
Protein Conformational Dynamics Underlie Selective Recognition of Thermophilic over Mesophilic Enzyme I by a Substrate Analogue
 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
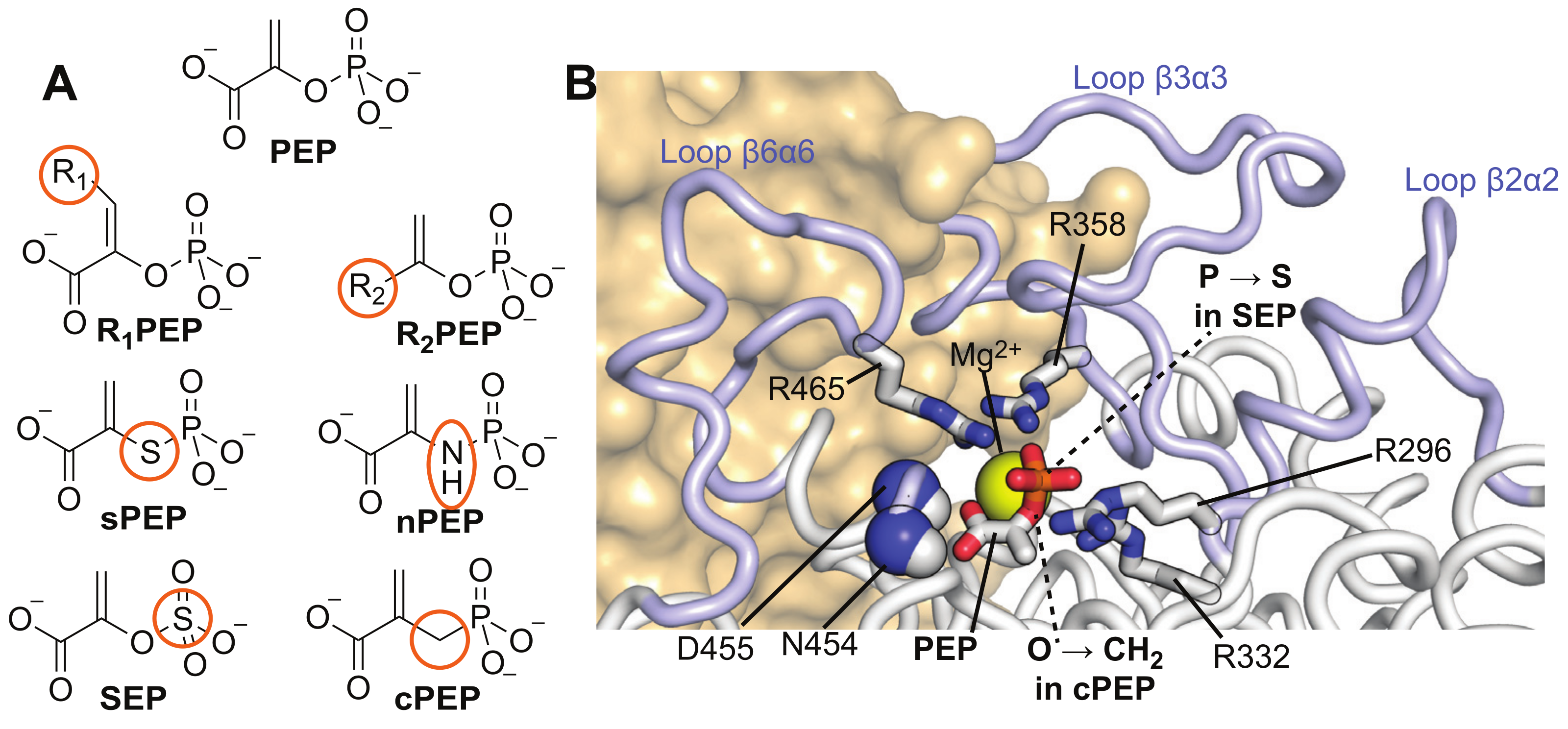
2.1. Selection of PEP Analogs
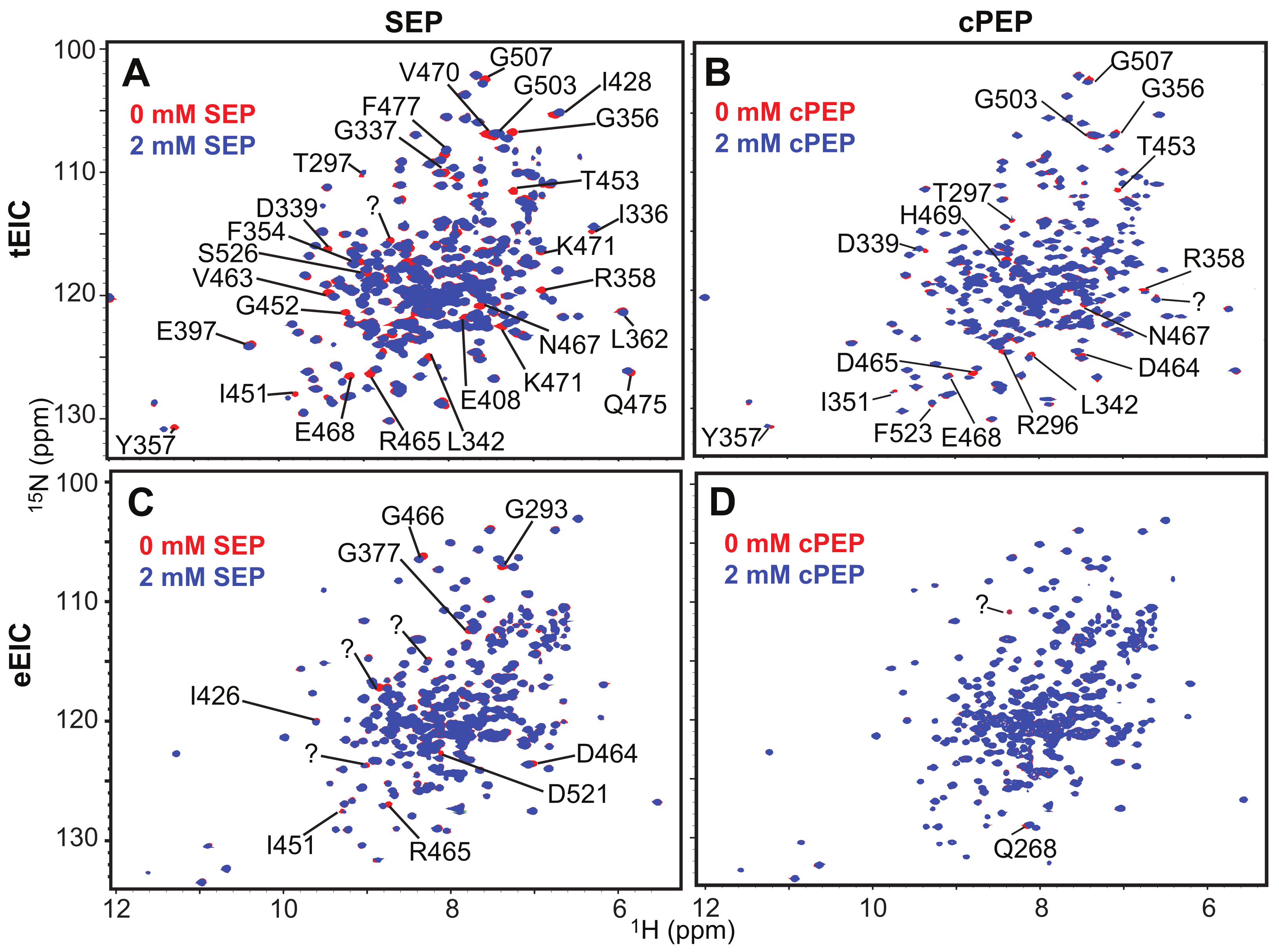
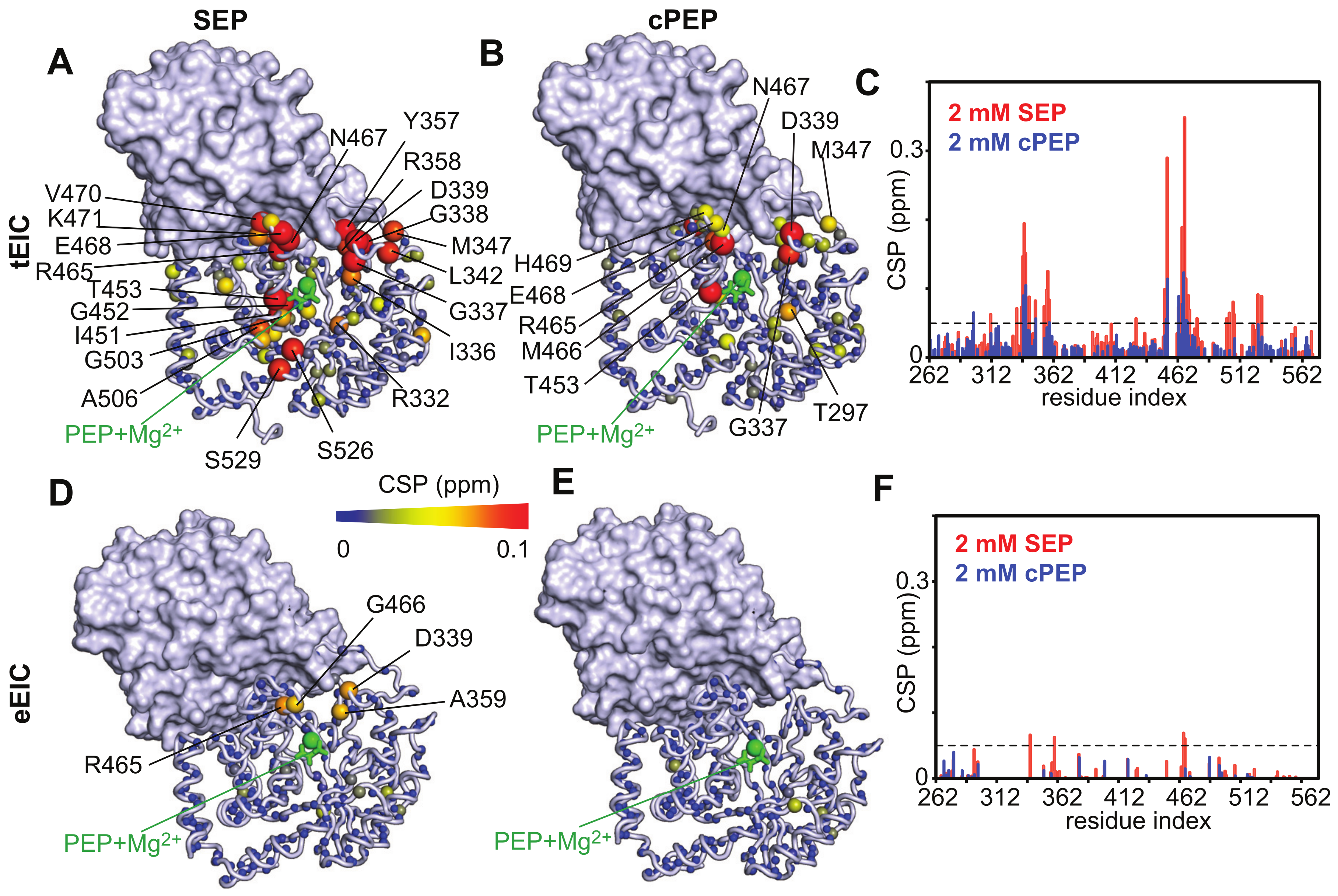
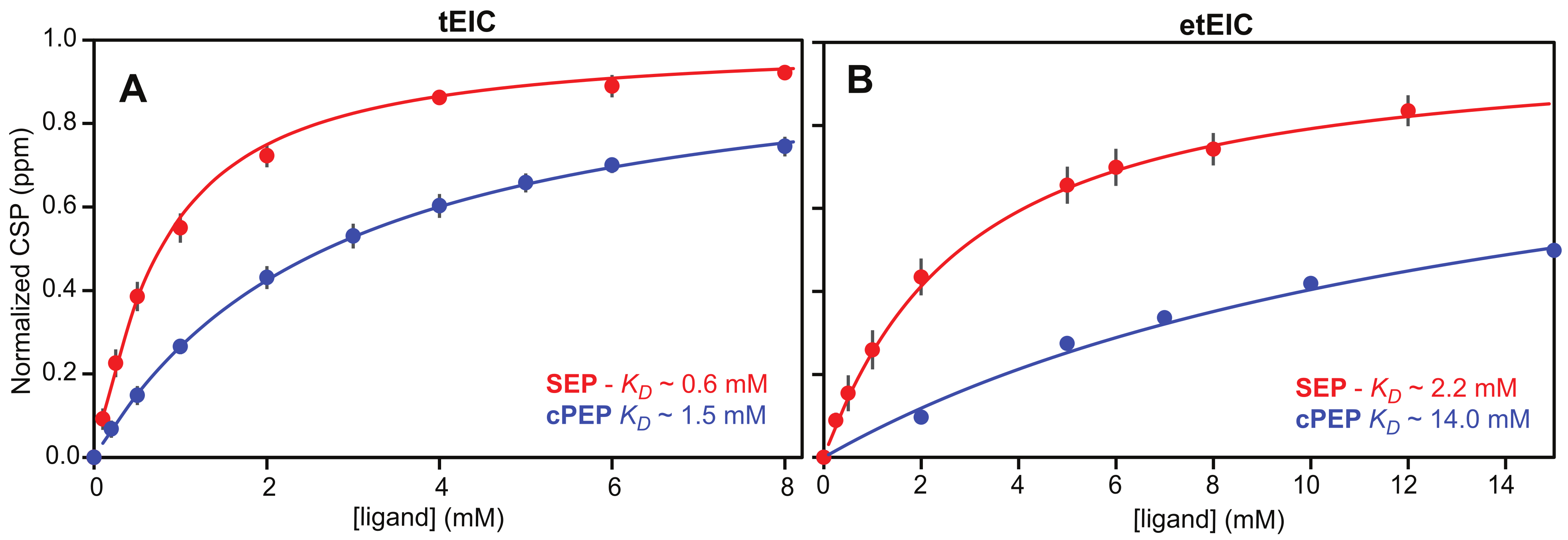
2.2. SEP and cPEP Recognize Thermophilic over Mesophilic EI
2.3. Binding of SEP and cPEP to Chimeric Thermophilic/Mesophilic Constructs
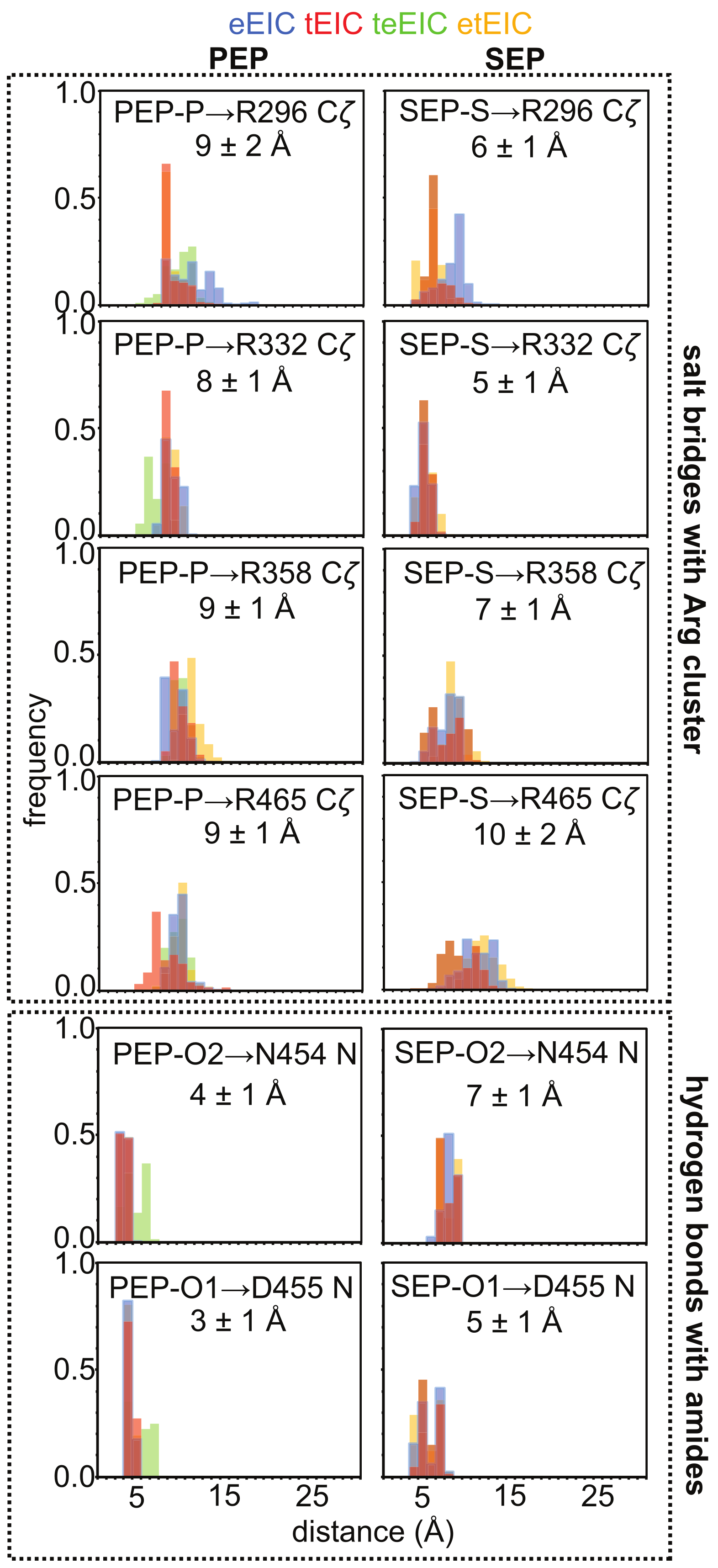
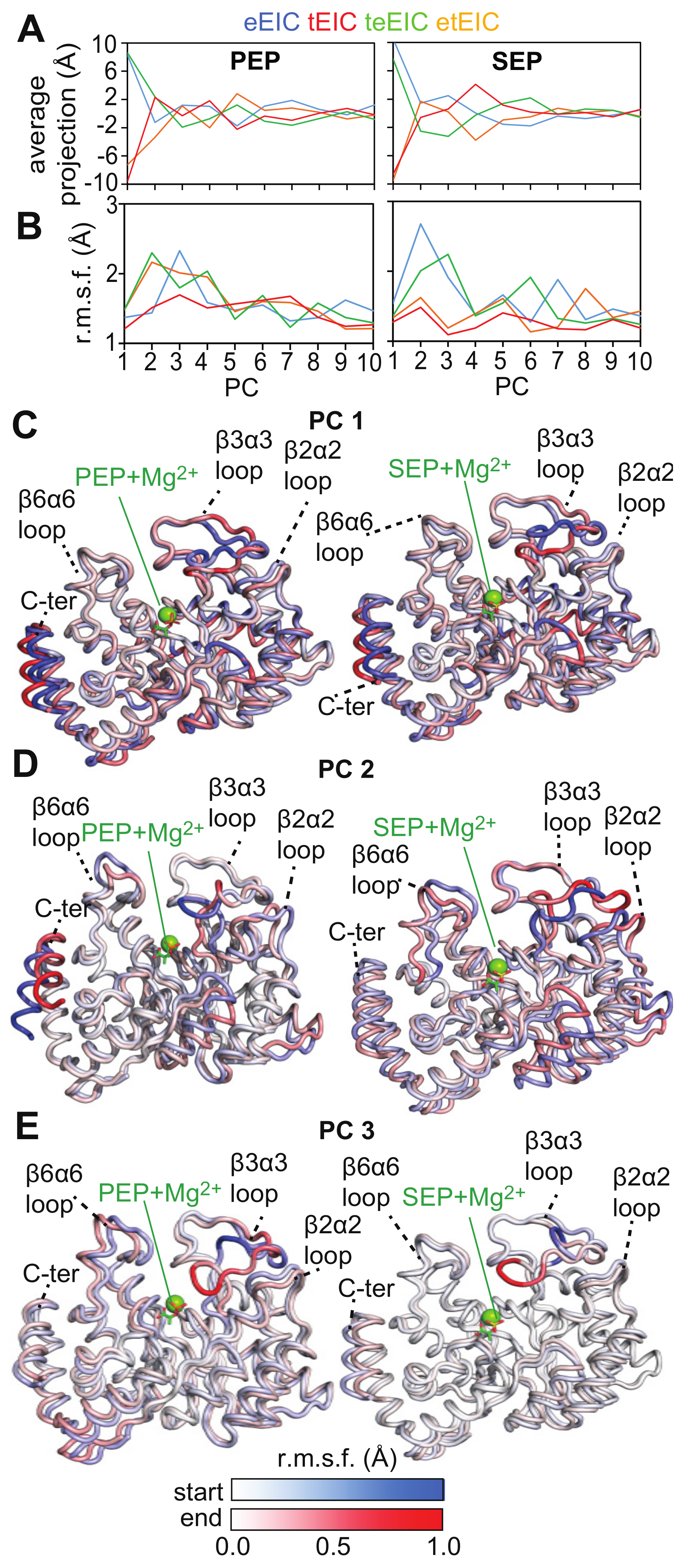
2.4. MD Simulations of the EIC–PEP and EIC–SEP Complexes
3. Discussion
4. Materials and Methods
4.1. cPEP and SEP Synthesis and Protein Expression and Purification
4.2. NMR Spectroscopy
4.3. MD Simulations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kundig, W.; Ghosh, S.; Roseman, S. Phosphate bound to histidine in a protein as an intermediate in a novel phospho-transferase system. Proc. Natl. Acad. Sci. USA 1964, 52, 1067–1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clore, G.M.; Venditti, V. Structure, dynamics and biophysics of the cytoplasmic protein–protein complexes of the bacterial phosphoenolpyruvate: Sugar phosphotransferase system. Trends Biochem. Sci. 2013, 38, 515–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deutscher, J.; Aké, F.M.D.; Derkaoui, M.; Zébré, A.C.; Cao, T.N.; Bouraoui, H.; Kentache, T.; Mokhtari, A.; Milohanic, E.; Joyet, P. The bacterial phosphoenolpyruvate:carbohydrate phosphotransferase system: Regulation by protein phosphorylation and phosphorylation-dependent protein-protein interactions. Microbiol. Mol. Biol. Rev. 2014, 78, 231–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Postma, P.W.; Lengeler, J.W.; Jacobson, G.R. Phosphoenolpyruvate: Carbohydrate phosphotransferase systems of bacteria. Microbiol. Rev. 1993, 57, 543–594. [Google Scholar] [CrossRef]
- Doucette, C.D.; Schwab, D.J.; Wingreen, N.S.; Rabinowitz, J.D. α-Ketoglutarate coordinates carbon and nitrogen utilization via enzyme I inhibition. Nat. Chem. Biol. 2011, 7, 894–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venditti, V.; Ghirlando, R.; Clore, G.M. Structural basis for enzyme i inhibition by α-ketoglutarate. ACS Chem. Biol. 2013, 8, 1232–1240. [Google Scholar] [CrossRef] [Green Version]
- Hogema, B.M.; Arents, J.C.; Bader, R.; Eijkemans, K.; Yoshida, H.; Takahashi, H.; Aiba, H.; Postma, P.W. Inducer exclusion in Escherichia coli by non-PTS substrates: The role of the PEP to pyruvate ratio in determining the phosphorylation state of enzyme IIAGlc. Mol. Microbiol. 1998, 30, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Roux, B. Automated Force Field Parameterization for Nonpolarizable and Polarizable Atomic Models Based on Ab Initio Target Data. J. Chem. Theory Comput. 2013, 9, 3543–3556. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.T.; Venditti, V. An allosteric pocket for inhibition of bacterial Enzyme I identified by NMR-based fragment screening. J. Struct. Biol. X 2020, 4, 100034. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Ghirlando, R.; Roche, J.; Venditti, V. Structure elucidation of the elusive Enzyme I monomer reveals the molecular mechanisms linking oligomerization and enzymatic activity. Proc. Natl. Acad. Sci. USA 2021, 118, e2100298118. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Ghirlando, R.; Venditti, V. The oligomerization state of bacterial enzyme I (EI) determines EI’s allosteric stimulation or competitive inhibition by α-ketoglutarate. J. Biol. Chem. 2018, 293, 2631–2639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chauvin, F.; Brand, L.; Roseman, S. Enzyme I: The first protein and potential regulator of the bacterial phosphoenolpyruvate: Glycose phosphotransferase system. Res. Microbiol. 1996, 147, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Venditti, V.; Clore, G.M. Conformational selection and substrate binding regulate the monomer/dimer equilibrium of the C-terminal domain of Escherichia coli enzyme I. J. Biol. Chem. 2012, 287, 26989–26998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, H.V.; Vyas, K.A.; Savtchenko, R.; Roseman, S. The Monomer/Dimer Transition of Enzyme I of the Escherichia coli Phosphotransferase System. J. Biol. Chem. 2006, 281, 17570–17578. [Google Scholar] [CrossRef] [Green Version]
- Venditti, V.; Tugarinov, V.; Schwieters, C.D.; Grishaev, A.; Clore, G.M. Large interdomain rearrangement triggered by suppression of micro- to millisecond dynamics in bacterial Enzyme I. Nat. Commun. 2015, 6, 5960. [Google Scholar] [CrossRef] [Green Version]
- Dotas, R.R.; Nguyen, T.T.; Stewart, C.E.; Ghirlando, R.; Potoyan, D.A.; Venditti, V. Hybrid Thermophilic/Mesophilic Enzymes Reveal a Role for Conformational Disorder in Regulation of Bacterial Enzyme I. J. Mol. Biol. 2020, 432, 4481–4498. [Google Scholar] [CrossRef]
- Suh, J.-Y.; Cai, M.; Clore, G.M. Impact of Phosphorylation on Structure and Thermodynamics of the Interaction between the N-terminal Domain of Enzyme I and the Histidine Phosphocarrier Protein of the Bacterial Phosphotransferase System. J. Biol. Chem. 2008, 283, 18980–18989. [Google Scholar] [CrossRef] [Green Version]
- Purslow, J.; Thimmesch, J.; Sivo, V.; Nguyen, T.; Khatiwada, B.; Dotas, R.; Venditti, V. A Single Point Mutation Controls the Rate of Interconversion Between the g (+) and g (−) Rotamers of the Histidine 189 chi2 Angle That Activates Bacterial Enzyme I for Catalysis. Front. Mol. Biosci. 2021, 8, 699203. [Google Scholar] [CrossRef]
- Schwieters, C.D.; Suh, J.-Y.; Grishaev, A.; Ghirlando, R.; Takayama, Y.; Clore, G.M. Solution Structure of the 128 kDa Enzyme I Dimer from Escherichia coli and Its 146 kDa Complex with HPr Using Residual Dipolar Couplings and Small- and Wide-Angle X-ray Scattering. J. Am. Chem. Soc. 2010, 132, 13026–13045. [Google Scholar] [CrossRef] [Green Version]
- Teplyakov, A.; Lim, K.; Zhu, P.-P.; Kapadia, G.; Chen, C.C.H.; Schwartz, J.; Howard, A.; Reddy, P.T.; Peterkofsky, A.; Herzberg, O. Structure of phosphorylated enzyme I, the phosphoenolpyruvate:sugar phosphotransferase system sugar translocation signal protein. Proc. Natl. Acad. Sci. USA 2006, 103, 16218. [Google Scholar] [CrossRef]
- Venditti, V.; Schwieters, C.D.; Grishaev, A.; Clore, G.M. Dynamic equilibrium between closed and partially closed states of the bacterial Enzyme I unveiled by solution NMR and X-ray scattering. Proc. Natl. Acad. Sci. USA 2015, 112, 11565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deutscher, J.; Francke, C.; Postma, P.W. How phosphotransferase system-related protein phosphorylation regulates carbohydrate metabolism in bacteria. Microbiol. Mol. Biol. Rev. 2006, 70, 939–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meza, E.; Becker, J.; Bolivar, F.; Gosset, G.; Wittmann, C. Consequences of phosphoenolpyruvate:sugar phosphotranferase system and pyruvate kinase isozymes inactivation in central carbon metabolism flux distribution in Escherichia coli. Microb. Cell Factories 2012, 11, 127. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Alles, L.F.; Flükiger, K.; Hewel, J.; Gutknecht, R.; Siebold, C.; Schürch, S.; Erni, B. Mechanism-based inhibition of enzyme I of the Escherichia coli phosphotransferase system. Cysteine 502 is an essential residue. J. Biol. Chem. 2002, 277, 6934–6942. [Google Scholar] [CrossRef] [Green Version]
- Purslow, J.A.; Khatiwada, B.; Bayro, M.J.; Venditti, V. NMR Methods for Structural Characterization of Protein-Protein Complexes. Front. Mol. Biosci. 2020, 7, 9. [Google Scholar] [CrossRef]
- Navdaeva, V.; Zurbriggen, A.; Waltersperger, S.; Schneider, P.; Oberholzer, A.E.; Bähler, P.; Bächler, C.; Grieder, A.; Baumann, U.; Erni, B. Phosphoenolpyruvate: Sugar phosphotransferase system from the hyperthermophilic Thermoanaerobacter tengcongensis. Biochemistry 2011, 50, 1184–1193. [Google Scholar] [CrossRef] [PubMed]
- Van Aalten, D.M.F.; Amadei, A.; Linssen, A.B.M.; Eijsink, V.G.H.; Vriend, G.; Berendsen, H.J.C. The essential dynamics of thermolysin: Confirmation of the hinge-bending motion and comparison of simulations in vacuum and water. Proteins Struct. Funct. Bioinform. 1995, 22, 45–54. [Google Scholar] [CrossRef]
- Purslow, J.A.; Nguyen, T.T.; Khatiwada, B.; Singh, A.; Venditti, V. N6-methyladenosine binding induces a metal-centered rearrangement that activates the human RNA demethylase Alkbh5. Sci. Adv. 2021, 7, eabi8215. [Google Scholar] [CrossRef]
- Sikkema, K.D.; O’Leary, M.H. Synthesis and study of phosphoenolthiopyruvate. Biochemistry 1988, 27, 1342–1347. [Google Scholar] [CrossRef]
- Walker, S.R.; Cumming, H.; Parker, E.J. Substrate and reaction intermediate mimics as inhibitors of 3-deoxy-d-arabino-heptulosonate 7-phosphate synthase. Org. Biomol. Chem. 2009, 7, 3031–3035. [Google Scholar] [CrossRef]
- Medgen, T.; Scholz, T.; Klein, C.D. Structure–activity relationships of tulipalines, tuliposides, and related compounds as inhibitors of MurA. Bioorg. Med. Chem. Lett. 2010, 20, 5757–5762. [Google Scholar] [CrossRef] [PubMed]
- Dotas, R.R.; Venditti, V. 1H, 15N, 13C backbone resonance assignment of the C-terminal domain of enzyme I from Thermoanaerobacter tengcongensis. Biomol. NMR Assign. 2018, 12, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Delaglio, F.; Grzesiek, S.; Vuister, G.W.; Zhu, G.; Pfeifer, J.; Bax, A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 1995, 6, 277–293. [Google Scholar] [CrossRef]
- Pervushin, K.; Riek, R.; Wider, G.; Wüthrich, K. Attenuated T2 relaxation by mutual cancellation of dipole–dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc. Natl. Acad. Sci. USA 1997, 94, 12366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulder, F.A.A.; Schipper, D.; Bott, R.; Boelens, R. Altered flexibility in the substrate-binding site of related native and engineered high-alkaline Bacillus subtilisins. J. Mol. Biol. 1999, 292, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Granot, J. Determination of dissociation constants of 1:1 complexes from NMR data. Optimization of the experimental setup by statistical analysis of simulated experiments. J. Magn. Reson. 1983, 55, 216–224. [Google Scholar] [CrossRef]
- Eastman, P.; Swails, J.; Chodera, J.D.; McGibbon, R.T.; Beauchamp, K.A.; Wang, L.-P.; Simmonett, A.C.; Harrigan, M.P.; Stern, C.D.; Wiewiora, R.P.; et al. Pande OpenMM 7: Rapid development of high performance algorithms for molecular dynamics. PLoS Comput Biol. 2017, 13, e1005659. [Google Scholar] [CrossRef] [Green Version]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Tian, C.; Kasavajhala, K.; Belfon, K.A.A.; Raguette, L.; Huang, H.; Migues, A.N.; Bickel, J.; Wang, Y.; Pincay, J.; Wu, Q.; et al. ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution. J. Chem. Theory Comput. 2020, 16, 528–552. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef]
- Mark, P.; Nilsson, L. Structure and Dynamics of the TIP3P, SPC, and SPC/E Water Models at 298 K. J. Phys. Chem. 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Liu, D.C.; Nocedal, J. On the limited memory BFGS method for large scale optimization. Math. Program. 1989, 45, 503–528. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Michaud-Agrawal, N.; Denning, E.; Woolf, T.; Beckstein, O. MDAnalysis: A toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 2011, 32, 2319–2327. [Google Scholar] [CrossRef]
- Gowers, R.J.; Linke, M.; Barnoud, J.; Reddy, T.J.E.; Melo, M.N.; Seyler, S.L.; Domanski, J.; Dotson, D.L.; Buchoux, S.; Kenney, I.M.; et al. MDAnalysis: A Python Package for the Rapid Analysis of Molecular Dynamics Simulations; Los Alamos National Lab.(LANL): Los Alamos, NM, USA, 2019. [Google Scholar]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, A.; Burns, D.; Sedinkin, S.L.; Van Veller, B.; Potoyan, D.A.; Venditti, V. Protein Conformational Dynamics Underlie Selective Recognition of Thermophilic over Mesophilic Enzyme I by a Substrate Analogue. Biomolecules 2023, 13, 160. https://doi.org/10.3390/biom13010160
Singh A, Burns D, Sedinkin SL, Van Veller B, Potoyan DA, Venditti V. Protein Conformational Dynamics Underlie Selective Recognition of Thermophilic over Mesophilic Enzyme I by a Substrate Analogue. Biomolecules. 2023; 13(1):160. https://doi.org/10.3390/biom13010160
Chicago/Turabian StyleSingh, Aayushi, Daniel Burns, Sergey L. Sedinkin, Brett Van Veller, Davit A. Potoyan, and Vincenzo Venditti. 2023. "Protein Conformational Dynamics Underlie Selective Recognition of Thermophilic over Mesophilic Enzyme I by a Substrate Analogue" Biomolecules 13, no. 1: 160. https://doi.org/10.3390/biom13010160