




Biomolecules 2024, 14(3), 311; https://doi.org/10.3390/biom14030311 - 06 Mar 2024
Viewed by 896
Abstract
►
Show Figures
A Type I reaction center (RC) (Fe-S type, ferredoxin reducing) is found in several phyla containing anoxygenic phototrophic bacteria. These include the heliobacteria (HB), the green sulfur bacteria (GSB), and the chloracidobacteria (CB), for which high-resolution homodimeric RC-photosystem (PS) structures have recently appeared.
[...] Read more.
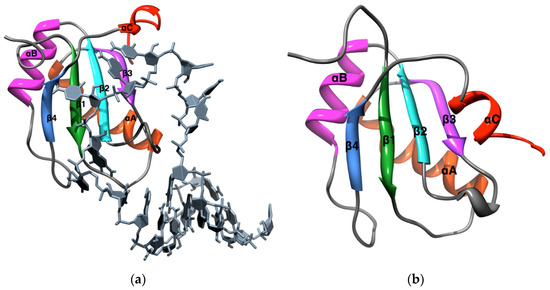
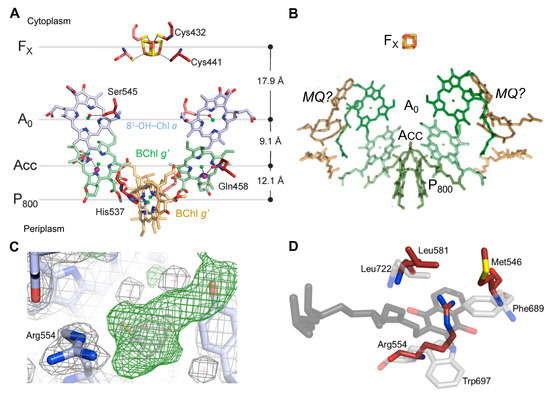
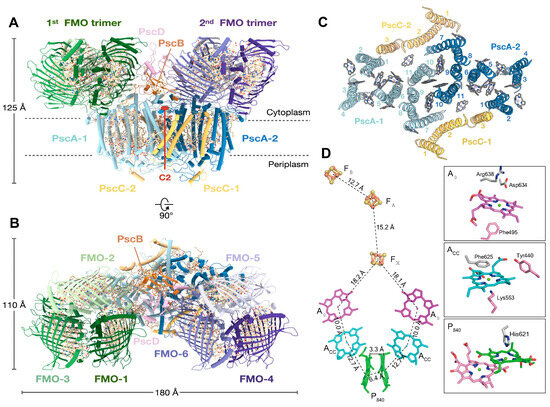
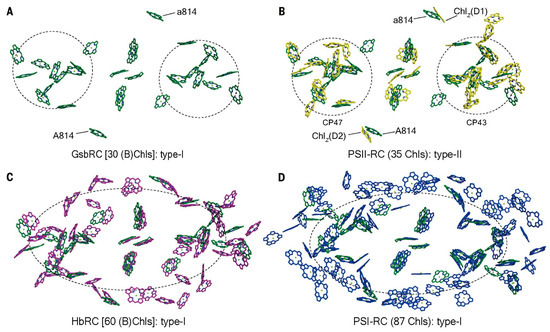
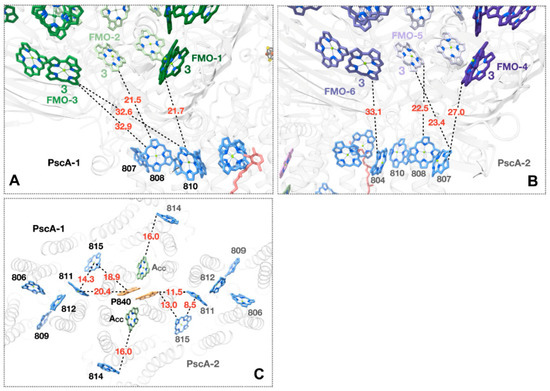
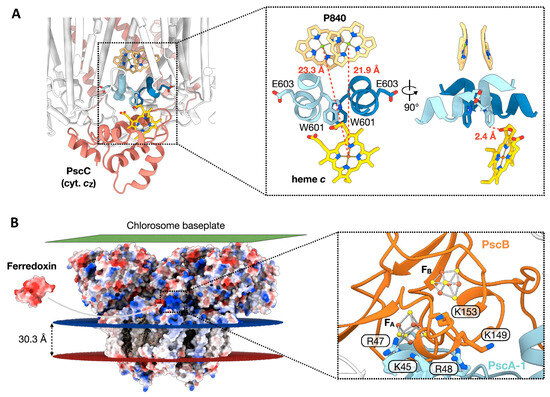
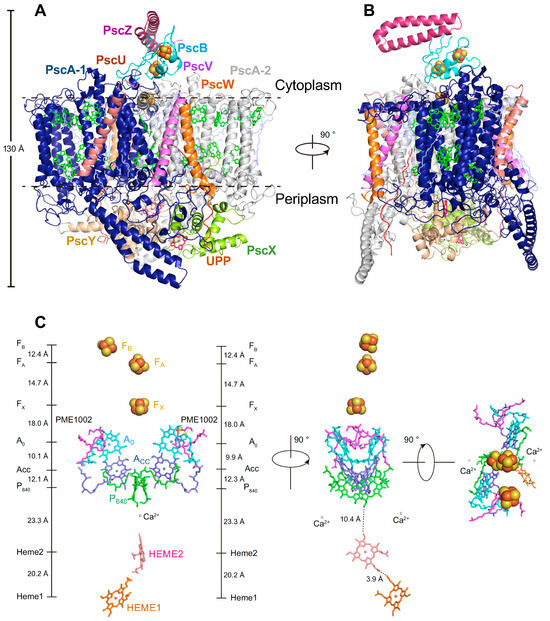
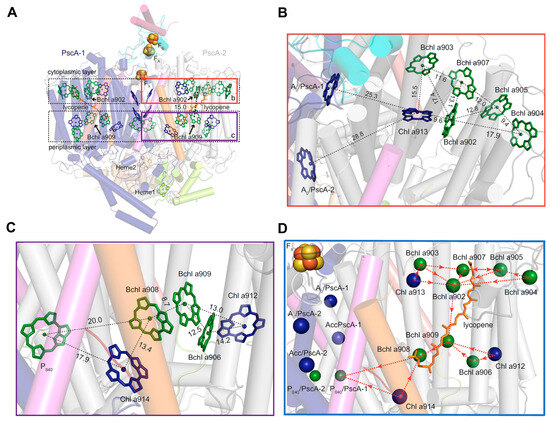
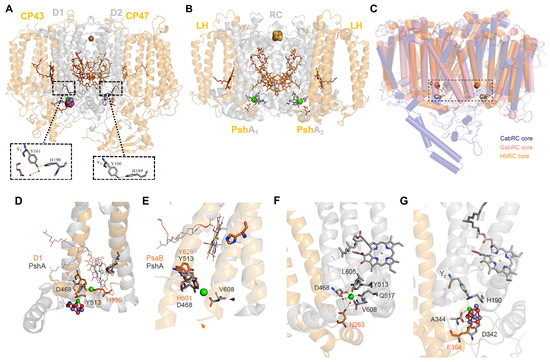
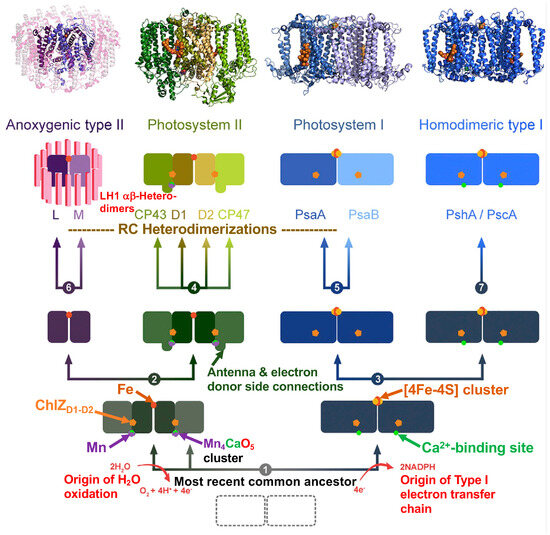
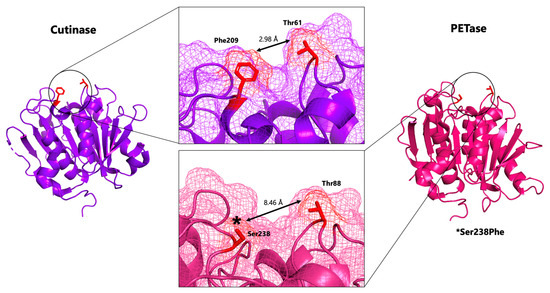
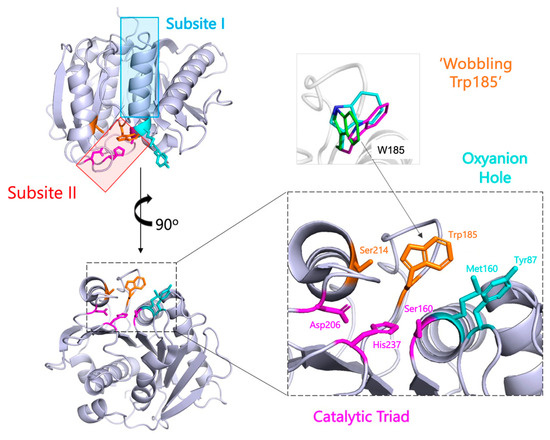
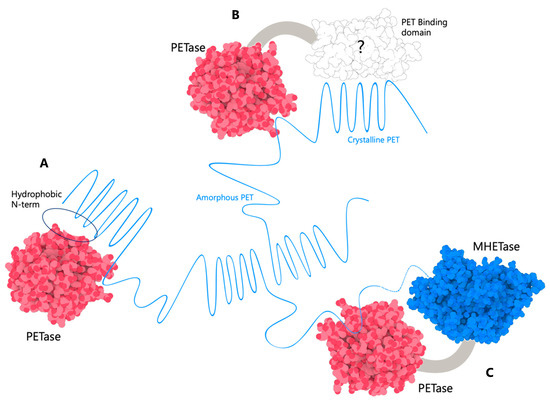
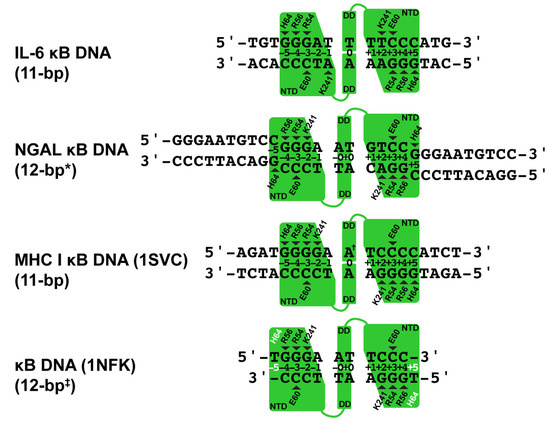
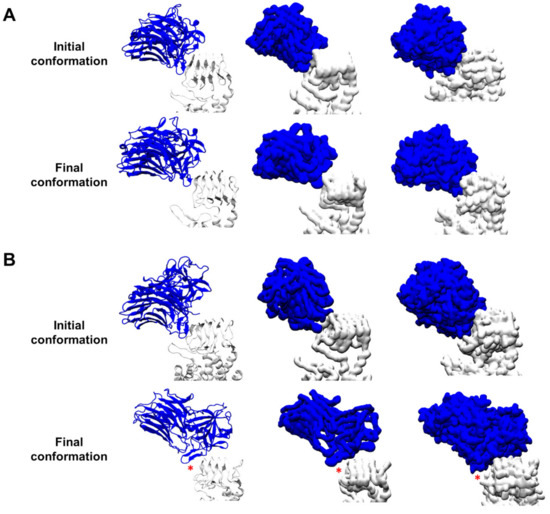
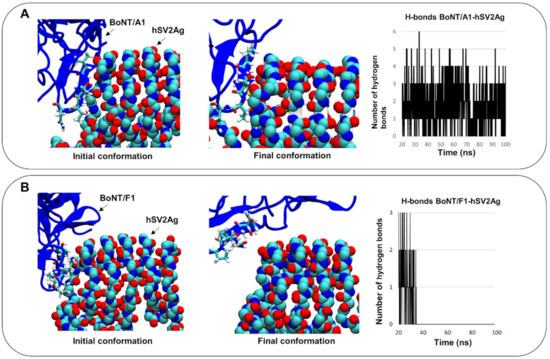
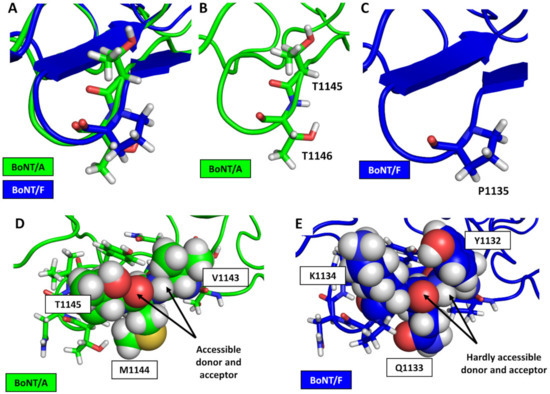
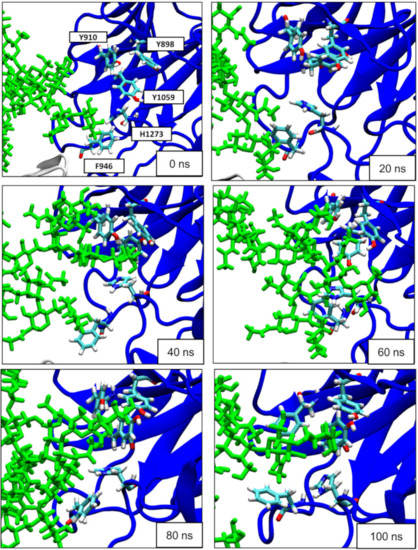
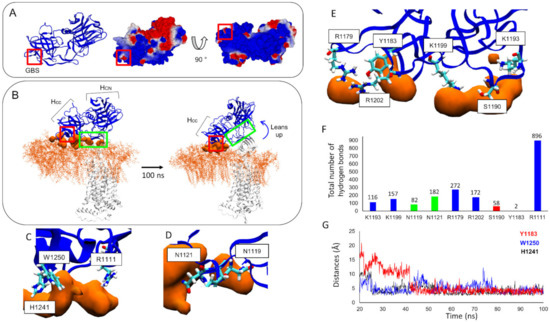
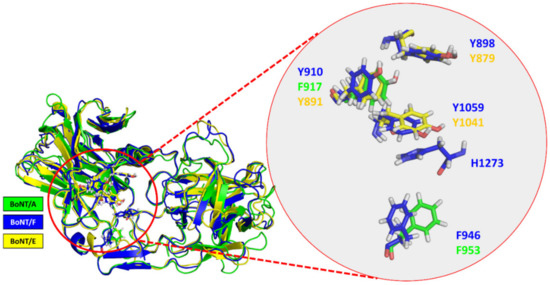
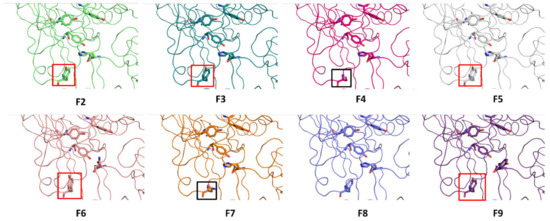
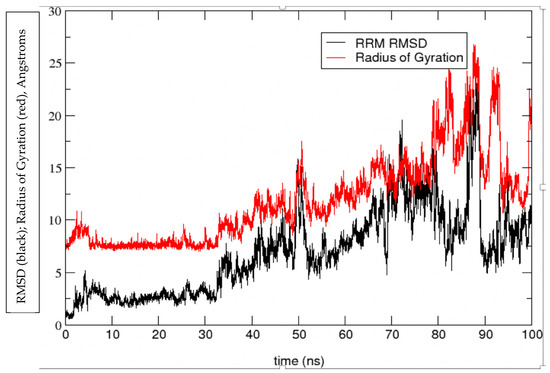
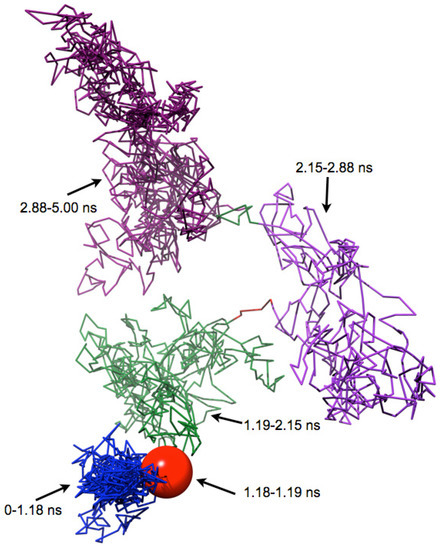
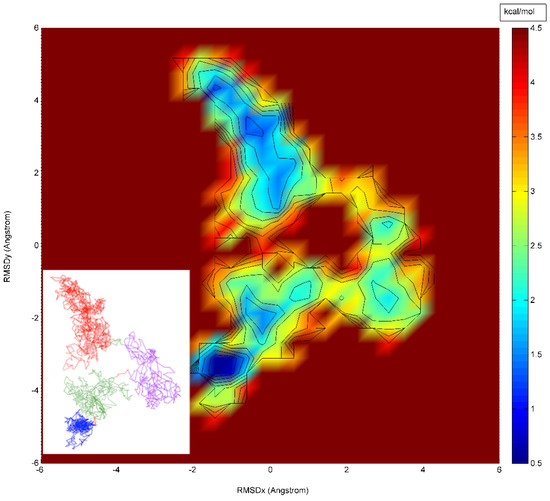
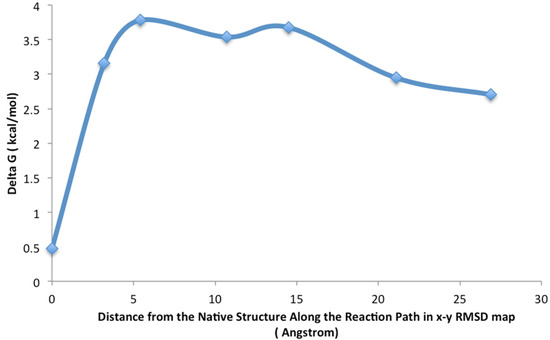
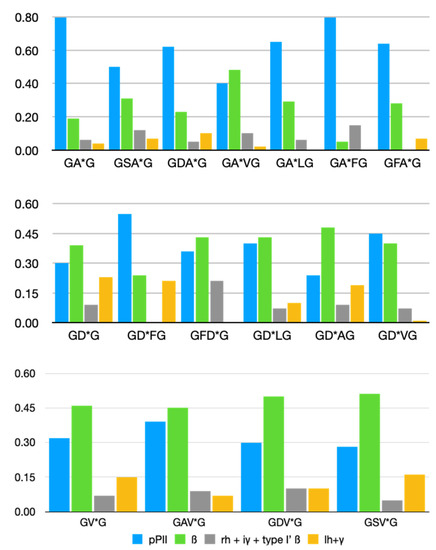
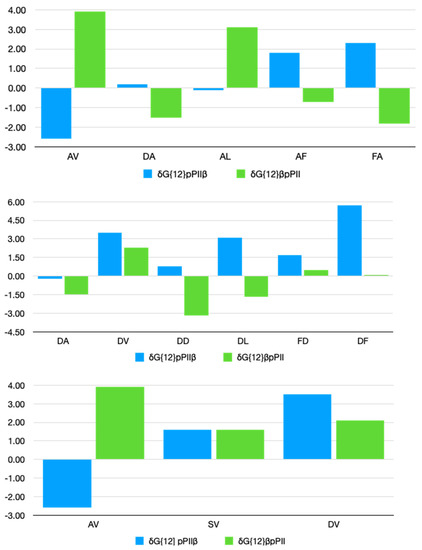
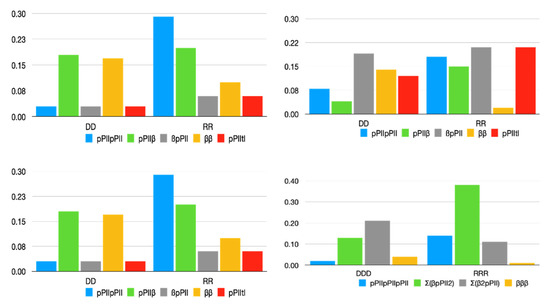
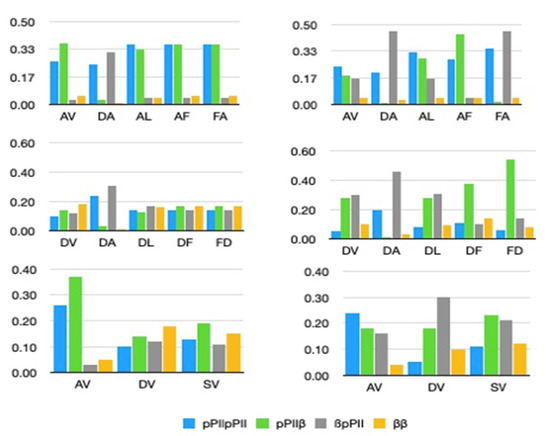
A Type I reaction center (RC) (Fe-S type, ferredoxin reducing) is found in several phyla containing anoxygenic phototrophic bacteria. These include the heliobacteria (HB), the green sulfur bacteria (GSB), and the chloracidobacteria (CB), for which high-resolution homodimeric RC-photosystem (PS) structures have recently appeared. The 2.2-Å X-ray structure of the RC-PS of Heliomicrobium modesticaldum revealed that the core PshA apoprotein (PshA-1 and PshA-2 homodimeric pair) exhibits a structurally conserved PSI arrangement comprising five C-terminal transmembrane α-helices (TMHs) forming the RC domain and six N-terminal TMHs coordinating the light-harvesting (LH) pigments. The Hmi. modesticaldum structure lacked quinone molecules, indicating that electrons were transferred directly from the A0 (81-OH-chlorophyll (Chl) a) acceptor to the FX [4Fe-4S] component, serving as the terminal RC acceptor. A pair of additional TMHs designated as Psh X were also found that function as a low-energy antenna. The 2.5-Å resolution cryo-electron microscopy (cryo-EM) structure for the RC-PS of the green sulfur bacterium Chlorobaculum tepidum included a pair of Fenna–Matthews–Olson protein (FMO) antennae, which transfer excitations from the chlorosomes to the RC-PS (PscA-1 and PscA-2) core. A pair of cytochromes cZ (PscC) molecules was also revealed, acting as electron donors to the RC bacteriochlorophyll (BChl) a’ special pair, as well as PscB, housing the [4Fe-4S] cluster FA and FB, and the associated PscD protein. While the FMO components were missing from the 2.6-Å cryo-EM structure of the Zn- (BChl) a’ special pair containing RC-PS of Chloracidobacterium thermophilum, a unique architecture was revealed that besides the (PscA)2 core, consisted of seven additional subunits including PscZ in place of PscD, the PscX and PscY cytochrome c serial electron donors and four low mol. wt. subunits of unknown function. Overall, these diverse structures have revealed that (i) the HB RC-PS is the simplest light–energy transducing complex yet isolated and represents the closest known homolog to a common homodimeric RC-PS ancestor; (ii) the symmetrically localized Ca2+-binding sites found in each of the Type I homodimeric RC-PS structures likely gave rise to the analogously positioned Mn4CaO5 cluster of the PSII RC and the TyrZ RC donor site; (iii) a close relationship between the GSB RC-PS and the PSII Chl proteins (CP)43 and CP47 was demonstrated by their strongly conserved LH-(B)Chl localizations; (iv) LH-BChls of the GSB-RC-PS are also localized in the conserved RC-associated positions of the PSII ChlZ-D1 and ChlZ-D2 sites; (v) glycosylated carotenoids of the GSB RC-PS are located in the homologous carotenoid-containing positions of PSII, reflecting an O2-tolerance mechanism capable of sustaining early stages in the evolution of oxygenic photosynthesis. In addition to the close relationships found between the homodimeric RC-PS and PSII, duplication of the gene encoding the ancestral Type I RC apoprotein, followed by genetic divergence, may well account for the appearance of the heterodimeric Type I and Type II RCs of the extant oxygenic phototrophs. Accordingly, the long-held view that PSII arose from the anoxygenic Type II RC is now found to be contrary to the new evidence provided by Type I RC-PS homodimer structures, indicating that the evolutionary origins of anoxygenic Type II RCs, along with their distinct antenna rings are likely to have been preceded by the events that gave rise to their oxygenic counterparts.
Full article
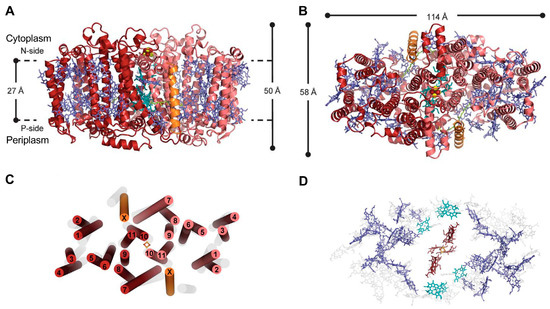
Figure 1
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}