
The Challenges and Prospects of p53-Based Therapies in Ovarian Cancer
Abstract
:1. Introduction
2. Ovarian Cancer Types
3. Origin of Ovarian Cancer
4. Biology and Genetic Landscape of Ovarian Cancer
5. Current Standard of Care for Ovarian Cancer
6. p53 and Cancer
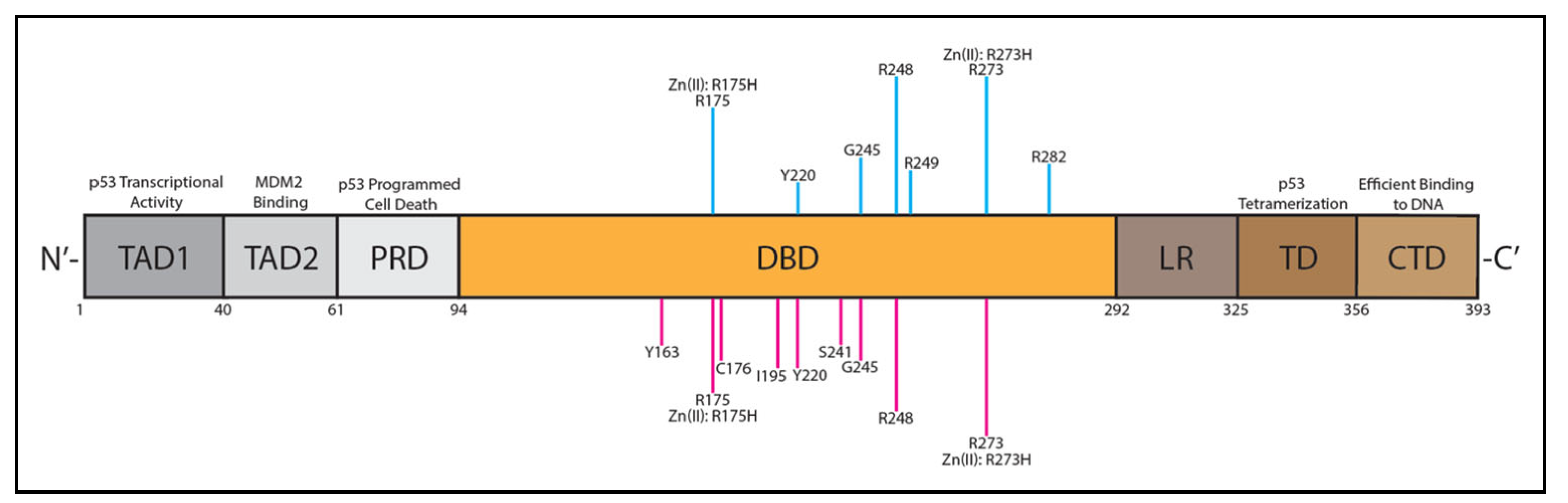
7. p53 Structure and Dysfunction in HGSOC
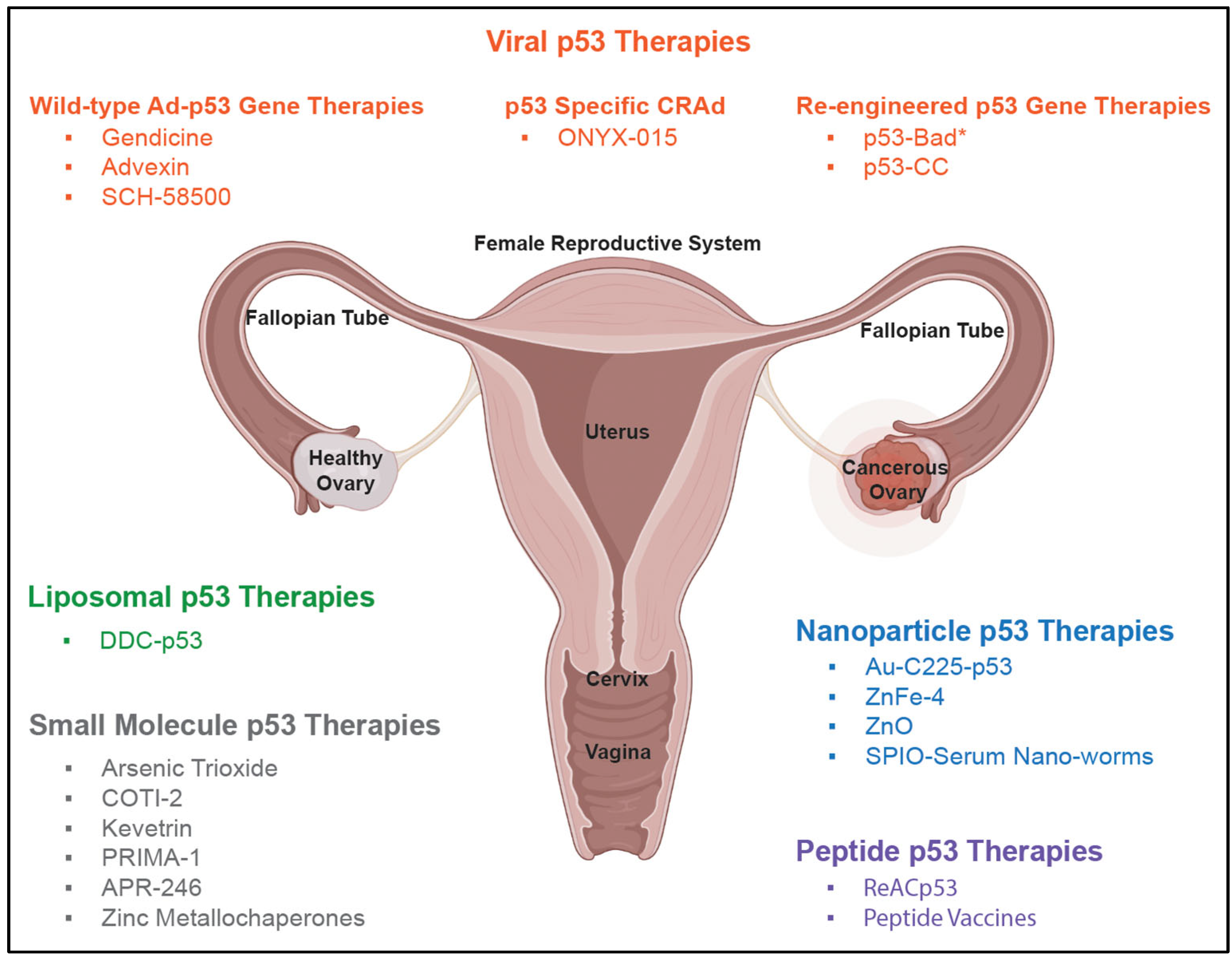
8. Early p53-Based Therapies
8.1. Wild-Type Ad-p53 Therapies: Gendicine, Advexin, and SCH-58500
8.2. Liposomal WT p53 Therapies
8.3. p53-Specific CRAd: ONYX-015
9. The Failure of Early p53-Based Therapies in HGSOC
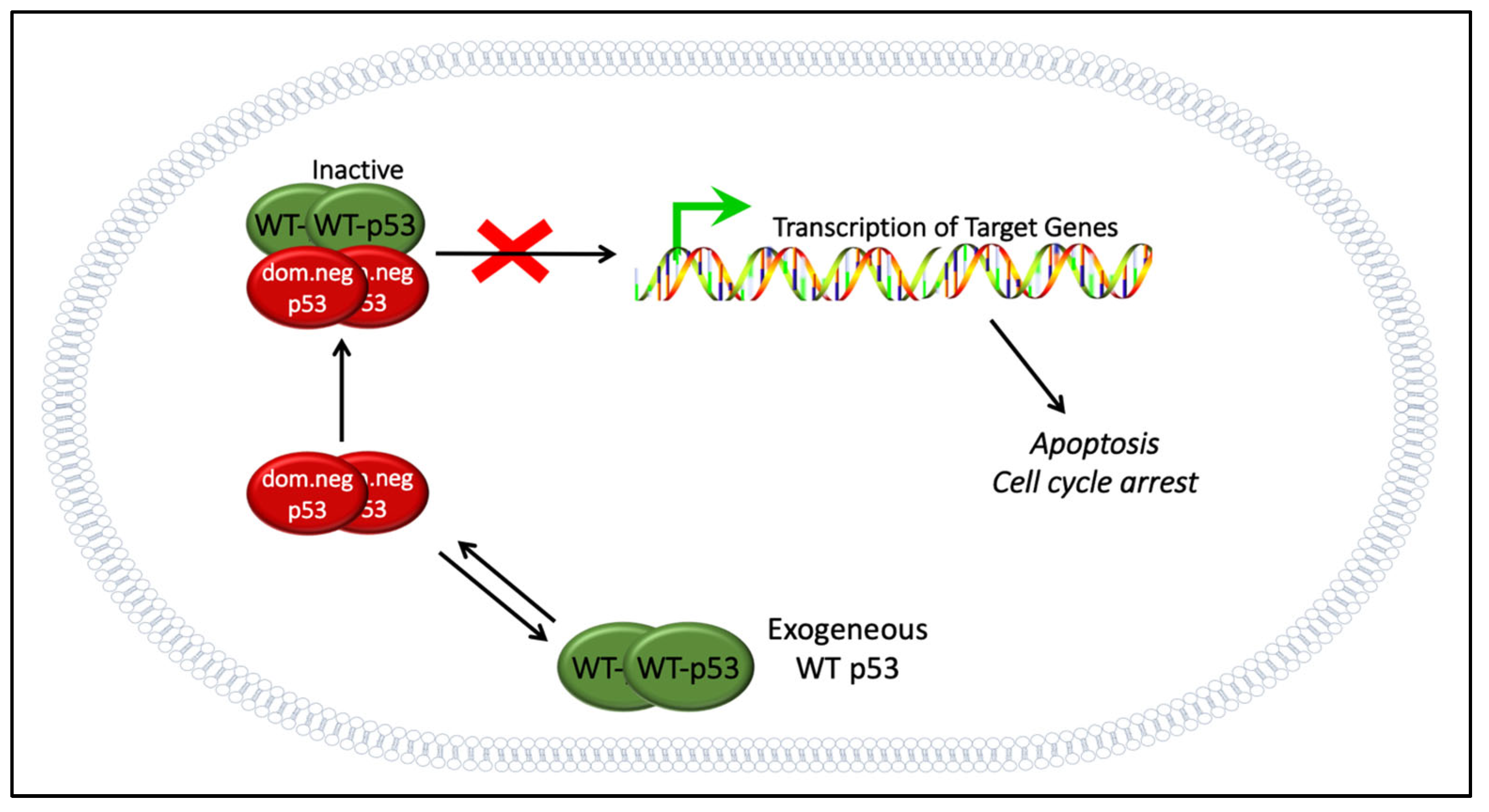
9.1. The Dominant Negative Effect
9.2. Delivery Challenges
9.3. The Potential of WT p53 Gene Therapy
10. Next-Generation p53-Based Therapies
10.1. Next-Generation Viral Therapies
10.2. Re-Engineered p53 Gene Therapies: p53-MTS, p53-Bad*, and p53-CC
10.3. Nanoparticle p53 Therapies
10.4. Peptide p53 Therapies
10.5. Small Molecule p53 Therapies
11. The Future of p53-Based Therapies for HGSOC
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Torre, L.A.; Trabert, B.; Desantis, C.E.; Miller, K.D.; Samimi, G.; Runowicz, C.D.; Gaudet, M.M.; Jemal, A.; Siegel, R.L. Ovarian cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 284–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrenson, K.; Gayther, S.A. Ovarian Cancer: A Clinical Challenge That Needs Some Basic Answers. PLoS Med. 2009, 6, e1000025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corney, D.C.; Flesken-Nikitin, A.; Choi, J.; Nikitin, A.Y. Role of p53 and Rb in Ovarian Cancer; Springer: New York, NY, USA, 2008; pp. 99–117. [Google Scholar]
- Vinyals, A.; Peinado, M.A.; Gonzalez-Garrigues, M.; Monzó, M.; Bonfil, R.D.; Fabra, A. Failure of wild-type p53 gene therapy in human cancer cells expressing a mutant p53 protein. Gene Ther. 1999, 6, 22–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeimet, A.G.; Marth, C. Why did p53 gene therapy fail in ovarian cancer? Lancet Oncol. 2003, 4, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Jayson, G.C.; Kohn, E.C.; Kitchener, H.C.; Ledermann, J.A. Ovarian cancer. Lancet 2014, 384, 1376–1388. [Google Scholar] [CrossRef] [PubMed]
- Bast, R.C.; Hennessy, B.; Mills, G.B. The biology of ovarian cancer: New opportunities for translation. Nat. Rev. Cancer 2009, 9, 415–428. [Google Scholar] [CrossRef]
- Jacob, F.; Nixdorf, S.; Hacker, N.F.; Heinzelmann-Schwarz, V.A. Reliable in vitro studies require appropriate ovarian cancer cell lines. J. Ovarian Res. 2014, 7, 60. [Google Scholar] [CrossRef] [Green Version]
- Sambasivan, S. Epithelial ovarian cancer: Review article. Cancer Treat. Res. Commun. 2022, 33, 100629. [Google Scholar] [CrossRef]
- Matulonis, U.A.; Sood, A.K.; Fallowfield, L.; Howitt, B.E.; Sehouli, J.; Karlan, B.Y. Ovarian cancer. Nat. Rev. Dis. Prim. 2016, 2, 16061. [Google Scholar] [CrossRef]
- Desai, A. Epithelial ovarian cancer: An overview. World J. Transl. Med. 2014, 3, 1. [Google Scholar] [CrossRef]
- Shih, I.-M.; Wang, Y.; Wang, T.-L. The Origin of Ovarian Cancer Species and Precancerous Landscape. Am. J. Pathol. 2021, 191, 26–39. [Google Scholar] [CrossRef] [PubMed]
- Vang, R.; Shih Ie, M.; Kurman, R.J. Ovarian low-grade and high-grade serous carcinoma: Pathogenesis, clinicopathologic and molecular biologic features, and diagnostic problems. Adv. Anat. Pathol. 2009, 16, 267–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero, I.; Sun, C.C.; Wong, K.K.; Bast, R.C.; Gershenson, D.M. Low-grade serous carcinoma: New concepts and emerging therapies. Gynecol. Oncol. 2013, 130, 660–666. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.A.; Etemadmoghadam, D.; Temple, J.; Lynch, A.G.; Riad, M.; Sharma, R.; Stewart, C.; Fereday, S.; Caldas, C.; deFazio, A.; et al. Driver mutations in TP53 are ubiquitous in high grade serous carcinoma of the ovary. J. Pathol. 2010, 221, 49–56. [Google Scholar] [CrossRef] [Green Version]
- Network, T.C.G.A.R. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef] [Green Version]
- Auersperg, N. The stem-cell profile of ovarian surface epithelium is reproduced in the oviductal fimbriae, with increased stem-cell marker density in distal parts of the fimbriae. Int. J. Gynecol. Pathol. 2013, 32, 444–453. [Google Scholar] [CrossRef]
- Zhang, S.; Dolgalev, I.; Zhang, T.; Ran, H.; Levine, D.A.; Neel, B.G. Both fallopian tube and ovarian surface epithelium are cells-of-origin for high-grade serous ovarian carcinoma. Nat. Commun. 2019, 10, 5367. [Google Scholar] [CrossRef] [Green Version]
- Auersperg, N. The origin of ovarian carcinomas: A unifying hypothesis. Int. J. Gynecol. Pathol. 2011, 30, 12–21. [Google Scholar] [CrossRef]
- Lawrenson, K.; Fonseca, M.A.S.; Liu, A.Y.; Segato Dezem, F.; Lee, J.M.; Lin, X.; Corona, R.I.; Abbasi, F.; Vavra, K.C.; Dinh, H.Q.; et al. A Study of High-Grade Serous Ovarian Cancer Origins Implicates the SOX18 Transcription Factor in Tumor Development. Cell Rep. 2019, 29, 3726–3735.e3724. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Padilla, I.; Malpica, A.L.; Minig, L.; Chiva, L.M.; Gershenson, D.M.; Gonzalez-Martin, A. Ovarian low-grade serous carcinoma: A comprehensive update. Gynecol. Oncol. 2012, 126, 279–285. [Google Scholar] [CrossRef]
- Boylan, K.L.M.; Petersen, A.; Starr, T.K.; Pu, X.; Geller, M.A.; Bast, R.C., Jr.; Lu, K.H.; Cavallaro, U.; Connolly, D.C.; Elias, K.M.; et al. Development of a Multiprotein Classifier for the Detection of Early Stage Ovarian Cancer. Cancers 2022, 14, 3077. [Google Scholar] [CrossRef] [PubMed]
- Charkhchi, P.; Cybulski, C.; Gronwald, J.; Wong, F.O.; Narod, S.A.; Akbari, M.R. CA125 and Ovarian Cancer: A Comprehensive Review. Cancers 2020, 12, 3730. [Google Scholar] [CrossRef] [PubMed]
- Huh, S.; Kang, C.; Park, J.E.; Nam, D.; Kim, S.I.; Seol, A.; Choi, K.; Hwang, D.; Yu, M.-H.; Chung, H.H.; et al. Novel Diagnostic Biomarkers for High-Grade Serous Ovarian Cancer Uncovered by Data-Independent Acquisition Mass Spectrometry. J. Proteome Res. 2022, 21, 2146–2159. [Google Scholar] [CrossRef] [PubMed]
- Romero, I.; Bast, R.C., Jr. Minireview: Human ovarian cancer: Biology, current management, and paths to personalizing therapy. Endocrinology 2012, 153, 1593–1602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayano, T.; Yokota, Y.; Hosomichi, K.; Nakaoka, H.; Yoshihara, K.; Adachi, S.; Kashima, K.; Tsuda, H.; Moriya, T.; Tanaka, K.; et al. Molecular Characterization of an Intact p53 Pathway Subtype in High-Grade Serous Ovarian Cancer. PLoS ONE 2014, 9, e114491. [Google Scholar] [CrossRef]
- Phelan, C.M.; Kuchenbaecker, K.B.; Tyrer, J.P.; Kar, S.P.; Lawrenson, K.; Winham, S.J.; Dennis, J.; Pirie, A.; Riggan, M.J.; Chornokur, G.; et al. Identification of 12 new susceptibility loci for different histotypes of epithelial ovarian cancer. Nat. Genet. 2017, 49, 680–691. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.m.; Ledermann, J.A.; Kohn, E.C. PARP Inhibitors for BRCA1/2 mutation-associated and BRCA-like malignancies. Ann. Oncol. 2014, 25, 32–40. [Google Scholar] [CrossRef]
- Guppy, A.E.; Nathan, P.D.; Rustin, G.J. Epithelial ovarian cancer: A review of current management. Clin. Oncol. (R. Coll. Radiol.) 2005, 17, 399–411. [Google Scholar] [CrossRef]
- Garcia, A.; Singh, H. Bevacizumab and ovarian cancer. Ther. Adv. Med. Oncol. 2013, 5, 133–141. [Google Scholar] [CrossRef] [Green Version]
- Lisio, M.A.; Fu, L.; Goyeneche, A.; Gao, Z.H.; Telleria, C. High-Grade Serous Ovarian Cancer: Basic Sciences, Clinical and Therapeutic Standpoints. Int. J. Mol. Sci. 2019, 20, 952. [Google Scholar] [CrossRef]
- Ramraj, S.K.; Elayapillai, S.P.; Pelikan, R.C.; Zhao, Y.D.; Isingizwe, Z.R.; Kennedy, A.L.; Lightfoot, S.A.; Benbrook, D.M. Novel ovarian cancer maintenance therapy targeted at mortalin and mutant p53. Int. J. Cancer 2020, 147, 1086–1097. [Google Scholar] [CrossRef]
- Jiang, X.; Li, X.; Li, W.; Bai, H.; Zhang, Z. PARP inhibitors in ovarian cancer: Sensitivity prediction and resistance mechanisms. J. Cell. Mol. Med. 2019, 23, 2303–2313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neff, R.T.; Senter, L.; Salani, R. BRCA mutation in ovarian cancer: Testing, implications and treatment considerations. Ther. Adv. Med. Oncol. 2017, 9, 519–531. [Google Scholar] [CrossRef] [Green Version]
- Nougaret, S.; Lakhman, Y.; Gönen, M.; Goldman, D.A.; Miccò, M.; D’Anastasi, M.; Johnson, S.A.; Juluru, K.; Arnold, A.G.; Sosa, R.E.; et al. High-Grade Serous Ovarian Cancer: Associations between BRCA Mutation Status, CT Imaging Phenotypes, and Clinical Outcomes. Radiology 2017, 285, 472–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baugh, E.H.; Ke, H.; Levine, A.J.; Bonneau, R.A.; Chan, C.S. Why are there hotspot mutations in the TP53 gene in human cancers? Cell Death Differ. 2018, 25, 154–160. [Google Scholar] [CrossRef]
- Kastan, M.B.; Berkovich, E. p53: A two-faced cancer gene. Nat. Cell Biol. 2007, 9, 489–491. [Google Scholar] [CrossRef]
- Levine, A.J.; Oren, M. The first 30 years of p53: Growing ever more complex. Nat. Rev. Cancer 2009, 9, 749–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wojnarowicz, P.M.; Oros, K.K.; Quinn, M.C.J.; Arcand, S.L.; Gambaro, K.; Madore, J.; Birch, A.H.; de Ladurantaye, M.; Rahimi, K.; Provencher, D.M.; et al. The Genomic Landscape of TP53 and p53 Annotated High Grade Ovarian Serous Carcinomas from a Defined Founder Population Associated with Patient Outcome. PLoS ONE 2012, 7, e45484. [Google Scholar] [CrossRef] [PubMed]
- Laptenko, O.; Prives, C. Transcriptional regulation by p53: One protein, many possibilities. Cell Death Differ. 2006, 13, 951–961. [Google Scholar] [CrossRef] [Green Version]
- Brady, C.A.; Attardi, L.D. p53 at a glance. J. Cell Sci. 2010, 123, 2527. [Google Scholar] [CrossRef]
- Wei, H.; Qu, L.; Dai, S.; Li, Y.; Wang, H.; Feng, Y.; Chen, X.; Jiang, L.; Guo, M.; Li, J.; et al. Structural insight into the molecular mechanism of p53-mediated mitochondrial apoptosis. Nat. Commun. 2021, 12, 2280. [Google Scholar] [CrossRef]
- Leu, J.I.J.; Dumont, P.; Hafey, M.; Murphy, M.E.; George, D.L. Mitochondrial p53 activates Bak and causes disruption of a Bak–Mcl1 complex. Nat. Cell Biol. 2004, 6, 443–450. [Google Scholar] [CrossRef]
- Heyne, K.; Schmitt, K.; Mueller, D.; Armbruester, V.; Mestres, P.; Roemer, K. Resistance of mitochondrial p53 to dominant inhibition. Mol. Cancer 2008, 7, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joerger, A.C.; Fersht, A.R. The tumor suppressor p53: From structures to drug discovery. Cold Spring Harb. Perspect. Biol. 2010, 2, a000919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duffy, M.J.; Synnott, N.C.; O’Grady, S.; Crown, J. Targeting p53 for the treatment of cancer. Semin. Cancer Biol. 2022, 79, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Cole, A.J.; Dwight, T.; Gill, A.J.; Dickson, K.-A.; Zhu, Y.; Clarkson, A.; Gard, G.B.; Maidens, J.; Valmadre, S.; Clifton-Bligh, R.; et al. Assessing mutant p53 in primary high-grade serous ovarian cancer using immunohistochemistry and massively parallel sequencing. Sci. Rep. 2016, 6, 26191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, G.; Pan, C.; Bei, J.-X.; Li, B.; Liang, C.; Xu, Y.; Fu, X. Mutant p53 in Cancer Progression and Targeted Therapies. Front. Oncol. 2020, 10, 595187. [Google Scholar] [CrossRef]
- Tuna, M.; Ju, Z.; Yoshihara, K.; Amos, C.I.; Tanyi, J.L.; Mills, G.B. Clinical relevance of TP53 hotspot mutations in high-grade serous ovarian cancers. Br. J. Cancer 2020, 122, 405–412. [Google Scholar] [CrossRef]
- Kamaraj, B.; Bogaerts, A. Structure and Function of p53-DNA Complexes with Inactivation and Rescue Mutations: A Molecular Dynamics Simulation Study. PLoS ONE 2015, 10, e0134638. [Google Scholar] [CrossRef]
- Flemming, A. Mutant p53 rescued by aggregation inhibitor. Nat. Rev. Drug Discov. 2016, 15, 85. [Google Scholar] [CrossRef]
- Matissek, K.J.; Okal, A.; Mossalam, M.; Lim, C.S. Delivery of a monomeric p53 subdomain with mitochondrial targeting signals from pro-apoptotic Bak or Bax. Pharm. Res. 2014, 31, 2503–2515. [Google Scholar] [CrossRef] [Green Version]
- Koo, N.; Sharma, A.K.; Narayan, S. Therapeutics Targeting p53-MDM2 Interaction to Induce Cancer Cell Death. Int. J. Mol. Sci. 2022, 23, 5005. [Google Scholar] [CrossRef]
- Okal, A.; Cornillie, S.; Matissek, S.J.; Matissek, K.J.; Cheatham, T.E.; Lim, C.S. Re-Engineered p53 Chimera with Enhanced Homo-Oligomerization That Maintains Tumor Suppressor Activity. Mol. Pharm. 2014, 11, 2442–2452. [Google Scholar] [CrossRef] [PubMed]
- Okal, A.; Matissek, K.J.; Matissek, S.J.; Price, R.; Salama, M.E.; Janát-Amsbury, M.M.; Lim, C.S. Re-engineered p53 Activates Apoptosis In Vivo and Causes Primary Tumor Regression in A Dominant Negative Breast Cancer Xenograft Model. Gene Ther. 2014, 21, 903–912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Astanehe, A.; Arenillas, D.; Wasserman, W.W.; Leung, P.C.K.; Dunn, S.E.; Davies, B.R.; Mills, G.B.; Auersperg, N. Mechanisms underlying p53 regulation of PIK3CA transcription in ovarian surface epithelium and in ovarian cancer. J. Cell Sci. 2008, 121, 664–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toledo, F.; Wahl, G.M. MDM2 and MDM4: p53 regulators as targets in anticancer therapy. Int. J. Biochem. Cell Biol. 2007, 39, 1476–1482. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Cavazza, E.; Barlier, C.; Salleron, J.; Filhine-Tresarrieu, P.; Gavoilles, C.; Merlin, J.L.; Harlé, A. Beside P53 and PTEN: Identification of molecular alterations of the RAS/MAPK and PI3K/AKT signaling pathways in high-grade serous ovarian carcinomas to determine potential novel therapeutic targets. Oncol. Lett. 2016, 12, 3264–3272. [Google Scholar] [CrossRef] [Green Version]
- Hu, L.; Zaloudek, C.; Mills, G.B.; Gray, J.; Jaffe, R.B. In vivo and in vitro ovarian carcinoma growth inhibition by a phosphatidylinositol 3-kinase inhibitor (LY294002). Clin. Cancer Res. 2000, 6, 880–886. [Google Scholar]
- Buller, R.E.; Runnebaum, I.B.; Karlan, B.Y.; Horowitz, J.A.; Shahin, M.; Buekers, T.; Petrauskas, S.; Kreienberg, R.; Slamon, D.; Pegram, M. A phase I/II trial of rAd/p53 (SCH 58500) gene replacement in recurrent ovarian cancer. Cancer Gene Ther. 2002, 9, 553–566. [Google Scholar] [CrossRef]
- Buller, R.E.; Shahin, M.S.; Horowitz, J.A.; Runnebaum, I.B.; Mahavni, V.; Petrauskas, S.; Kreienberg, R.; Karlan, B.; Slamon, D.; Pegram, M. Long term follow-up of patients with recurrent ovarian cancer after Ad p53 gene replacement with SCH 58500. Cancer Gene Ther. 2002, 9, 567–572. [Google Scholar] [CrossRef]
- Faramarzi, L.; Dadashpour, M.; Sadeghzadeh, H.; Mahdavi, M.; Zarghami, N. Enhanced anti-proliferative and pro-apoptotic effects of metformin encapsulated PLGA-PEG nanoparticles on SKOV3 human ovarian carcinoma cells. Artif. Cells Nanomed. Biotechnol. 2019, 47, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Fang, Z.; Zhang, M.; Yang, D.; Wang, S.; Liu, K. A Co-Delivery System of Curcumin and p53 for Enhancing the Sensitivity of Drug-Resistant Ovarian Cancer Cells to Cisplatin. Molecules 2020, 25, 2621. [Google Scholar] [CrossRef] [PubMed]
- Gurnani, M.; Lipari, P.; Dell, J.; Shi, B.; Nielsen, L.L. Adenovirus-mediated p53 gene therapy has greater efficacy when combined with chemotherapy against human head and neck, ovarian, prostate, and breast cancer. Cancer Chemother. Pharmacol. 1999, 44, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Gurunathan, S.; Zhang, X.-F. Combination of salinomycin and silver nanoparticles enhances apoptosis and autophagy in human ovarian cancer cells: An effective anticancer therapy. Int. J. Nanomed. 2016, 11, 3655–3675. [Google Scholar] [CrossRef] [Green Version]
- Isayeva, T.; Ren, C.; Ponnazhagan, S. Intraperitoneal gene therapy by rAAV provides long-term survival against epithelial ovarian cancer independently of survivin pathway. Gene Ther. 2007, 14, 138–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanamori, Y.; Kigawa, J.; Minagawa, Y.; Irie, T.; Oishi, T.; Shimada, M.; Takahashi, M.; Nakamura, T.; Sato, K.; Terakawa, N. A newly developed adenovirus-mediated transfer of a wild-type p53 gene increases sensitivity to cis-diamminedichloroplatinum (II) in p53-deleted ovarian cancer cells. Eur. J. Cancer 1998, 34, 1802–1806. [Google Scholar] [CrossRef]
- Kareem, S.H.; Naji, A.M.; Taqi, Z.J.; Jabir, M.S. Polyvinylpyrrolidone Loaded-MnZnFe2O4 Magnetic Nanocomposites Induce Apoptosis in Cancer Cells Through Mitochondrial Damage and P53 Pathway. J. Inorg. Organomet. Polym. Mater. 2020, 30, 5009–5023. [Google Scholar] [CrossRef]
- Kigawa, J.; Terakawa, N. Adenovirus-mediated Transfer of a p53 Gene in Ovarian Cancer. In Cancer Gene Therapy: Past Achievements and Future Challenges; Habib, N.A., Ed.; Springer: New York, NY, USA, 2002; pp. 207–214. [Google Scholar]
- Kim, C.-K.; Choi, E.-J.; Choi, S.-H.; Park, J.-S.; Haider, K.H.; Ahn, W.S. Enhanced p53 gene transfer to human ovarian cancer cells using the cationic nonviral vector, DDC. Gynecol. Oncol. 2003, 90, 265–272. [Google Scholar] [CrossRef]
- Kim, J.; Hwang, E.S.; Kim, J.S.; You, E.H.; Lee, S.H.; Lee, J.H. Intraperitoneal gene therapy with adenoviral-mediated p53 tumor suppressor gene for ovarian cancer model in nude mouse. Cancer Gene Ther. 1999, 6, 172–178. [Google Scholar] [CrossRef] [Green Version]
- Koch, M.; Krieger, M.L.; Stölting, D.; Brenner, N.; Beier, M.; Jaehde, U.; Wiese, M.; Royer, H.-D.; Bendas, G. Overcoming chemotherapy resistance of ovarian cancer cells by liposomal cisplatin: Molecular mechanisms unveiled by gene expression profiling. Biochem. Pharmacol. 2013, 85, 1077–1090. [Google Scholar] [CrossRef]
- Kotcherlakota, R.; Vydiam, K.; Jeyalakshmi Srinivasan, D.; Mukherjee, S.; Roy, A.; Kuncha, M.; Rao, T.N.; Sistla, R.; Gopal, V.; Patra, C.R. Restoration of p53 Function in Ovarian Cancer Mediated by Gold Nanoparticle-Based EGFR Targeted Gene Delivery System. ACS Biomater. Sci. Eng. 2019, 5, 3631–3644. [Google Scholar] [CrossRef] [PubMed]
- Leffers, N.; Lambeck, A.J.A.; Gooden, M.J.M.; Hoogeboom, B.N.; Wolf, R.; Hamming, I.E.; Hepkema, B.G.; Willemse, P.H.B.; Molmans, B.H.W.; Hollema, H.; et al. Immunization with a P53 synthetic long peptide vaccine induces P53-specific immune responses in ovarian cancer patients, a phase II trial. Int. J. Cancer 2009, 125, 2104–2113. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, A.; Kaushik, M.; Niranjan, R.; Richards, J.S.; Ebright, B.; Venkatasubbu, G.D. Zinc oxide nanoparticles induce oxidative and proteotoxic stress in ovarian cancer cells and trigger apoptosis independent of p53-mutation status. Appl. Surf. Sci. 2019, 487, 807–818. [Google Scholar] [CrossRef]
- Qian, J.; Zhang, W.; Wei, P.; Yao, G.; Yi, T.; Zhang, H.; Ding, H.; Huang, X.; Wang, M.; Song, Y.; et al. Enhancing Chemotherapy of p53-Mutated Cancer through Ubiquitination-Dependent Proteasomal Degradation of Mutant p53 Proteins by Engineered ZnFe-4 Nanoparticles. Adv. Funct. Mater. 2020, 30, 2001994. [Google Scholar] [CrossRef]
- Ramezani, T.; Nabiuni, M.; Baharara, J.; Parivar, K.; Namvar, F. Sensitization of Resistance Ovarian Cancer Cells to Cisplatin by Biogenic Synthesized Silver Nanoparticles through p53 Activation. Iran. J. Pharm Res. 2019, 18, 222–231. [Google Scholar]
- Santoso, J.T.; Tang, D.-C.; Lane, S.B.; Hung, J.; Reed, D.J.; Muller, C.Y.; Carbone, D.P.; Lucciiii, J.A.; Scottmiller, D.; Mathis, M.J. Adenovirus-Based p53 Gene Therapy in Ovarian Cancer. Gynecol. Oncol. 1995, 59, 171–178. [Google Scholar] [CrossRef]
- Soragni, A.; Janzen, D.M.; Johnson, L.M.; Lindgren, A.G.; Nguyen, A.T.-Q.; Tiourin, E.; Soriaga, A.B.; Lu, J.; Jiang, L.; Faull, K.F.; et al. A Designed Inhibitor of p53 Aggregation Rescues p53 Tumor Suppression in Ovarian Carcinomas. Cancer Cell 2016, 29, 90–103. [Google Scholar] [CrossRef] [Green Version]
- STÖLTING, D.P.; BORRMANN, M.; KOCH, M.; WIESE, M.; ROYER, H.-D.; BENDAS, G. How Liposomal Cisplatin Overcomes Chemoresistance in Ovarian Tumour Cells. Anticancer Res. 2014, 34, 525–530. [Google Scholar]
- Drakopoulou, E.; Anagnou, N.P.; Pappa, K.I. Gene Therapy for Malignant and Benign Gynaecological Disorders: A Systematic Review of an Emerging Success Story. Cancers 2022, 14, 3238. [Google Scholar] [CrossRef]
- Zhang, W.-W.; Li, L.; Li, D.; Liu, J.; Li, X.; Li, W.; Xu, X.; Zhang, M.J.; Chandler, L.A.; Lin, H.; et al. The First Approved Gene Therapy Product for Cancer Ad-p53 (Gendicine): 12 Years in the Clinic. Hum. Gene Ther. 2018, 29, 160–179. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Guo, W.; Li, X.; Zhang, J.; Sun, M.; Tang, Z.; Ran, W.; Yang, K.; Huang, G.; Li, L. Expert consensus on the clinical application of recombinant adenovirus human p53 for head and neck cancers. Int. J. Oral Sci. 2021, 13, 38. [Google Scholar] [CrossRef]
- Sobol, R.E.; Menander, K.B.; Chada, S.; Wiederhold, D.; Sellman, B.; Talbott, M.; Nemunaitis, J.J. Analysis of Adenoviral p53 Gene Therapy Clinical Trials in Recurrent Head and Neck Squamous Cell Carcinoma. Front. Oncol. 2021, 11, 1223. [Google Scholar] [CrossRef]
- Tazawa, H.; Kagawa, S.; Fujiwara, T. Advances in adenovirus-mediated p53 cancer gene therapy. Expert Opin. Biol. Ther. 2013, 13, 1569–1583. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z. Current status of gendicine in China: Recombinant human Ad-p53 agent for treatment of cancers. Hum. Gene Ther. 2005, 16, 1016–1027. [Google Scholar] [CrossRef] [PubMed]
- Ma, G.; Shimada, H.; Hiroshima, K.; Tada, Y.; Suzuki, N.; Tagawa, M. Gene medicine for cancer treatment: Commercially available medicine and accumulated clinical data in China. Drug Des. Devel. Ther. 2009, 2, 115–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrissey, R.E.; Horvath, C.; Snyder, E.A.; Patrick, J.; Collins, N.; Evans, E.; MacDonald, J.S. Porcine toxicology studies of SCH 58500, an adenoviral vector for the p53 gene. Toxicol. Sci. 2002, 65, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Áyen, Á.; Jiménez Martínez, Y.; Marchal, J.A.; Boulaiz, H. Recent Progress in Gene Therapy for Ovarian Cancer. Int. J. Mol. Sci. 2018, 19, 1930. [Google Scholar] [CrossRef] [Green Version]
- INGN 201: Ad-p53, Ad5CMV-p53, Adenoviral p53, INGN 101, p53 gene therapy--Introgen, RPR/INGN 201. Biodrugs 2003, 17, 216–222. [CrossRef]
- Wolf, J.K.; Bodurka, D.C.; Gano, J.B.; Deavers, M.; Ramondetta, L.; Ramirez, P.T.; Levenback, C.; Gershenson, D.M. A phase I study of Adp53 (INGN 201; ADVEXIN) for patients with platinum- and paclitaxel-resistant epithelial ovarian cancer. Gynecol. Oncol. 2004, 94, 442–448. [Google Scholar] [CrossRef]
- Samad, A.; Sultana, Y.; Aqil, M. Liposomal Drug Delivery Systems: An Update Review. Curr. Drug Deliv. 2007, 4, 297–305. [Google Scholar] [CrossRef]
- Hortobagyi, G.N.; Ueno, N.T.; Xia, W.; Zhang, S.; Wolf, J.K.; Putnam, J.B.; Weiden, P.L.; Willey, J.S.; Carey, M.; Branham, D.L.; et al. Cationic Liposome-Mediated E1A Gene Transfer to Human Breast and Ovarian Cancer Cells and Its Biologic Effects: A Phase I Clinical Trial. J. Clin. Oncol. 2001, 19, 3422–3433. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Ahn, H.J. PEGylated DC-Chol/DOPE cationic liposomes containing KSP siRNA as a systemic siRNA delivery Carrier for ovarian cancer therapy. Biochem. Biophys. Res. Commun. 2018, 503, 1716–1722. [Google Scholar] [CrossRef] [PubMed]
- Ries, S.; Korn, W.M. ONYX-015: Mechanisms of action and clinical potential of a replication-selective adenovirus. Br. J. Cancer 2002, 86, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Pastor, R.; Goedegebuure, P.S.; Curiel, D.T. Understanding and addressing barriers to successful adenovirus-based virotherapy for ovarian cancer. Cancer Gene Ther. 2021, 28, 375–389. [Google Scholar] [CrossRef]
- Nemunaitis, J.; Ganly, I.; Khuri, F.; Arseneau, J.; Kuhn, J.; McCarty, T.; Landers, S.; Maples, P.; Romel, L.; Randlev, B.; et al. Selective replication and oncolysis in p53 mutant tumors with ONYX-015, an E1B-55kD gene-deleted adenovirus, in patients with advanced head and neck cancer: A phase II trial. Cancer Res. 2000, 60, 6359–6366. [Google Scholar]
- Bischoff, J.R.; Kirn, D.H.; Williams, A.; Heise, C.; Horn, S.; Muna, M.; Ng, L.; Nye, J.A.; Sampson-Johannes, A.; Fattaey, A.; et al. An Adenovirus Mutant That Replicates Selectively in p53- Deficient Human Tumor Cells. Science 1996, 274, 373. [Google Scholar] [CrossRef]
- Rothmann, T.; Hengstermann, A.; Whitaker, N.J.; Scheffner, M.; Hausen, H.z. Replication of ONYX-015, a Potential Anticancer Adenovirus, Is Independent of p53 Status in Tumor Cells. J. Virol. 1998, 72, 9470–9478. [Google Scholar] [CrossRef] [Green Version]
- Cook, M.; Chauhan, A. Clinical Application of Oncolytic Viruses: A Systematic Review. Int. J. Mol. Sci. 2020, 21, 7505. [Google Scholar] [CrossRef]
- Reid, T.R.; Freeman, S.; Post, L.; McCormick, F.; Sze, D.Y. Effects of Onyx-015 among metastatic colorectal cancer patients that have failed prior treatment with 5-FU/leucovorin. Cancer Gene Ther. 2005, 12, 673–681. [Google Scholar] [CrossRef]
- Vasey, P.A.; Shulman, L.N.; Campos, S.; Davis, J.; Gore, M.; Johnston, S.; Kirn, D.H.; O’Neill, V.; Siddiqui, N.; Seiden, M.V.; et al. Phase I trial of intraperitoneal injection of the E1B-55-kd-gene-deleted adenovirus ONYX-015 (dl1520) given on days 1 through 5 every 3 weeks in patients with recurrent/refractory epithelial ovarian cancer. J. Clin. Oncol. 2002, 20, 1562–1569. [Google Scholar] [CrossRef]
- Chen, G.X.; Zhang, S.; He, X.H.; Liu, S.Y.; Ma, C.; Zou, X.P. Clinical utility of recombinant adenoviral human p53 gene therapy: Current perspectives. Onco Targets Ther. 2014, 7, 1901–1909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, P.; Redd Bowman, K.E.; Brown, S.M.; Joklik-Mcleod, M.; Vander Mause, E.R.; Nguyen, H.T.N.; Lim, C.S. p53-Bad: A novel tumor suppressor/proapoptotic factor hybrid directed to the mitochondria for ovarian cancer gene therapy. Mol. Pharm. 2019, 16, 3386–3398. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Vander Mause, E.R.; Redd Bowman, K.E.; Brown, S.M.; Ahne, L.; Lim, C.S. Mitochondrially targeted p53 or DBD subdomain is superior to wild type p53 in ovarian cancer cells even with strong dominant negative mutant p53.(Report). J. Ovarian Res. 2019, 12, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Mossalam, M.; Matissek, K.J.; Okal, A.; Constance, J.E.; Lim, C.S. Direct induction of apoptosis using an optimal mitochondrially targeted p53. Mol. Pharm. 2012, 9, 1449–1458. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Liu, Z.; Myers, J.N. TP53 Mutations in Head and Neck Squamous Cell Carcinoma and Their Impact on Disease Progression and Treatment Response. J. Cell. Biochem. 2016, 117, 2682–2692. [Google Scholar] [CrossRef] [Green Version]
- Bouvet, M.; Bold, R.J.; Lee, J.; Evans, D.B.; Abbruzzese, J.L.; Chiao, P.J.; McConkey, D.J.; Chandra, J.; Chada, S.; Fang, B.; et al. Adenovirus-mediated wild-type p53 tumor suppressor gene therapy induces apoptosis and suppresses growth of human pancreatic cancer [seecomments]. Ann. Surg. Oncol. 1998, 5, 681–688. [Google Scholar] [CrossRef]
- Spitz, F.R.; Nguyen, D.; Skibber, J.M.; Cusack, J.; Roth, J.A.; Cristiano, R.J. In vivo adenovirus-mediated p53 tumor suppressor gene therapy for colorectal cancer. Anticancer Res. 1996, 16, 3415–3422. [Google Scholar]
- von Gruenigen, V.E.; Santoso, J.T.; Coleman, R.L.; Muller, C.Y.; Miller, D.S.; Mathis, J.M. In vivo studies of adenovirus-based p53 gene therapy for ovarian cancer. Gynecol. Oncol. 1998, 69, 197–204. [Google Scholar] [CrossRef]
- Choi, J.W.; Lee, Y.S.; Yun, C.O.; Kim, S.W. Polymeric oncolytic adenovirus for cancer gene therapy. J. Control. Release 2015, 219, 181–191. [Google Scholar] [CrossRef] [Green Version]
- Parker, A.L.; Waddington, S.N.; Buckley, S.M.; Custers, J.; Havenga, M.J.; van Rooijen, N.; Goudsmit, J.; McVey, J.H.; Nicklin, S.A.; Baker, A.H. Effect of neutralizing sera on factor x-mediated adenovirus serotype 5 gene transfer. J. Virol. 2009, 83, 479–483. [Google Scholar] [CrossRef] [Green Version]
- Stallwood, Y.; Fisher, K.D.; Gallimore, P.H.; Mautner, V. Neutralisation of adenovirus infectivity by ascitic fluid from ovarian cancer patients. Gene Ther. 2000, 7, 637–643. [Google Scholar] [CrossRef] [Green Version]
- Nwanegbo, E.; Vardas, E.; Gao, W.; Whittle, H.; Sun, H.; Rowe, D.; Robbins, P.D.; Gambotto, A. Prevalence of neutralizing antibodies to adenoviral serotypes 5 and 35 in the adult populations of The Gambia, South Africa, and the United States. Clin. Diagn. Lab. Immunol. 2004, 11, 351–357. [Google Scholar] [CrossRef] [Green Version]
- Voysey, M.; Clemens, S.A.C.; Madhi, S.A.; Weckx, L.Y.; Folegatti, P.M.; Aley, P.K.; Angus, B.; Baillie, V.L.; Barnabas, S.L.; Bhorat, Q.E.; et al. Safety and efficacy of the ChAdOx1 nCoV-19 vaccine (AZD1222) against SARS-CoV-2: An interim analysis of four randomised controlled trials in Brazil, South Africa, and the UK. Lancet 2021, 397, 99–111. [Google Scholar] [CrossRef]
- Ghosh, S.; Brown, A.M.; Jenkins, C.; Campbell, K. Viral Vector Systems for Gene Therapy: A Comprehensive Literature Review of Progress and Biosafety Challenges. Appl. Biosaf. 2020, 25, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Zegadło, J.S.; Kowalska, M.; Spiewankiewicz, B.; Smiertka, W.; Gawrychowski, K.; Małecki, M. Anti Adeno-Associated Virus 2 (AAV) Antibody Profile in Ovarian Cancer Ascitic Fluid: Implications for AAV Intraperitoneal Gene Therapy. J. Clin. Exp. Oncol. 2017, 6. [Google Scholar] [CrossRef]
- Reeh, M.; Bockhorn, M.; Gorgens, D.; Vieth, M.; Hoffmann, T.; Simon, R.; Izbicki, J.R.; Sauter, G.; Schumacher, U.; Anders, M. Presence of the coxsackievirus and adenovirus receptor (CAR) in human neoplasms: A multitumour array analysis. Br. J. Cancer 2013, 109, 1848–1858. [Google Scholar] [CrossRef] [PubMed]
- You, Z.; Fischer, D.C.; Tong, X.; Hasenburg, A.; Aguilar-Cordova, E.; Kieback, D.G. Coxsackievirus-adenovirus receptor expression in ovarian cancer cell lines is associated with increased adenovirus transduction efficiency and transgene expression. Cancer Gene Ther. 2001, 8, 168–175. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Nam, H.Y.; Choi, J.W.; Yun, C.O.; Kim, S.W. Efficient lung orthotopic tumor-growth suppression of oncolytic adenovirus complexed with RGD-targeted bioreducible polymer. Gene Ther. 2014, 21, 476–483. [Google Scholar] [CrossRef]
- Gamble, L.J.; Borovjagin, A.V.; Matthews, Q.L. Role of RGD-containing ligands in targeting cellular integrins: Applications for ovarian cancer virotherapy (Review). Exp. Ther. Med. 2010, 1, 233–240. [Google Scholar] [CrossRef] [Green Version]
- Hulin-Curtis, S.L.; Davies, J.A.; Nestic, D.; Bates, E.A.; Baker, A.T.; Cunliffe, T.G.; Majhen, D.; Chester, J.D.; Parker, A.L. Identification of folate receptor alpha (FRalpha) binding oligopeptides and their evaluation for targeted virotherapy applications. Cancer Gene Ther. 2020, 27, 785–798. [Google Scholar] [CrossRef] [Green Version]
- Lang, F.F.; Conrad, C.; Gomez-Manzano, C.; Yung, W.K.A.; Sawaya, R.; Weinberg, J.S.; Prabhu, S.S.; Rao, G.; Fuller, G.N.; Aldape, K.D.; et al. Phase I Study of DNX-2401 (Delta-24-RGD) Oncolytic Adenovirus: Replication and Immunotherapeutic Effects in Recurrent Malignant Glioma. J. Clin. Oncol. 2018, 36, 1419–1427. [Google Scholar] [CrossRef]
- Sasaki, H.; Sheng, Y.; Kotsuji, F.; Tsang, B.K. Down-regulation of X-linked inhibitor of apoptosis protein induces apoptosis in chemoresistant human ovarian cancer cells. Cancer Res. 2000, 60, 5659–5666. [Google Scholar]
- Kigawa, J.; Sato, S.; Shimada, M.; Kanamori, Y.; Itamochi, H.; Terakawa, N. Effect of p53 gene transfer and cisplatin in a peritonitis carcinomatosa model with p53-deficient ovarian cancer cells. Gynecol. Oncol. 2002, 84, 210–215. [Google Scholar] [CrossRef]
- Salameh, J.W.; Zhou, L.; Ward, S.M.; Santa Chalarca, C.F.; Emrick, T.; Figueiredo, M.L. Polymer-mediated gene therapy: Recent advances and merging of delivery techniques. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2020, 12, e1598. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Velez, N.; Garcia-Moure, M.; Marigil, M.; Gonzalez-Huarriz, M.; Puigdelloses, M.; Gallego Perez-Larraya, J.; Zalacain, M.; Marrodan, L.; Varela-Guruceaga, M.; Laspidea, V.; et al. The oncolytic virus Delta-24-RGD elicits an antitumor effect in pediatric glioma and DIPG mouse models. Nat. Commun. 2019, 10, 2235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, P.H.; Kim, J.; Kim, T.I.; Nam, H.Y.; Yockman, J.W.; Kim, M.; Kim, S.W.; Yun, C.O. Bioreducible polymer-conjugated oncolytic adenovirus for hepatoma-specific therapy via systemic administration. Biomaterials 2011, 32, 9328–9342. [Google Scholar] [CrossRef]
- Lockley, M.; Fernandez, M.; Wang, Y.; Li, N.F.; Conroy, S.; Lemoine, N.; McNeish, I. Activity of the adenoviral E1A deletion mutant dl922-947 in ovarian cancer: Comparison with E1A wild-type viruses, bioluminescence monitoring, and intraperitoneal delivery in icodextrin. Cancer Res. 2006, 66, 989–998. [Google Scholar] [CrossRef] [Green Version]
- Kimball, K.J.; Preuss, M.A.; Barnes, M.N.; Wang, M.; Siegal, G.P.; Wan, W.; Kuo, H.; Saddekni, S.; Stockard, C.R.; Grizzle, W.E.; et al. A phase I study of a tropism-modified conditionally replicative adenovirus for recurrent malignant gynecologic diseases. Clin. Cancer Res. 2010, 16, 5277–5287. [Google Scholar] [CrossRef] [Green Version]
- Bai, D.P.; Zhang, X.F.; Zhang, G.L.; Huang, Y.F.; Gurunathan, S. Zinc oxide nanoparticles induce apoptosis and autophagy in human ovarian cancer cells. Int. J. Nanomed. 2017, 12, 6521–6535. [Google Scholar] [CrossRef] [Green Version]
- Divita, G.; Czuba, E.; Grunenberger, A.; Guidetti, M.; Josserand, V.; Desai, N. p53 mRNA rescue of tumor suppressor function prevents tumor growth and restores PARPi sensitivity in p53-deficient cancers in vitro and in vivo. Eur. J. Cancer 2022, 174, S21–S22. [Google Scholar] [CrossRef]
- Xu, J.; Reumers, J.; Couceiro, J.R.; De Smet, F.; Gallardo, R.; Rudyak, S.; Cornelis, A.; Rozenski, J.; Zwolinska, A.; Marine, J.-C.; et al. Gain of function of mutant p53 by coaggregation with multiple tumor suppressors. Nat. Chem. Biol. 2011, 7, 285–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leffers, N.; Vermeij, R.; Hoogeboom, B.-N.; Schulze, U.R.; Wolf, R.; Hamming, I.E.; van der Zee, A.G.; Melief, K.J.; van der Burg, S.H.; Daemen, T.; et al. Long-term clinical and immunological effects of p53-SLP® vaccine in patients with ovarian cancer. Int. J. Cancer 2012, 130, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Fan, C.; Zeng, Z.; Young, K.H.; Li, Y. Clinical and Immunological Effects of p53-Targeting Vaccines. Front. Cell Dev. Biol. 2021, 9, 762796. [Google Scholar] [CrossRef] [PubMed]
- Vermeij, R.; Leffers, N.; Hoogeboom, B.-N.; Hamming, I.L.E.; Wolf, R.; Reyners, A.K.L.; Molmans, B.H.W.; Hollema, H.; Bart, J.; Drijfhout, J.W.; et al. Potentiation of a p53-SLP vaccine by cyclophosphamide in ovarian cancer: A single-arm phase II study. Int. J. Cancer 2012, 131, E670–E680. [Google Scholar] [CrossRef] [PubMed]
- Dijkgraaf, E.M.; Santegoets, S.J.; Reyners, A.K.; Goedemans, R.; Nijman, H.W.; van Poelgeest, M.I.; van Erkel, A.R.; Smit, V.T.; Daemen, T.A.; van der Hoeven, J.J.; et al. A phase 1/2 study combining gemcitabine, Pegintron and p53 SLP vaccine in patients with platinum-resistant ovarian cancer. Oncotarget 2015, 6, 32228–32243. [Google Scholar] [CrossRef] [Green Version]
- Rahma, O.E.; Ashtar, E.; Czystowska, M.; Szajnik, M.E.; Wieckowski, E.; Bernstein, S.; Herrin, V.E.; Shams, M.A.; Steinberg, S.M.; Merino, M.; et al. A gynecologic oncology group phase II trial of two p53 peptide vaccine approaches: Subcutaneous injection and intravenous pulsed dendritic cells in high recurrence risk ovarian cancer patients. Cancer Immunol. Immunother. 2012, 61, 373–384. [Google Scholar] [CrossRef]
- Chen, S.; Wu, J.-L.; Liang, Y.; Tang, Y.-G.; Song, H.-X.; Wu, L.-L.; Xing, Y.-F.; Yan, N.; Li, Y.-T.; Wang, Z.-Y.; et al. Arsenic Trioxide Rescues Structural p53 Mutations through a Cryptic Allosteric Site. Cancer Cell 2021, 39, 225–239.e8. [Google Scholar] [CrossRef]
- Byun, J.M.; Lee, D.S.; Landen, C.N.; Kim, D.H.; Kim, Y.N.; Lee, K.B.; Sung, M.S.; Park, S.G.; Jeong, D.H. Arsenic trioxide and tetraarsenic oxide induce cytotoxicity and have a synergistic effect with cisplatin in paclitaxel-resistant ovarian cancer cells. Acta Oncol. 2019, 58, 1594–1602. [Google Scholar] [CrossRef]
- Tang, S.; Shen, Y.; Wei, X.; Shen, Z.; Lu, W.; Xu, J. Olaparib synergizes with arsenic trioxide by promoting apoptosis and ferroptosis in platinum-resistant ovarian cancer. Cell Death Dis. 2022, 13, 826. [Google Scholar] [CrossRef]
- Yang, Y.; Li, X.; Wang, Y.; Shen, X.; Zhao, L.; Wu, Y.; Li, Y.; Wang, J.; Wei, L. Application of Arsenic Trioxide-Based Combined Sequential Chemotherapy in Recurrent Resistant and Refractory Ovarian Cancers: A Single-Center, Open Phase II Clinical Study. J. Oncol. 2022, 2022, 6243165. [Google Scholar] [CrossRef]
- Synnott, N.C.; O’Connell, D.; Crown, J.; Duffy, M.J. COTI-2 reactivates mutant p53 and inhibits growth of triple-negative breast cancer cells. Breast Cancer Res. Treat. 2020, 179, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Lindemann, A.; Patel, A.A.; Silver, N.L.; Tang, L.; Liu, Z.; Wang, L.; Tanaka, N.; Rao, X.; Takahashi, H.; Maduka, N.K.; et al. COTI-2, A Novel Thiosemicarbazone Derivative, Exhibits Antitumor Activity in HNSCC through p53-dependent and -independent Mechanisms. Clin. Cancer Res. 2019, 25, 5650–5662. [Google Scholar] [CrossRef] [Green Version]
- Pósa, V.; Stefanelli, A.; Nunes, J.H.B.; Hager, S.; Mathuber, M.; May, N.V.; Berger, W.; Keppler, B.K.; Kowol, C.R.; Enyedy, É.A.; et al. Thiosemicarbazone Derivatives Developed to Overcome COTI-2 Resistance. Cancers 2022, 14, 4455. [Google Scholar] [CrossRef] [PubMed]
- Bormio Nunes, J.H.; Hager, S.; Mathuber, M.; Pósa, V.; Roller, A.; Enyedy, É.A.; Stefanelli, A.; Berger, W.; Keppler, B.K.; Heffeter, P.; et al. Cancer Cell Resistance Against the Clinically Investigated Thiosemicarbazone COTI-2 Is Based on Formation of Intracellular Copper Complex Glutathione Adducts and ABCC1-Mediated Efflux. J. Med. Chem. 2020, 63, 13719–13732. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, R.; De Matteis, S.; Carloni, S.; Bruno, S.; Abbati, G.; Capelli, L.; Ghetti, M.; Bochicchio, M.T.; Liverani, C.; Mercatali, L.; et al. Kevetrin induces apoptosis in TP53 wild-type and mutant acute myeloid leukemia cells. Oncol. Rep. 2020, 44, 1561–1573. [Google Scholar] [CrossRef]
- Rangel, L.P.; Ferretti, G.D.S.; Costa, C.L.; Andrade, S.; Carvalho, R.S.; Costa, D.C.F.; Silva, J.L. p53 reactivation with induction of massive apoptosis-1 (PRIMA-1) inhibits amyloid aggregation of mutant p53 in cancer cells. J. Biol. Chem. 2019, 294, 3670–3682. [Google Scholar] [CrossRef]
- Kobayashi, N.; Abedini, M.; Sakuragi, N.; Tsang, B.K. PRIMA-1 increases cisplatin sensitivity in chemoresistant ovarian cancer cells with p53 mutation: A requirement for Akt down-regulation. J. Ovarian Res. 2013, 6, 7. [Google Scholar] [CrossRef] [Green Version]
- Kukut Hatipoglu, M.; Mahjabeen, S.; Garcia-Contreras, L. Development and validation of a reverse phase HPLC method for SHetA2, a novel anti-cancer drug, in mouse biological samples. J. Pharm. Biomed. Anal. 2019, 170, 124–131. [Google Scholar] [CrossRef]
- Benbrook, D.M.; Nammalwar, B.; Long, A.; Matsumoto, H.; Singh, A.; Bunce, R.A.; Berlin, K.D. SHetA2 interference with mortalin binding to p66shc and p53 identified using drug-conjugated magnetic microspheres. Investig. New Drugs 2014, 32, 412–423. [Google Scholar] [CrossRef] [Green Version]
- Yoshikawa, N.; Kajiyama, H.; Nakamura, K.; Utsumi, F.; Niimi, K.; Mitsui, H.; Sekiya, R.; Suzuki, S.; Shibata, K.; Callen, D.; et al. PRIMA-1MET induces apoptosis through accumulation of intracellular reactive oxygen species irrespective of p53 status and chemo-sensitivity in epithelial ovarian cancer cells. Oncol. Rep. 2016, 35, 2543–2552. [Google Scholar] [CrossRef] [Green Version]
- Ha, J.H.; Prela, O.; Carpizo, D.R.; Loh, S.N. p53 and Zinc: A Malleable Relationship. Front. Mol. Biosci. 2022, 9, 895887. [Google Scholar] [CrossRef]
- Cvrljevic, A.N.; Butt, U.; Huhtinen, K.; Grönroos, T.J.; Böckelman, C.; Lassus, H.; Butzow, R.; Haglund, C.; Kaipio, K.; Arsiola, T.; et al. Ovarian Cancers with Low CIP2A Tumor Expression Constitute an APR-246–Sensitive Disease Subtype. Mol. Cancer Ther. 2022, 21, 1236–1245. [Google Scholar] [CrossRef]
- Elayapillai, S.; Ramraj, S.; Benbrook, D.M.; Bieniasz, M.; Wang, L.; Pathuri, G.; Isingizwe, Z.R.; Kennedy, A.L.; Zhao, Y.D.; Lightfoot, S.; et al. Potential and mechanism of mebendazole for treatment and maintenance of ovarian cancer. Gynecol. Oncol. 2021, 160, 302–311. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Kogan, S.; Chen, Y.; Tsang, A.T.; Withers, T.; Lin, H.; Gilleran, J.; Buckley, B.; Moore, D.; Bertino, J.; et al. Zinc Metallochaperones Reactivate Mutant p53 Using an ON/OFF Switch Mechanism: A New Paradigm in Cancer Therapeutics. Clin. Cancer Res. 2018, 24, 4505–4517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kogan, S.; Carpizo, D.R. Zinc Metallochaperones as Mutant p53 Reactivators: A New Paradigm in Cancer Therapeutics. Cancers 2018, 10, 166. [Google Scholar] [CrossRef] [Green Version]
- Blanden, A.R.; Yu, X.; Wolfe, A.J.; Gilleran, J.A.; Augeri, D.J.; O’Dell, R.S.; Olson, E.C.; Kimball, S.D.; Emge, T.J.; Movileanu, L.; et al. Synthetic Metallochaperone ZMC1 Rescues Mutant p53 Conformation by Transporting Zinc into Cells as an Ionophore. Mol. Pharmacol. 2015, 87, 825–831. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Vazquez, A.; Levine, A.J.; Carpizo, D.R. Allele-specific p53 mutant reactivation. Cancer Cell 2012, 21, 614–625. [Google Scholar] [CrossRef] [Green Version]
- Zaman, S.; Yu, X.; Bencivenga, A.F.; Blanden, A.R.; Liu, Y.; Withers, T.; Na, B.; Blayney, A.J.; Gilleran, J.; Boothman, D.A.; et al. Combinatorial Therapy of Zinc Metallochaperones with Mutant p53 Reactivation and Diminished Copper Binding. Mol. Cancer Ther. 2019, 18, 1355–1365. [Google Scholar] [CrossRef]
- Yu, X.; Blanden, A.; Tsang, A.T.; Zaman, S.; Liu, Y.; Gilleran, J.; Bencivenga, A.F.; Kimball, S.D.; Loh, S.N.; Carpizo, D.R. Thiosemicarbazones Functioning as Zinc Metallochaperones to Reactivate Mutant p53. Mol. Pharmacol. 2017, 91, 567–575. [Google Scholar] [CrossRef] [Green Version]
- Gilleran, J.A.; Yu, X.; Blayney, A.J.; Bencivenga, A.F.; Na, B.; Augeri, D.J.; Blanden, A.R.; Kimball, S.D.; Loh, S.N.; Roberge, J.Y.; et al. Benzothiazolyl and Benzoxazolyl Hydrazones Function as Zinc Metallochaperones to Reactivate Mutant p53. J. Med. Chem. 2021, 64, 2024–2045. [Google Scholar] [CrossRef]
- Dumbrava, E.E.; Johnson, M.L.; Tolcher, A.W.; Shapiro, G.; Thompson, J.A.; El-Khoueiry, A.B.; Vandross, A.L.; Kummar, S.; Parikh, A.R.; Munster, P.N.; et al. First-in-human study of PC14586, a small molecule structural corrector of Y220C mutant p53, in patients with advanced solid tumors harboring a TP53 Y220C mutation. J. Clin. Oncol. 2022, 40, 3003. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. The Evaluation of PC14586 in Patients with Advanced Solid Tumors Harboring a p53 Y220C Mutation. Available online: https://clinicaltrials.gov/ct2/show/NCT04585750 (accessed on 12 September 2022).
- Matissek, K.J.; Mossalam, M.; Okal, A.; Lim, C.S. The DNA binding domain of p53 is sufficient to trigger a potent apoptotic response at the mitochondria. Mol. Pharm. 2013, 10, 3592–3602. [Google Scholar] [CrossRef] [PubMed]
- Bowman, K.E.R.; Ahne, L.; O’Brien, L.; Vander Mause, E.R.; Lu, P.; Wallis, B.; Evason, K.J.; Lim, C.S. p53-Bad* Fusion Gene Therapy Induces Apoptosis In Vitro and Reduces Zebrafish Tumor Burden in Hepatocellular Carcinoma. Mol. Pharm. 2023, 20, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Okal, A.; Mossalam, M.; Matissek, K.J.; Dixon, A.S.; Moos, P.J.; Lim, C.S. A Chimeric p53 Evades Mutant p53 Transdominant Inhibition in Cancer Cells. Mol. Pharm. 2013, 10, 3922–3933. [Google Scholar] [CrossRef] [PubMed]
- Okal, A.; Mossalam, M.; Matissek, K.J.; Lim, C.S. Abstract 1176: Bypassing the dominant-negative effect of mutant p53 in cancer cells. Cancer Res. 2012, 72, 1176. [Google Scholar] [CrossRef]
- Mehner, C.; Oberg, A.L.; Goergen, K.M.; Kalli, K.R.; Maurer, M.J.; Nassar, A.; Goode, E.L.; Keeney, G.L.; Jatoi, A.; Radisky, D.C.; et al. EGFR as a prognostic biomarker and therapeutic target in ovarian cancer: Evaluation of patient cohort and literature review. Genes Cancer 2017, 8, 589–599. [Google Scholar] [CrossRef]
- Garufi, A.; Trisciuoglio, D.; Porru, M.; Leonetti, C.; Stoppacciaro, A.; D’Orazi, V.; Avantaggiati, M.; Crispini, A.; Pucci, D.; D’Orazi, G. A fluorescent curcumin-based Zn(II)-complex reactivates mutant (R175H and R273H) p53 in cancer cells. J. Exp. Clin. Cancer Res. 2013, 32, 72. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Xia, M.; Zhou, Z.; Hu, X.; Wang, J.; Zhang, M.; Li, Y.; Sun, L.; Chen, F.; Yu, H. p53 Promoted Ferroptosis in Ovarian Cancer Cells Treated with Human Serum Incubated-Superparamagnetic Iron Oxides. Int. J. Nanomed. 2021, 16, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Martin Lluesma, S.; Wolfer, A.; Harari, A.; Kandalaft, L.E. Cancer Vaccines in Ovarian Cancer: How Can We Improve? Biomedicines 2016, 4, 10. [Google Scholar] [CrossRef]
- Xu, J.; Shen, Y.; Wang, C.; Tang, S.; Hong, S.; Lu, W.; Xie, X.; Cheng, X. Arsenic compound sensitizes homologous recombination proficient ovarian cancer to PARP inhibitors. Cell Death Discov. 2021, 7, 259. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Arsenic Trioxide in Recurrent and Metastatic Ovarian Cancer and Endometrial Cancer with P53 Mutation. Available online: https://clinicaltrials.gov/ct2/show/NCT04489706?term=p53&cond=ovarian+cancer&draw=2&rank=3 (accessed on 11 September 2022).
- ClinicalTrials.gov. A Phase 2 Study of Kevetrin in Subjects with Ovarian Cancer. Available online: https://clinicaltrials.gov/ct2/show/results/NCT03042702 (accessed on 11 October 2022).
- Yoon, A.-R.; Wadhwa, R.; Kaul, S.C.; Yun, C.-O. Why is Mortalin a Potential Therapeutic Target for Cancer? Front. Cell Dev. Biol. 2022, 10, 914540. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Advanced or Recurrent Ovarian, Cervical, and Endometrial Cancer Treated with SHetA2 (Okgyn1). Available online: https://clinicaltrials.gov/ct2/show/NCT04928508 (accessed on 11 October 2022).
- ClinicalTrials.gov. p53 Activation in Platinum-Resistant High Grade Serous Ovarian Cancer, a Study of PLD with APR-246. Available online: https://clinicaltrials.gov/ct2/show/results/NCT03268382 (accessed on 11 October 2022).
- ClinicalTrials.gov. p53 Suppressor Activation in Recurrent High Grade Serous Ovarian Cancer, a Phase Ib/II Study of Systemic Carboplatin Combination Chemotherapy with or without APR-246. Available online: https://www.clinicaltrials.gov/ct2/show/NCT02098343 (accessed on 11 October 2022).
- Chandra, A.; Pius, C.; Nabeel, M.; Nair, M.; Vishwanatha, J.K.; Ahmad, S.; Basha, R. Ovarian cancer: Current status and strategies for improving therapeutic outcomes. Cancer Med. 2019, 8, 7018–7031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghisoni, E.; Imbimbo, M.; Zimmermann, S.; Valabrega, G. Ovarian Cancer Immunotherapy: Turning up the Heat. Int. J. Mol. Sci. 2019, 20, 2927. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Name, Year, and Ref(s) | Gene Therapy, Small Molecule, or Peptide | Drug Delivery System | Target Population/ Mutation(s) | Function(s) | In Vitro, In Vivo, or Clinical Trial Phase | Response/ Outcome |
|---|---|---|---|---|---|---|
| Gendicine (2003) [5,82,84,85,86,87] | Gene Therapy | Ad5 | p53 mutations | Restore WT p53 induction of apoptosis | Approved in China | Positive in head and neck cancer, failed in ovarian cancer |
| Advexin (2004) [85,87,91] | Gene Therapy | Ad5 | Platinum and paclitaxel resistant; p53 mutations | Restore WT p53 induction of apoptosis | Phase I clinical trial | Well tolerated; dosing schedule and amount could not be deduced |
| SCH-58500 (2002) [60,85,96] | Gene Therapy | Ad5CMV-p53 ΔE1A | p53 mutations | Restore WT p53 induction of apoptosis | Phase I and II clinical trialsNCT00002960 | Well tolerated; Similar toxicity to traditional chemotherapy |
| ONYX-015 (2002) [87,102] | NA | CRAd, dl1520 with E1B 55-kd gene deletion | p53 mutations; cisplatin-resistant | Restore WT p53 induction of apoptosis | In vivo p-53-deficient nude mouse xenograft; phase I; clinical development halted | Efficacy only satisfactory when used with other anticancer agents; discontinued in U.S. |
| dl922-947 (2006) [129] | NA | CRAd dl922-947 | Rb and p53 mutations | Restore WT p53 induction of apoptosis | In vivo p-53 deficient nude mouse xenograft | Higher survival rate compared to WT ad5 or ONYX-015 treatments |
| CRAd, Ad5-Δ24-RGD [96,130] | NA | CRAd, Ad5-Δ24-RGD | Rb and p53 mutations | Restore WT p53 induction of apoptosis | Phase I clinical trial NCT00562003 | Future chemotherapy combination studies in development |
| p53-Bad* (2019) [104] | Gene Therapy | Ad | p53 mutations and WT p53 | Restore WT p53 induction of apoptosis | In vitro | Induction of apoptosis regardless of p53 status |
| Au-C225-p53 (2019) [73] | pCMV WT p53 Gene Therapy | Au NP/cetuximab | p53 mutations | Restore WT p53 induction of apoptosis | In vitro SKOV-3 cells and in vivo SKOV-3 xenograft mice | Positive; future clinical trials possible |
| ZeFe-4 NPs (2020) [76] | NA | ZeFe-4 NPs | Gain-of-function p53 mutations, loss of Zn(II) binding; R175H and R273H | Degrade gain-of-function p53 mutations | In vitro p53 S241F ES-2 ovarian cancer cells; In vivo orthotopically implanted p53 Y220C xenograft breast cancer model | Positive; minimal toxicity |
| SPIO-Serum Nanoworms (2017) [131] | NA | SPIO-serum Nanoworms | Overexpressed mutated p53 | Induce ferroptosis | In vitro | Unknown |
| Zinc Oxide NPs (2017) [75,131] | NA | ZnO NPs | p53 mutations | Induce apoptosis via size-dependent cytotoxicity | In vitro SKOV-3 cells | Unknown |
| ADGN-531-p53-mRNA (2022) [132] | Gene Therapy | Nanocarrier ADGN-531-p53-mRNA | p53 mutations | Restore WT p53 induction of apoptosis. Restore PAPR inhibitor sensitivity | In vitro and in vivo | Unknown |
| DDC-p53 Liposomes (2007) [70,92] | Gene Therapy | Cationic Liposomes: DDC-DOTAP-DOPE | p53 mutations | Restore WT p53 induction of apoptosis | In vitro OVCAR-3 cells In vivo nude mice | Unknown |
| ReACp53 (2011) [79,133] | Peptide | NA | R175H R282W | Restore WT p53 induction of apoptosis by eliminating p53 aggregation | In vivo xenograft model; combination with carboplatin | Minimal toxicity; increased efficacy of carboplatin in some patients |
| p53-SLP vaccine (p53 70:248) (2012) [134,135,136,137,138] | Overlapping Synthetic Long Peptides | Vaccine | Gain-of-function p53 mutations; overexpression of p53 | Restore WT p53 induction of apoptosis | Phase II clinical trial | No change in response to secondary chemotherapy; induced immune response; potent |
| p53 vaccine (2012) [134,135,136,137,138] | Short Peptide | Vaccine (WT p53: 264–272) | Gain-of-function p53 mutations; overexpression of p53 | Restore WT p53 induction of apoptosis | Tested in combination with interleukin 2 (IL-2) | Immune response; potent; some high-grade adverse events due to IL-2 |
| Arsenic Trioxide (2019) [139,140,141,142] | Small Molecule | NA | R175H R273H R248Q | Restore WT p53 induction of apoptosis by reactivating misfolded p53 mutants; increase efficacy of PARP inhibitors | Phase II clinical trial (NCT04489706) | Adverse myelosuppression |
| COTI-2 (2019) [143,144,145,146] | Small Molecule (thio-semicarbazone) | NA | p53 misfolded mutations | Reactivate misfolded mutant p53 | Phase I clinical trial NCT02433626 | Safe and well tolerated |
| Kevetrin (2020), [147] | Small Molecule | NA | p53 mutations | Induce cell cycle arrest and apoptosis | Phase II clinical trialNCT03042702 | Stable disease (n = 2) |
| PRIMA-1 (2019) [32,148,149,150,151] | Small Molecule (can be combined with SHetA2) | NA | p53 mutations | p53 reactivation and refolding with induction of apoptosis; reduces aggregation | Phase I clinical trial NCT04928508 | Currently recruiting; prevented ovarian cancer in 67% of mice |
| Eprenetapop-t APR-246 (PRIMA-1MET) (2016) [152,153,154,155] | Small Molecule (can be combined with carboplatin or mebendazole) | NA | Preferentially effective in CIP2A-deficient patients | Restore WT p53 folding and subsequent induction of apoptosis | >10 clinical trials 1. Phase II clinical trial NCT03268382 2. Phase Ib/II clinical trial NCT02098343 | 1. Disease control rate of 70%; 2. disease control rate of 75% |
| Zinc Metallochap-rones (2022) [153,156,157,158,159,160,161,162] | Small Molecule | NA | R175H p53 | Restore zinc binding; restore induction of apoptosis | In vitro In vivo | No synergistic effects with chemotherapies; toxic off-target effects due to zinc |
| PC14586 (2021-present) [46,163,164] | Small Molecule | NA-oral | Y220C p53 | Stabilize p53 and restore cell cycle arrest | Phase I/II clinical trials NCT04585750 | Preliminary results indicate good safety profile |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wallis, B.; Bowman, K.R.; Lu, P.; Lim, C.S. The Challenges and Prospects of p53-Based Therapies in Ovarian Cancer. Biomolecules 2023, 13, 159. https://doi.org/10.3390/biom13010159
Wallis B, Bowman KR, Lu P, Lim CS. The Challenges and Prospects of p53-Based Therapies in Ovarian Cancer. Biomolecules. 2023; 13(1):159. https://doi.org/10.3390/biom13010159
Chicago/Turabian StyleWallis, Bryce, Katherine Redd Bowman, Phong Lu, and Carol S. Lim. 2023. "The Challenges and Prospects of p53-Based Therapies in Ovarian Cancer" Biomolecules 13, no. 1: 159. https://doi.org/10.3390/biom13010159