
Integrative Molecular Analysis of DNA Methylation Dynamics Unveils Molecules with Prognostic Potential in Breast Cancer

Abstract
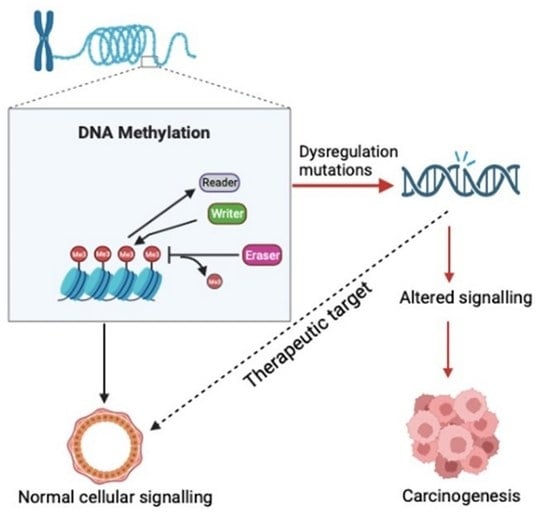
:
1. Introduction
2. Materials and Methods
2.1. Writers, Readers and Erasers of DNA Methylation
2.2. Genetic Alterations
2.3. Changes in the Transcriptome
2.4. Survival Analysis
3. Results
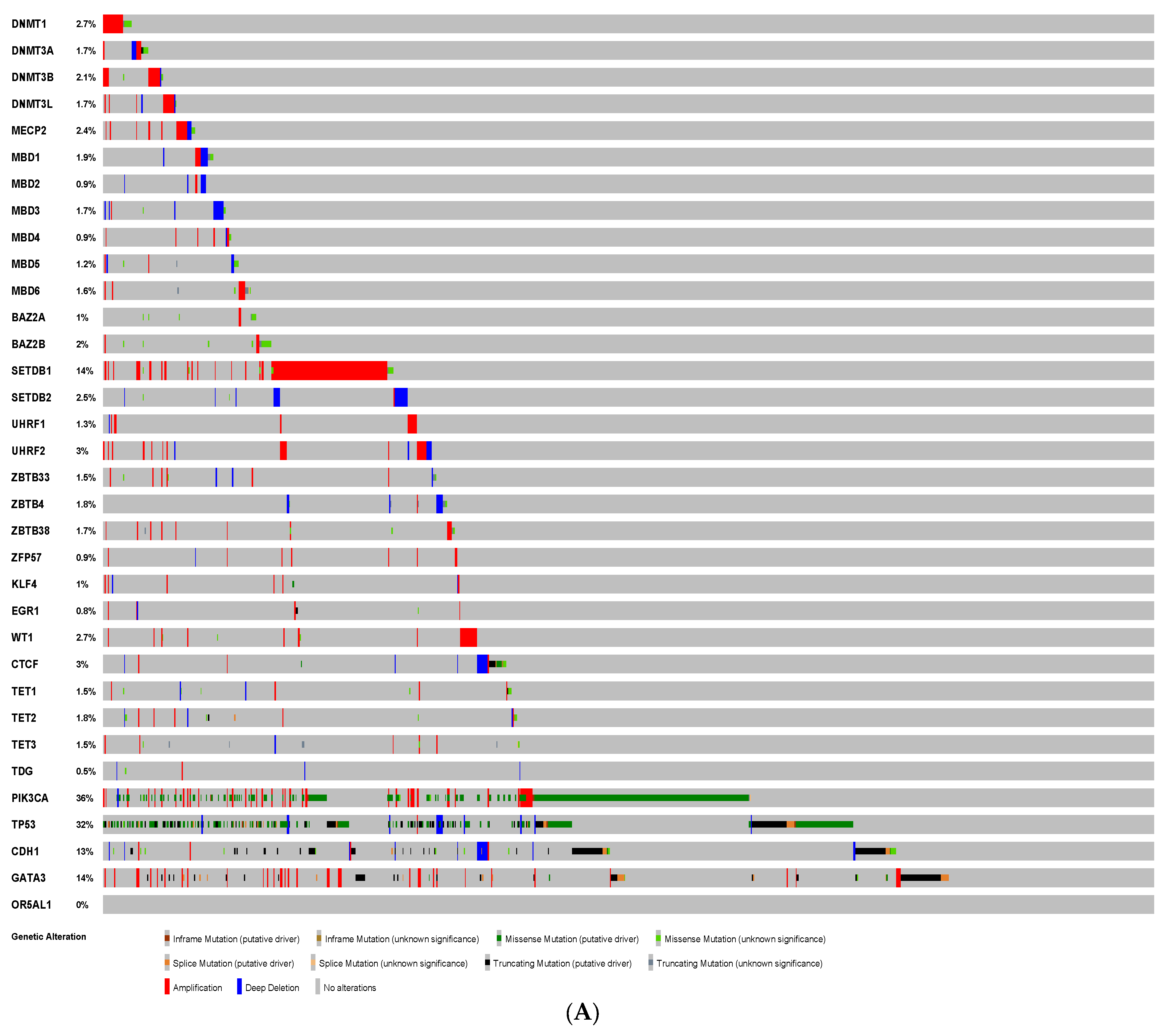
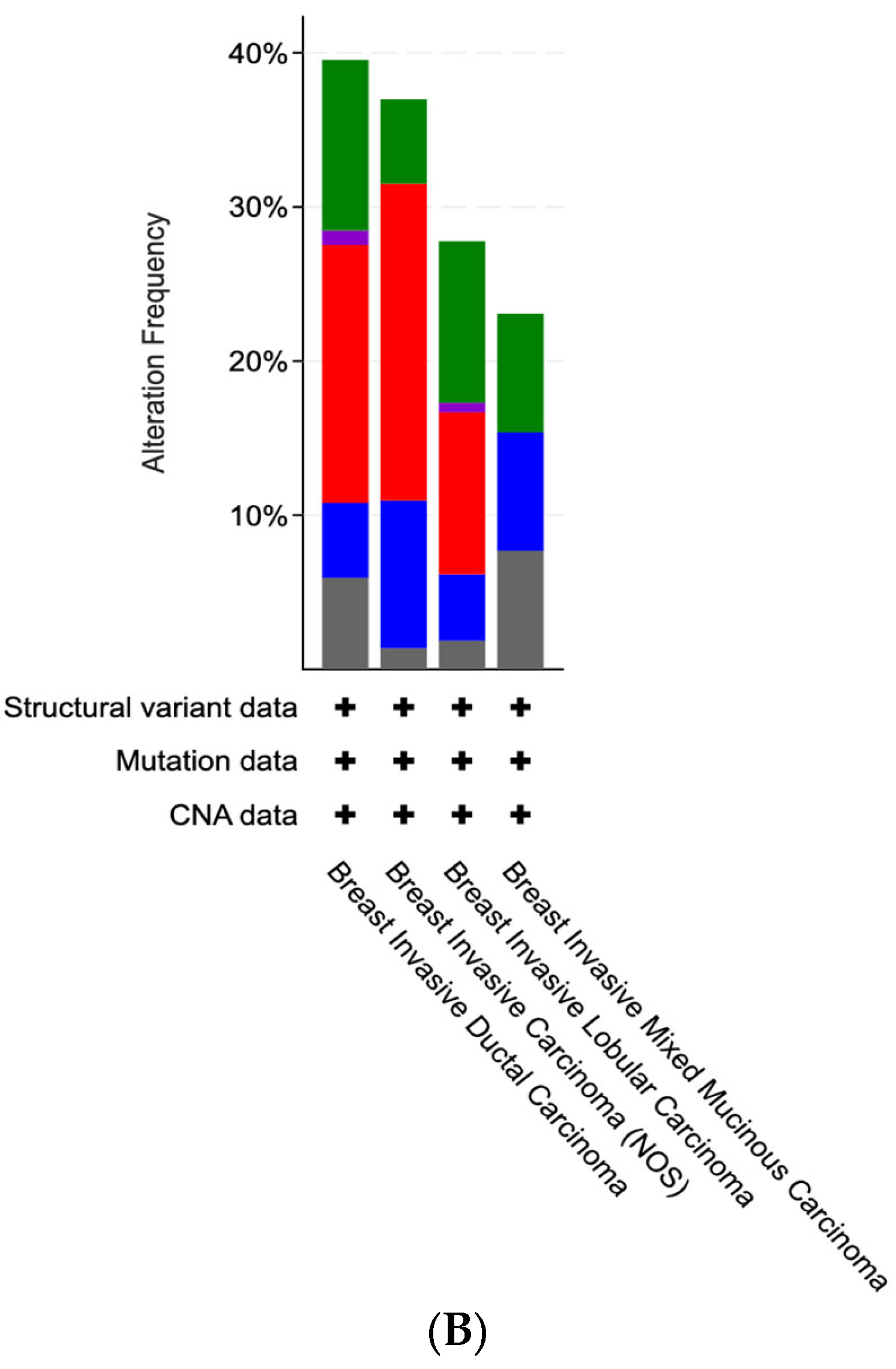
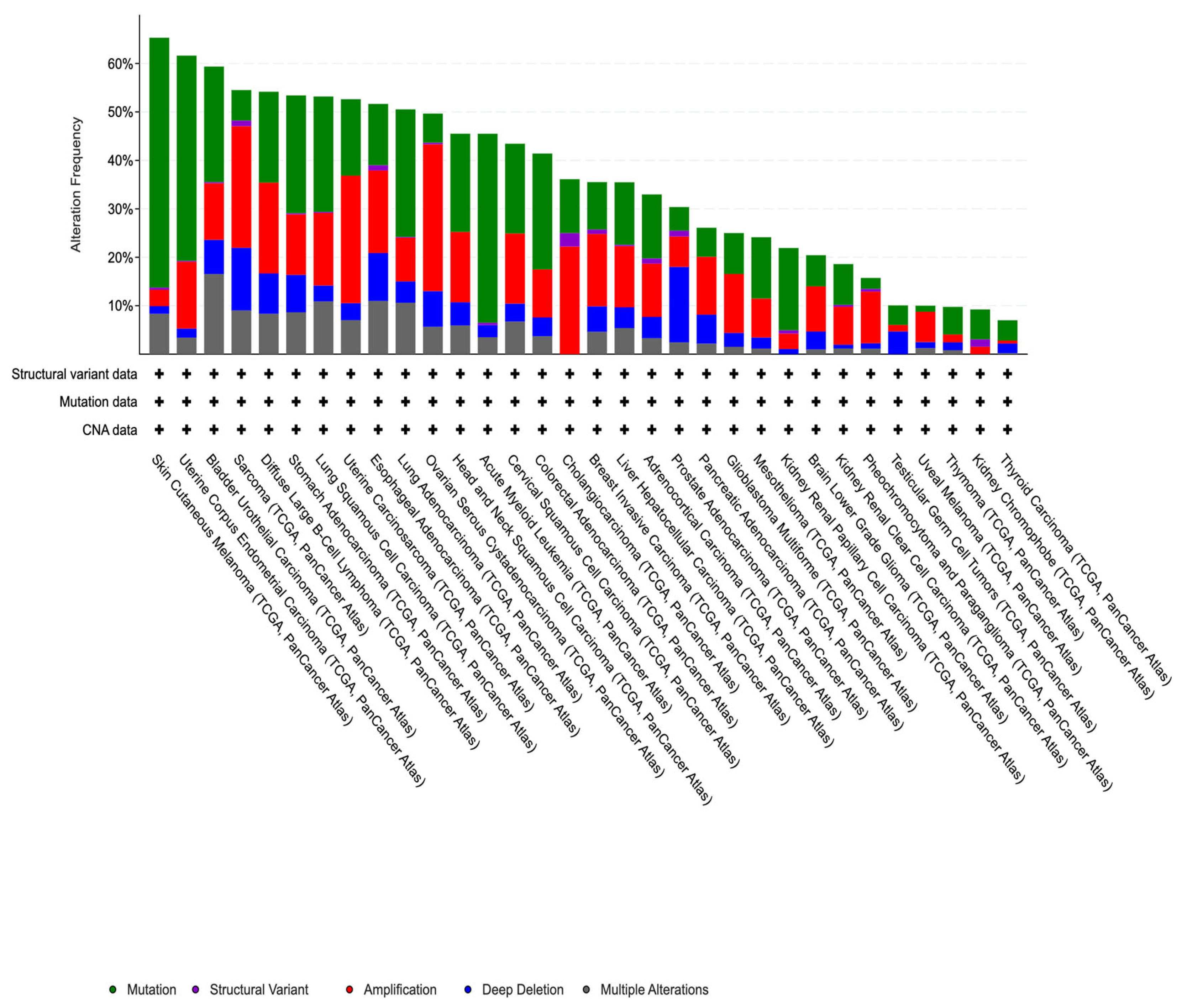
3.1. Mutational Analysis of DNA Methylation Regulators in Breast Cancer
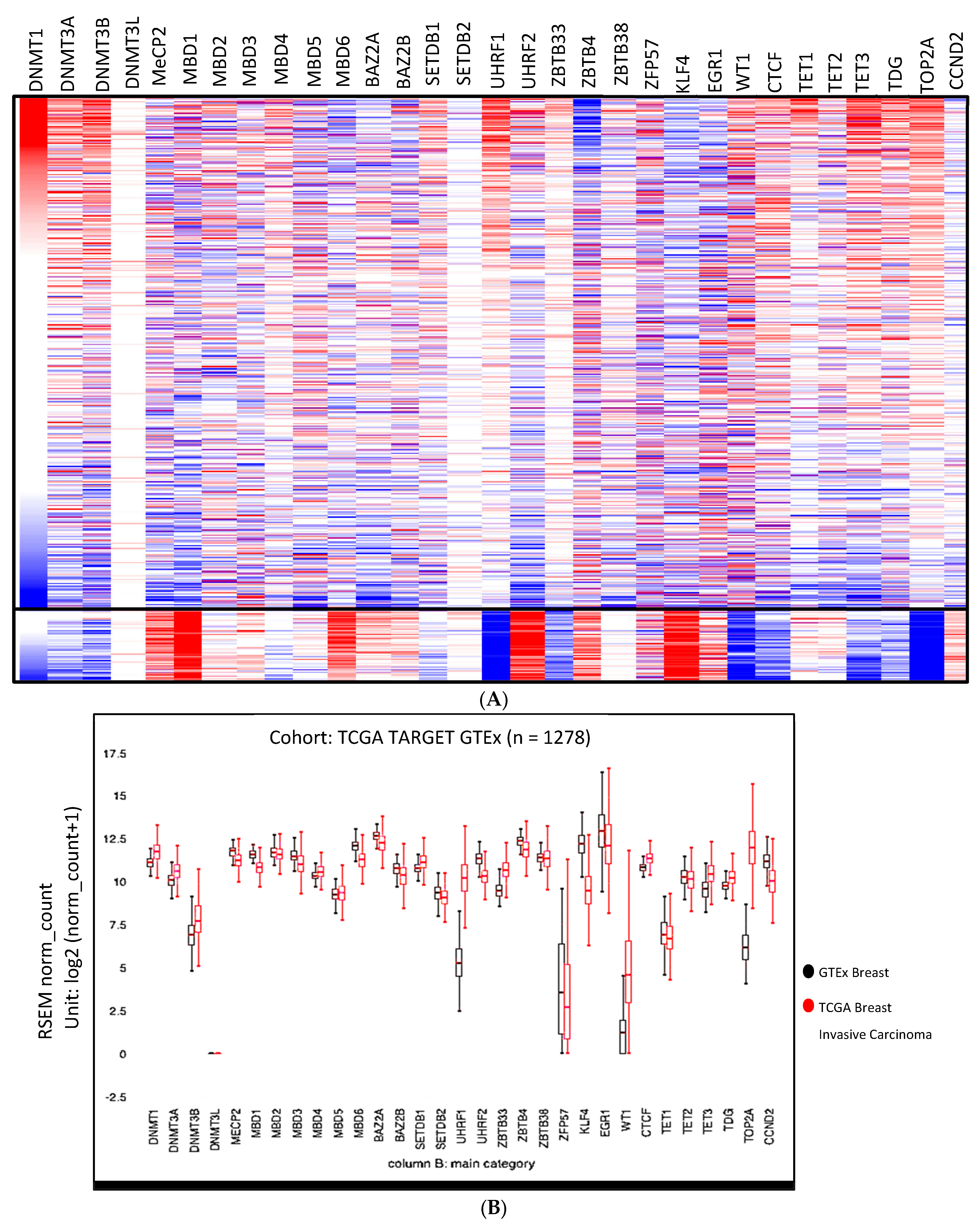
3.2. Expression Anomalies in Breast Cancer
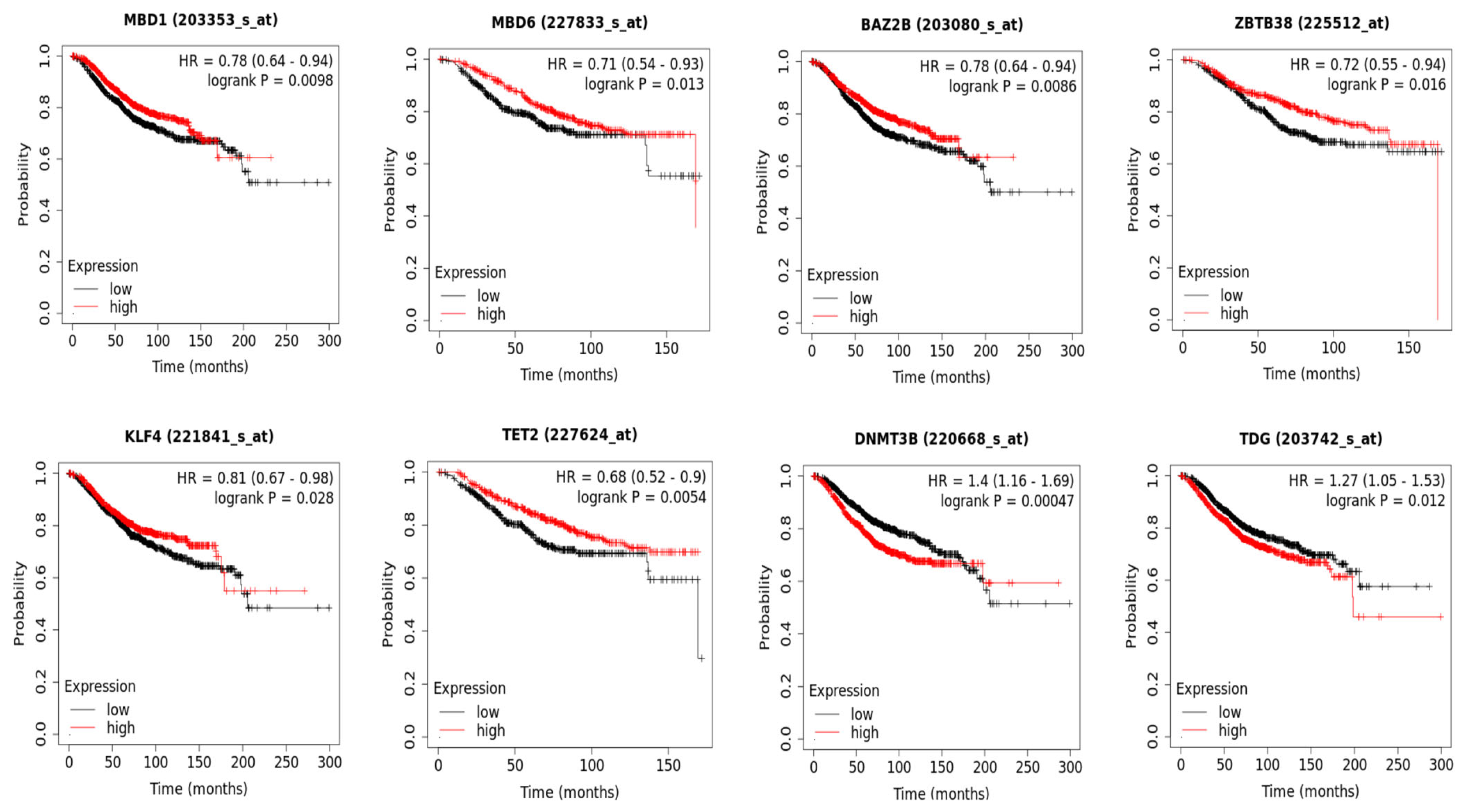
3.3. DNA Methylation and Patient Overall Survival
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Razin, A.; Cedar, H. DNA methylation and gene expression. Microbiol. Rev. 1991, 55, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Jaenisch, R.; Bird, A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat. Genet. 2003, 33, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Meehan, R.R.; Lewis, J.D.; McKay, S.; Kleiner, E.L.; Bird, A.P. Identification of a mammalian protein that binds specifically to DNA containing methylated CpGs. Cell 1989, 58, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Bestor, T.H. The DNA methyltransferases of mammals. Hum. Mol. Genet. 2000, 9, 2395–2402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, P.A.; Takai, D. The role of DNA methylation in mammalian epigenetics. Science 2001, 293, 1068–1070. [Google Scholar] [CrossRef] [PubMed]
- Bird, A. The essentials of DNA methylation. Cell 1992, 70, 5–8. [Google Scholar] [CrossRef]
- Martisova, A.; Holcakova, J.; Izadi, N.; Sebuyoya, R.; Hrstka, R.; Bartosik, M. DNA Methylation in Solid Tumors: Functions and Methods of Detection. Int. J. Mol. Sci. 2021, 22, 4247. [Google Scholar] [CrossRef]
- Guo, J.U.; Su, Y.; Shin, J.H.; Shin, J.; Li, H.; Xie, B.; Zhong, C.; Hu, S.; Le, T.; Fan, G.; et al. Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nat. Neurosci. 2014, 17, 215–222. [Google Scholar] [CrossRef] [Green Version]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.M.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef] [Green Version]
- Smith, Z.D.; Chan, M.M.; Humm, K.C.; Karnik, R.; Mekhoubad, S.; Regev, A.; Eggan, K.; Meissner, A. DNA methylation dynamics of the human preimplantation embryo. Nature 2014, 511, 611–615. [Google Scholar] [CrossRef] [Green Version]
- Okae, H.; Chiba, H.; Hiura, H.; Hamada, H.; Sato, A.; Utsunomiya, T.; Kikuchi, H.; Yoshida, H.; Tanaka, A.; Suyama, M.; et al. Genome-wide analysis of DNA methylation dynamics during early human development. PLoS Genet. 2014, 10, e1004868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egger, G.; Jeong, S.; Escobar, S.G.; Cortez, C.C.; Li, T.W.; Saito, Y.; Yoo, C.B.; Jones, P.A.; Liang, G. Identification of DNMT1 (DNA methyltransferase 1) hypomorphs in somatic knockouts suggests an essential role for DNMT1 in cell survival. Proc. Natl. Acad. Sci. USA 2006, 103, 14080–14085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fatemi, M.; Hermann, A.; Gowher, H.; Jeltsch, A. Dnmt3a and Dnmt1 functionally cooperate during de novo methylation of DNA. Eur. J. Biochem. 2002, 269, 4981–4984. [Google Scholar] [CrossRef] [PubMed]
- Parry, L.; Clarke, A.R. The Roles of the Methyl-CpG Binding Proteins in Cancer. Genes. Cancer 2011, 2, 618–630. [Google Scholar] [CrossRef] [Green Version]
- Nan, X.; Meehan, R.R.; Bird, A. Dissection of the methyl-CpG binding domain from the chromosomal protein MeCP2. Nucleic Acids Res. 1993, 21, 4886–4892. [Google Scholar] [CrossRef] [Green Version]
- Hendrich, B.; Bird, A. Identification and characterization of a family of mammalian methyl-CpG binding proteins. Mol. Cell. Biol. 1998, 18, 6538–6547. [Google Scholar] [CrossRef] [Green Version]
- Mahmood, N.; Rabbani, S.A. DNA Methylation Readers and Cancer: Mechanistic and Therapeutic Applications. Front. Oncol. 2019, 9, 489. [Google Scholar] [CrossRef] [Green Version]
- Du, Q.; Luu, P.L.; Stirzaker, C.; Clark, S.J. Methyl-CpG-binding domain proteins: Readers of the epigenome. Epigenomics 2015, 7, 1051–1073. [Google Scholar] [CrossRef]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [Green Version]
- He, Y.F.; Li, B.Z.; Li, Z.; Liu, P.; Wang, Y.; Tang, Q.; Ding, J.; Jia, Y.; Chen, Z.; Li, L.; et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 2011, 333, 1303–1307. [Google Scholar] [CrossRef] [Green Version]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef] [Green Version]
- Brinkman, A.B.; Nik-Zainal, S.; Simmer, F.; Rodriguez-Gonzalez, F.G.; Smid, M.; Alexandrov, L.B.; Butler, A.; Martin, S.; Davies, H.; Glodzik, D.; et al. Partially methylated domains are hypervariable in breast cancer and fuel widespread CpG island hypermethylation. Nat. Commun. 2019, 10, 1749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batra, R.N.; Lifshitz, A.; Vidakovic, A.T.; Chin, S.F.; Sati-Batra, A.; Sammut, S.J.; Provenzano, E.; Ali, H.R.; Dariush, A.; Bruna, A.; et al. DNA methylation landscapes of 1538 breast cancers reveal a replication-linked clock, epigenomic instability and cis-regulation. Nat. Commun. 2021, 12, 5406. [Google Scholar] [CrossRef] [PubMed]
- Umeh-Garcia, M.; O’Geen, H.; Simion, C.; Gephart, M.H.; Segal, D.J.; Sweeney, C.A. Aberrant promoter methylation contributes to LRIG1 silencing in basal/triple-negative breast cancer. Br. J. Cancer 2022, 127, 436–448. [Google Scholar] [CrossRef]
- Zhang, W.; Xu, J. DNA methyltransferases and their roles in tumorigenesis. Biomark. Res. 2017, 5, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasmussen, K.D.; Helin, K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 2016, 30, 733–750. [Google Scholar] [CrossRef] [Green Version]
- Robertson, K.D.; Uzvolgyi, E.; Liang, G.; Talmadge, C.; Sumegi, J.; Gonzales, F.A.; Jones, P.A. The human DNA methyltransferases (DNMTs) 1, 3a and 3b: Coordinate mRNA expression in normal tissues and overexpression in tumors. Nucleic Acids Res. 1999, 27, 2291–2298. [Google Scholar] [CrossRef] [Green Version]
- Saito, Y.; Kanai, Y.; Nakagawa, T.; Sakamoto, M.; Saito, H.; Ishii, H.; Hirohashi, S. Increased protein expression of DNA methyltransferase (DNMT) 1 is significantly correlated with the malignant potential and poor prognosis of human hepatocellular carcinomas. Int. J. Cancer 2003, 105, 527–532. [Google Scholar] [CrossRef]
- Zhao, Z.; Wu, Q.; Cheng, J.; Qiu, X.; Zhang, J.; Fan, H. Depletion of DNMT3A suppressed cell proliferation and restored PTEN in hepatocellular carcinoma cell. J. Biomed. Biotechnol. 2010, 2010, 737535. [Google Scholar] [CrossRef] [Green Version]
- Patra, S.K.; Patra, A.; Zhao, H.; Dahiya, R. DNA methyltransferase and demethylase in human prostate cancer. Mol. Carcinog. 2002, 33, 163–171. [Google Scholar] [CrossRef]
- Butcher, D.T.; Rodenhiser, D.I. Epigenetic inactivation of BRCA1 is associated with aberrant expression of CTCF and DNA methyltransferase (DNMT3B) in some sporadic breast tumours. Eur. J. Cancer 2007, 43, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Billard, L.M.; Magdinier, F.; Lenoir, G.M.; Frappart, L.; Dante, R. MeCP2 and MBD2 expression during normal and pathological growth of the human mammary gland. Oncogene 2002, 21, 2704–2712. [Google Scholar] [CrossRef] [PubMed]
- Man, X.; Li, Q.; Wang, B.; Zhang, H.; Zhang, S.; Li, Z. DNMT3A and DNMT3B in Breast Tumorigenesis and Potential Therapy. Front. Cell Dev. Biol. 2022, 10, 916725. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Wang, H.; Tan, L. Dysregulated TET Family Genes and Aberrant 5mC Oxidation in Breast Cancer: Causes and Consequences. Cancers 2021, 13, 6039. [Google Scholar] [CrossRef]
- Lan, J.; Hua, S.; He, X.; Zhang, Y. DNA methyltransferases and methyl-binding proteins of mammals. Acta Biochim. Biophys. Sin. 2010, 42, 243–252. [Google Scholar] [CrossRef] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repecka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef]
- Consortium, G.T. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef]
- Gyorffy, B. Survival analysis across the entire transcriptome identifies biomarkers with the highest prognostic power in breast cancer. Comput. Struct. Biotechnol. J. 2021, 19, 4101–4109. [Google Scholar] [CrossRef]
- Mehmood, R.; Jibiki, K.; Shibazaki, N.; Yasuhara, N. Molecular profiling of nucleocytoplasmic transport factor genes in breast cancer. Heliyon 2021, 7, e06039. [Google Scholar] [CrossRef]
- Jones, P.A.; Laird, P.W. Cancer epigenetics comes of age. Nat. Genet. 1999, 21, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Ballestar, E.; Esteller, M. Methyl-CpG-binding proteins in cancer: Blaming the DNA methylation messenger. Biochem. Cell. Biol. 2005, 83, 374–384. [Google Scholar] [CrossRef]
- Girault, I.; Tozlu, S.; Lidereau, R.; Bieche, I. Expression analysis of DNA methyltransferases 1, 3A, and 3B in sporadic breast carcinomas. Clin. Cancer Res. 2003, 9, 4415–4422. [Google Scholar]
- Oh, B.K.; Kim, H.; Park, H.J.; Shim, Y.H.; Choi, J.; Park, C.; Park, Y.N. DNA methyltransferase expression and DNA methylation in human hepatocellular carcinoma and their clinicopathological correlation. Int. J. Mol. Med. 2007, 20, 65–73. [Google Scholar] [CrossRef] [Green Version]
- Melki, J.R.; Warnecke, P.; Vincent, P.C.; Clark, S.J. Increased DNA methyltransferase expression in leukaemia. Leukemia 1998, 12, 311–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahluwalia, A.; Hurteau, J.A.; Bigsby, R.M.; Nephew, K.P. DNA methylation in ovarian cancer. II. Expression of DNA methyltransferases in ovarian cancer cell lines and normal ovarian epithelial cells. Gynecol. Oncol. 2001, 82, 299–304. [Google Scholar] [CrossRef]
- Shimbo, T.; Du, Y.; Grimm, S.A.; Dhasarathy, A.; Mav, D.; Shah, R.R.; Shi, H.; Wade, P.A. MBD3 localizes at promoters, gene bodies and enhancers of active genes. PLoS Genet. 2013, 9, e1004028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baubec, T.; Ivanek, R.; Lienert, F.; Schubeler, D. Methylation-dependent and -independent genomic targeting principles of the MBD protein family. Cell 2013, 153, 480–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Liu, Y.; Bai, F.; Zhang, J.-Y.; Ma, S.-H.; Liu, J.; Xu, Z.-D.; Zhu, H.-G.; Ling, Z.-Q.; Ye, D.; et al. Tumor development is associated with decrease of TET gene expression and 5-methylcytosine hydroxylation. Oncogene 2013, 32, 663–669. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.Y.; Luo, C.W.; Lai, Y.S.; Wu, C.C.; Hung, W.C. Lysine demethylase KDM2A inhibits TET2 to promote DNA methylation and silencing of tumor suppressor genes in breast cancer. Oncogenesis 2017, 6, e369. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Wang, G.; Liang, Z.; Yang, Y.; Cui, L.; Liu, C.Y. Loss of nuclear localization of TET2 in colorectal cancer. Clin. Epigenetics 2016, 8, 9. [Google Scholar] [CrossRef] [Green Version]
- He, Z.; Wang, X.; Huang, C.; Gao, Y.; Yang, C.; Zeng, P.; Chen, Z. The FENDRR/miR-214-3P/TET2 axis affects cell malignant activity via RASSF1A methylation in gastric cancer. Am. J. Transl. Res. 2018, 10, 3211–3223. [Google Scholar]
- Zhang, L.Y.; Li, P.L.; Wang, T.Z.; Zhang, X.C. Prognostic values of 5-hmC, 5-mC and TET2 in epithelial ovarian cancer. Arch. Gynecol. Obstet. 2015, 292, 891–897. [Google Scholar] [CrossRef] [PubMed]
- Nickerson, M.L.; Das, S.; Im, K.M.; Turan, S.; Berndt, S.I.; Li, H.; Lou, H.; Brodie, S.A.; Billaud, J.N.; Zhang, T.; et al. TET2 binds the androgen receptor and loss is associated with prostate cancer. Oncogene 2017, 36, 2172–2183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.; Liu, L.; Wang, M.; Xu, B.; Lyu, R.; Shi, Y.G.; Tan, L. TET2 Inhibits PD-L1 Gene Expression in Breast Cancer Cells through Histone Deacetylation. Cancers 2021, 13, 2207. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Um, S.J. Thymine-DNA glycosylase interacts with and functions as a coactivator of p53 family proteins. Biochem. Biophys. Res. Commun. 2008, 377, 838–842. [Google Scholar] [CrossRef]
- Thillainadesan, G.; Chitilian, J.M.; Isovic, M.; Ablack, J.N.; Mymryk, J.S.; Tini, M.; Torchia, J. TGF-beta-dependent active demethylation and expression of the p15ink4b tumor suppressor are impaired by the ZNF217/CoREST complex. Mol. Cell. 2012, 46, 636–649. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Yu, T.; Shi, J.; Chen, X.; Zhang, W.; Lin, T.; Liu, Z.; Wang, Y.; Zeng, Z.; Wang, C.; et al. Thymine DNA glycosylase is a positive regulator of Wnt signaling in colorectal cancer. J. Biol. Chem. 2014, 289, 8881–8890. [Google Scholar] [CrossRef] [Green Version]
- Mancuso, P.; Tricarico, R.; Bhattacharjee, V.; Cosentino, L.; Kadariya, Y.; Jelinek, J.; Nicolas, E.; Einarson, M.; Beeharry, N.; Devarajan, K.; et al. Thymine DNA glycosylase as a novel target for melanoma. Oncogene 2019, 38, 3710–3728. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Gene Symbol | Protein Function |
|---|---|---|
| Writer | DNMT1 | Maintenance methylation |
| DNMT3A | De novo methylation | |
| DNMT3B | De novo methylation | |
| DNMT3L | Assists the de novo methyltransferases | |
| Reader | MeCP2 | Methylated DNA binding |
| MBD1 | Methylated DNA binding | |
| MBD2 | Methylated DNA binding | |
| MBD3 | Methylated DNA binding | |
| MBD4 | Methylated DNA binding | |
| MBD5 | Methylated DNA binding | |
| MBD6 | Methylated DNA binding | |
| BAZ2A | Methylated DNA binding | |
| BAZ2B | Methylated DNA binding | |
| SETDB1 | Methylated DNA binding | |
| SETDB2 | Methylated DNA binding | |
| UHRF1 | Methylated DNA binding | |
| UHRF2 | Methylated DNA binding | |
| ZBTB33 | Methylated DNA binding | |
| ZBTB4 | Methylated DNA binding | |
| ZBTB38 | Methylated DNA binding | |
| ZFP57 | Methylated DNA binding | |
| KLF4 | Methylated DNA binding | |
| EGR1 | Methylated DNA binding | |
| WT1 | Methylated DNA binding | |
| CTCF | Methylated DNA binding | |
| Eraser | TET1 | DNA demethylation |
| TET2 | DNA demethylation | |
| TET3 | DNA demethylation | |
| TDG | DNA demethylation |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mehmood, R.; Alsaleh, A.; Want, M.Y.; Ahmad, I.; Siraj, S.; Ishtiaq, M.; Alshehri, F.A.; Naseem, M.; Yasuhara, N. Integrative Molecular Analysis of DNA Methylation Dynamics Unveils Molecules with Prognostic Potential in Breast Cancer. BioMedInformatics 2023, 3, 434-445. https://doi.org/10.3390/biomedinformatics3020029
Mehmood R, Alsaleh A, Want MY, Ahmad I, Siraj S, Ishtiaq M, Alshehri FA, Naseem M, Yasuhara N. Integrative Molecular Analysis of DNA Methylation Dynamics Unveils Molecules with Prognostic Potential in Breast Cancer. BioMedInformatics. 2023; 3(2):434-445. https://doi.org/10.3390/biomedinformatics3020029
Chicago/Turabian StyleMehmood, Rashid, Alanoud Alsaleh, Muzamil Y. Want, Ijaz Ahmad, Sami Siraj, Muhammad Ishtiaq, Faizah A. Alshehri, Muhammad Naseem, and Noriko Yasuhara. 2023. "Integrative Molecular Analysis of DNA Methylation Dynamics Unveils Molecules with Prognostic Potential in Breast Cancer" BioMedInformatics 3, no. 2: 434-445. https://doi.org/10.3390/biomedinformatics3020029