeland, K. Cell cycle regulation: p53-p21-RB signaling. Cell Death Differ. 2022, 29, 946–960. [Google Scholar] [CrossRef]
- Fischer, M.; Grossmann, P.; Padi, M.; DeCaprio, J.A. Integration of TP53, DREAM, MMB-FOXM1 and RB-E2F target gene analyses identifies cell cycle gene regulatory networks. Nucleic Acids Res. 2016, 44, 6070–6086. [Google Scholar] [CrossRef] [Green Version]
- Degtyarev, M.; De Mazière, A.; Orr, C.; Lin, J.; Lee, B.B.; Tien, J.Y.; Prior, W.W.; van Dijk, S.; Wu, H.; Gray, D.C.; et al. Akt inhibition promotes autophagy and sensitizes PTEN-null tumors to lysosomotropic agents. J. Cell Biol. 2008, 183, 101–116. [Google Scholar] [CrossRef] [Green Version]
- Mullen, P.J.; Yu, R.; Longo, J.; Archer, M.C.; Penn, L.Z. The interplay between cell signalling and the mevalonate pathway in cancer. Nat. Rev. Cancer 2016, 16, 718–731. [Google Scholar] [CrossRef] [PubMed]
- Laka, K.; Makgoo, L.; Mbita, Z. Cholesterol-Lowering Phytochemicals: Targeting the Mevalonate Pathway for Anticancer Interventions. Front. Genet. 2022, 13, 841639. [Google Scholar] [CrossRef] [PubMed]
- Sparks, A.; Dayal, S.; Das, J.; Robertson, P.; Menendez, S.; Saville, M.K. The degradation of p53 and its major E3 ligase Mdm2 is differentially dependent on the proteasomal ubiquitin receptor S5a. Oncogene 2014, 33, 4685–4696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carr, M.I.; Roderick, J.E.; Gannon, H.S.; Kelliher, M.A.; Jones, S.N. Mdm2 Phosphorylation Regulates Its Stability and Has Contrasting Effects on Oncogene and Radia-tion-Induced Tumorigenesis. Cell Rep. 2016, 16, 2618–2629. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, T.G.; Möller, A.; Sirma, H.; Zentgraf, H.; Taya, Y.; Dröge, W.; Will, H.; Schmitz, M.L. Regulation of p53 activity by its interaction with homeodomain-interacting protein kinase-2. Nature 2002, 4, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, T.G.; Glas, C.; Bitomsky, N. HIPK2: A tumour suppressor that controls DNA damage-induced cell fate and cytokinesis. BioEssays 2013, 35, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Conrad, E.; Polonio-Vallon, T.; Meister, M.; Matt, S.; Bitomsky, N.; Herbel, C.; Liebl, M.; Greiner, V.; Kriznik, B.; Schumacher, S.; et al. HIPK2 restricts SIRT1 activity upon severe DNA damage by a phosphorylation-controlled mechanism. Cell Death Differ. 2016, 23, 110–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palanichamy, K.; Patel, D.; Jacob, J.R.; Litzenberg, K.T.; Gordon, N.; Acus, K.; Noda, S.-E.; Chakravarti, A. Lack of Constitutively Active DNA Repair Sensitizes Glioblastomas to Akt Inhibition and Induces Synthetic Lethality with Radiation Treatment in a p53-Dependent Manner. Mol. Cancer Ther. 2018, 17, 336–346. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Turner, K.M.; Yung, W.K.A.; Chen, K.; Zhang, W. Role of AKT signaling in DNA repair and clinical response to cancer therapy. Neuro-Oncology 2014, 16, 1313–1323. [Google Scholar] [CrossRef] [Green Version]
- Alemi, F.; Sadigh, A.R.; Malakoti, F.; Elhaei, Y.; Ghaffari, S.H.; Maleki, M.; Asemi, Z.; Yousefi, B.; Targhazeh, N.; Majidinia, M. Molecular mechanisms involved in DNA repair in human cancers: An overview of PI3k/Akt signaling and PIKKs crosstalk. J. Cell. Physiol. 2022, 237, 313–328. [Google Scholar] [CrossRef]
- Han, X.; Xue, X.; Zhou, H.; Zhang, G. A molecular view of the radioresistance of gliomas. Oncotarget 2017, 8, 100931–100941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, M.Y.; Oliva, C.R.; Noman, A.S.M.; Allen, B.G.; Goswami, P.C.; Zakharia, Y.; Monga, V.; Spitz, D.R.; Buatti, J.M.; Griguer, C.E. Radioresistance in Glioblastoma and the Development of Radiosensitizers. Cancers 2020, 12, 2511. [Google Scholar] [CrossRef] [PubMed]
- Turner, K.M.; Sun, Y.; Ji, P.; Granberg, K.J.; Bernard, B.; Hu, L.; Cogdell, D.E.; Zhou, X.; Yli-Harja, O.; Nykter, M.; et al. Genomically amplified Akt3 activates DNA repair pathway and promotes glioma progression. Proc. Natl. Acad. Sci. USA 2015, 112, 3421–3426. [Google Scholar] [CrossRef] [Green Version]
- Hassin, O.; Nataraj, N.B.; Shreberk-Shaked, M.; Aylon, Y.; Yaeger, R.; Fontemaggi, G.; Mukherjee, S.; Maddalena, M.; Avioz, A.; Iancu, O.; et al. Different hotspot p53 mutants exert distinct phenotypes and predict outcome of colorectal cancer patients. Nat. Commun. 2022, 13, 2800. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.; Garraway, L.A.; Ashworth, A.; Weber, B. Synthetic lethality as an engine for cancer drug target discovery. Nat. Rev. Drug Discov. 2020, 19, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Kruiswijk, F.; Labuschagne, C.F.; Vousden, K.H. p53 in survival, death and metabolic health: A lifeguard with a licence to kill. Nat. Rev. Mol. Cell Biol. 2015, 16, 393–405. [Google Scholar] [CrossRef]
- Lukin, D.J.; Carvajal, L.A.; Liu, W.-J.; Resnick-Silverman, L.; Manfredi, J.J. p53 Promotes Cell Survival due to the Reversibility of Its Cell-Cycle Checkpoints. Mol. Cancer Res. 2015, 13, 16–28. [Google Scholar] [CrossRef] [Green Version]
- Kao, G.D.; Jiang, Z.; Fernandes, A.M.; Gupta, A.K.; Maity, A. Inhibition of Phosphatidylinositol-3-OH Kinase/Akt Signaling Impairs DNA Repair in Glioblastoma Cells following Ionizing Radiation. J. Biol. Chem. 2007, 282, 21206–21212. [Google Scholar] [CrossRef] [Green Version]
- Golding, S.E.; Morgan, R.N.; Adams, B.R.; Hawkins, A.J.; Povirk, L.F.; Valerie, K. Pro-survival AKT and ERK signaling from EGFR and mutant EGFRvIII enhances DNA double-strand break repair in human glioma cells. Cancer Biol. Ther. 2009, 8, 730–738. [Google Scholar] [CrossRef] [Green Version]
- Zhuo, S.; Chen, Z.; Yang, Y.; Zhang, J.; Tang, J.; Yang, K. Clinical and Biological Significances of a Ferroptosis-Related Gene Signature in Glioma. Front. Oncol. 2020, 10, 590861. [Google Scholar] [CrossRef]
- Goplen, D.; Bougnaud, S.; Rajcevic, U.; Bøe, S.O.; Skaftnesmo, K.O.; Voges, J.; Enger, P.; Wang, J.; Tysnes, B.B.; Laerum, O.D.; et al. αB-crystallin is elevated in highly infiltrative apoptosis-resistant glioblastoma cells. Am. J. Pathol. 2010, 177, 1618–1628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foltyn, M.; Luger, A.-L.; Lorenz, N.I.; Sauer, B.; Mittelbronn, M.; Harter, P.N.; Steinbach, J.P.; Ronellenfitsch, M.W. The physiological mTOR complex 1 inhibitor DDIT4 mediates therapy resistance in glioblastoma. Br. J. Cancer 2019, 120, 481–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, C.A.; Brault, C.; Peck, B.; Bensaad, K.; Griffiths, B.; Mitter, R.; Chakravarty, P.; East, P.; Dankworth, B.; Alibhai, D.; et al. SREBP maintains lipid biosynthesis and viability of cancer cells under lipid- and oxygen-deprived conditions and defines a gene signature associated with poor survival in glioblastoma multiforme. Oncogene 2015, 34, 5128–5140. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Chen, Y.; Hu, S.; Gao, L.; Tang, N.; Liu, R.; Qin, Y.; Ren, C.; Du, S. GDF15 expression in glioma is associated with malignant progression, immune microenvironment, and serves as a prognostic factor. CNS Neurosci. Ther. 2022, 28, 158–171. [Google Scholar] [CrossRef]
- Feng, X.; Zhang, L.; Ke, S.; Liu, T.; Hao, L.; Zhao, P.; Tu, W.; Cang, S. High expression of GPNMB indicates an unfavorable prognosis in glioma: Combination of data from the GEO and CGGA databases and validation in tissue microarray. Oncol. Lett. 2020, 20, 2356–2368. [Google Scholar] [CrossRef]
- Akçay, S.; Güven, E.; Afzal, M.; Kazmi, I. Non-negative matrix factorization and differential expression analyses identify hub genes linked to progression and prognosis of glioblastoma multiforme. Gene 2022, 824, 146395. [Google Scholar] [CrossRef]
- Delic, S.; Lottmann, N.; Jetschke, K.; Reifenberger, G.; Riemenschneider, M.J. Identification and functional validation of CDH11, PCSK6 and SH3GL3 as novel glioma invasion-associated candidate genes. Neuropathol. Appl. Neurobiol. 2012, 38, 201–212. [Google Scholar] [CrossRef]
- He, Z.; You, C.; Zhao, D. Long non-coding RNA UCA1/miR-182/PFKFB2 axis modulates glioblastoma-associated stromal cells-mediated glycolysis and invasion of glioma cells. Biochem. Biophys. Res. Commun. 2018, 500, 569–576. [Google Scholar] [CrossRef]
- Putthisen, S.; Silsirivanit, A.; Panawan, O.; Niibori-Nambu, A.; Nishiyama-Ikeda, Y.; Ma-In, P.; Luang, S.; Ohta, K.; Muisuk, K.; Wongkham, S.; et al. Targeting alpha2,3-sialylated glycan in glioma stem-like cells by Maackia amurensis lectin-II: A promising strategy for glioma treatment. Exp. Cell Res. 2022, 410, 112949. [Google Scholar] [CrossRef]
- Mariani, L.; Beaudry, C.; McDonough, W.S.; Hoelzinger, D.B.; Demuth, T.; Ross, K.R.; Berens, T.; Coons, S.W.; Watts, G.; Trent, J.M.; et al. Glioma Cell Motility is Associated with Reduced Transcription of Proapoptotic and Proliferation Genes: A cDNA Microarray Analysis. J. Neurooncol. 2001, 53, 161–176. [Google Scholar] [CrossRef]
- Sachdeva, R.; Wu, M.; Smiljanic, S.; Kaskun, O.; Ghannad-Zadeh, K.; Celebre, A.; Isaev, K.; Morrissy, A.S.; Guan, J.; Tong, J.; et al. ID1 Is Critical for Tumorigenesis and Regulates Chemoresistance in Glioblastoma. Cancer Res. 2019, 79, 4057–4071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, R.; Teo, W.S.; Mittal, V.; Swarbrick, A. ID proteins regulate diverse aspects of cancer progression and provide novel therapeutic opportunities. Mol. Ther. 2014, 22, 1407–1415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, C.; Zhang, J.; Fang, S.; Liu, F. IGFBP5 increases cell invasion and inhibits cell proliferation by EMT and Akt signaling pathway in Glioblastoma multiforme cells. Cell Div. 2020, 15, 4. [Google Scholar] [CrossRef] [PubMed]
- Serafim, R.B.; da Silva, P.; Cardoso, C.; Di Cristofaro, L.F.M.; Netto, R.P.; de Almeida, R.; Navegante, G.; Storti, C.B.; de Sousa, J.F.; de Souza, F.C.; et al. Expression Profiling of Glioblastoma Cell Lines Reveals Novel Extracellular Matrix-Receptor Genes Correlated With the Responsiveness of Glioma Patients to Ionizing Radiation. Front. Oncol. 2021, 11, 668090. [Google Scholar] [CrossRef] [PubMed]
- Hjelmeland, A.B.; Wu, Q.; Wickman, S.; Eyler, C.; Heddleston, J.; Shi, Q.; Lathia, J.D.; Macswords, J.; Lee, J.; McLendon, R.E.; et al. Targeting A20 decreases glioma stem cell survival and tumor growth. PLoS Biol. 2010, 8, e1000319. [Google Scholar] [CrossRef] [Green Version]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ClQ_DEGs_#993 (wtp53) | ||
|---|---|---|
| p53RGs (80) * p21-DREAM | down-regulated anln *; arhgap11a *; arhgap11b *; aspm *; aurka *; bub1 *; bub1b *; casc5 *; c11orf82 *; c12orf48 *; ccdc18 *; ccna2 *; ccnb1 *; ccnb2 *; ccne2; cdc2; cdca2 *; cdca3 *; cdca8 *; cdc25c *; cdkn3 *; cenpe *; cit *; ckap2l *; dc; depdc1 *; depdc1b *; dlgap5 *; esco2 *; exo1 *, fam64a *; fancb *; fancd2 **; fanci **; gas2l3 *; gtse1 *; hist1h2bm *; hjurp *; hmmr *; kif2c *; kif4a *; kif11 *; kif14 *; kif15 *; kif18a *; kif20a *; kif20b *; kif23 *; kif24 *; mad2l1 *; melk *; mki67 *; ncapg *; ncapg2 *; ncaph *; ndc80 *; neil3 *; nuf2 *; nusap1 *; plk1 *; plk4 *; polq *; prc1 *; prr11 *; pttg1 *; racgap1 *; rrm2; rtkn2 *; sema3a; sgol1 *; sgol2 *; shcbp1 *; spag5 *; spc25 *; stil *; top2a *; tpx2 *; troap *; ttk *; ube2c *; xrcc2 * | GO Terms: cell cycle mitosis cytokinesis DDR |
| p53RGs (12) ** MVA | up-regulated acat2 **; dhcr7 **; dhcr24 **; fasn **; fdft1 **; fdps **; lpin1 **; lss **; mvd **; nsdhl **; sc4mol **; tm7sf2 ** | GO Terms: lipid metabolism cholesterol- biosynthesis |
| ClQ_DEGs_G112 (R273H-p53) | ||
|---|---|---|
| p53RGs (9) * p21-DREAM | down-regulated: ccnb1 *; dlgap5 *; exo1 *, kif4a *; kif20a *; kif23 *; neil3 *; sema3a; top2a * | GO Terms: cell cycle mitosis cytokinesis DDR |
| p53RGs (7) ** MVA | up-regulated: dhcr7 **; fasn **; fdps **; hmgcr **; hmgcs1 **; lss **; sc4mol ** | GO Terms: lipid metabolism cholesterol- biosynthesis |
| ClQ_DEGs_#1095 (p53 null) | ||
|---|---|---|
| p53RGs (0) * GB promotion | down-regulated: arhgap29 *; id1 *; id3 *; igfbp5 *; itga3 * tnfaip3 *; trdc | GO Terms: receptor activity migration |
| p53RGs (0) * GB promotion | up-regulated: acsl6 *; bhlhe41; cryab *; ddit4l *; fabp3 *; fn3k; gdf15 *; gpnmb *; lipg; lrrc39; nckap5 *; pcsk6 *; pfkfb2 *; pi15; pnliprp3; serinc5; st3gal5 * | GO Terms: proliferation metabolism |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Müller, A.; Weyerhäuser, P.; Berte, N.; Jonin, F.; Lyubarskyy, B.; Sprang, B.; Kantelhardt, S.R.; Salinas, G.; Opitz, L.; Schulz-Schaeffer, W.; et al. Concurrent Activation of Both Survival-Promoting and Death-Inducing Signaling by Chloroquine in Glioblastoma Stem Cells: Implications for Potential Risks and Benefits of Using Chloroquine as Radiosensitizer. Cells 2023, 12, 1290. https://doi.org/10.3390/cells12091290
Müller A, Weyerhäuser P, Berte N, Jonin F, Lyubarskyy B, Sprang B, Kantelhardt SR, Salinas G, Opitz L, Schulz-Schaeffer W, et al. Concurrent Activation of Both Survival-Promoting and Death-Inducing Signaling by Chloroquine in Glioblastoma Stem Cells: Implications for Potential Risks and Benefits of Using Chloroquine as Radiosensitizer. Cells. 2023; 12(9):1290. https://doi.org/10.3390/cells12091290
Chicago/Turabian StyleMüller, Andreas, Patrick Weyerhäuser, Nancy Berte, Fitriasari Jonin, Bogdan Lyubarskyy, Bettina Sprang, Sven Rainer Kantelhardt, Gabriela Salinas, Lennart Opitz, Walter Schulz-Schaeffer, and et al. 2023. "Concurrent Activation of Both Survival-Promoting and Death-Inducing Signaling by Chloroquine in Glioblastoma Stem Cells: Implications for Potential Risks and Benefits of Using Chloroquine as Radiosensitizer" Cells 12, no. 9: 1290. https://doi.org/10.3390/cells12091290