
Design of Inhibitors That Target the Menin–Mixed-Lineage Leukemia Interaction
 , ,
, ,  , and
, and
Abstract
:1. Introduction
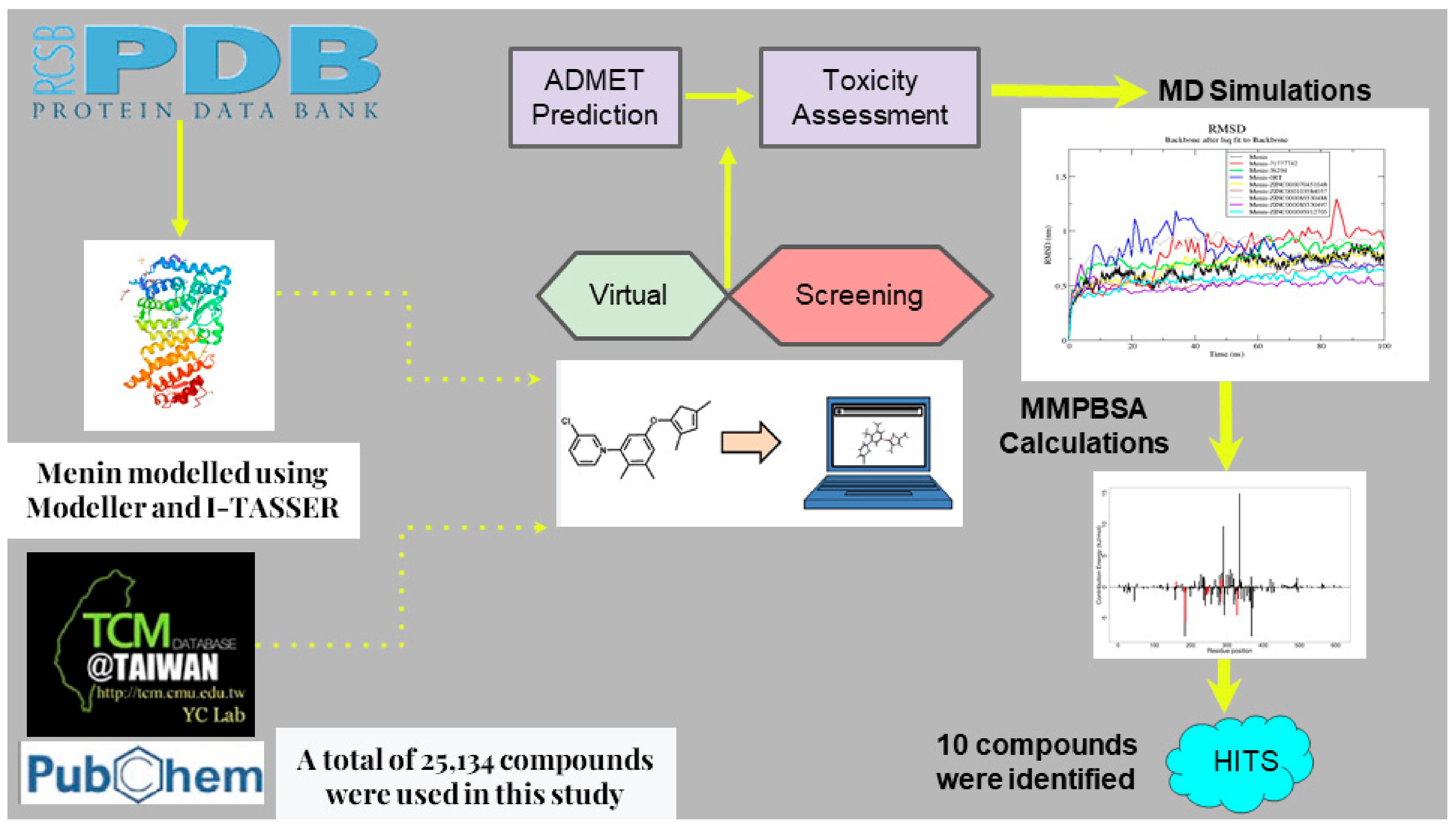
2. Materials and Methods
2.1. Obtaining Protein Structure and Sequence
2.2. Rebuilding the Menin Structure
2.3. Model Quality Assessment
2.4. Obtaining Compounds
2.5. Protein and Ligand Library Preparation
2.6. Virtual Screening
2.7. SwissADME Screening of Ligands
2.8. Prediction of Biological Activity of Compounds
2.9. Determining Binding Interactions
2.10. MD Simulations of Protein–Ligand Complexes
2.11. MM/PBSA Calculations of Protein–Ligand Complex
3. Results
3.1. Primary Structure Analysis
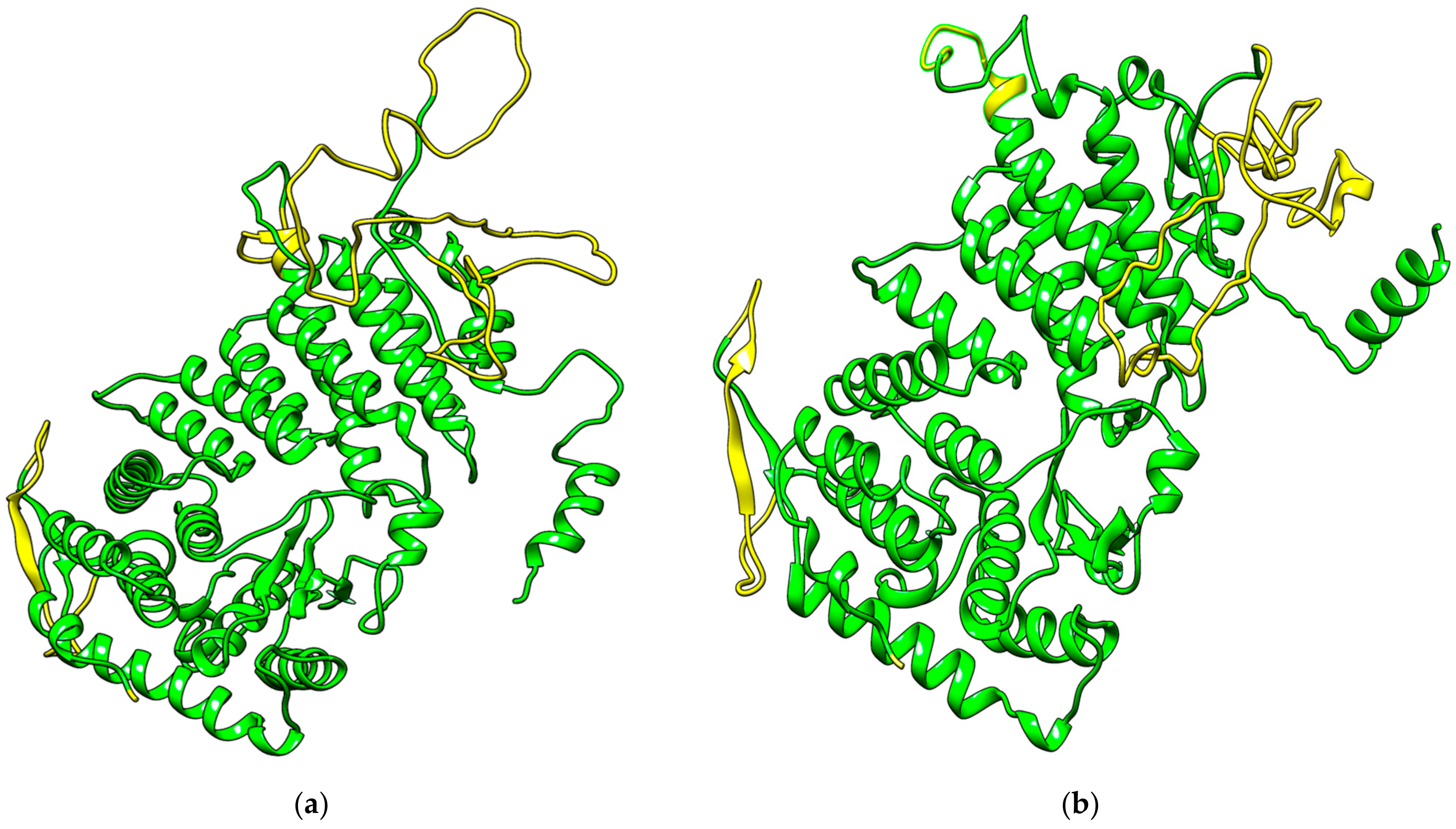
3.2. Remodeling Menin Structure
3.2.1. Protein Structure Identification
3.2.2. Model Rebuilding Using EasyModeller
3.2.3. Structure Prediction Using I-TASSER
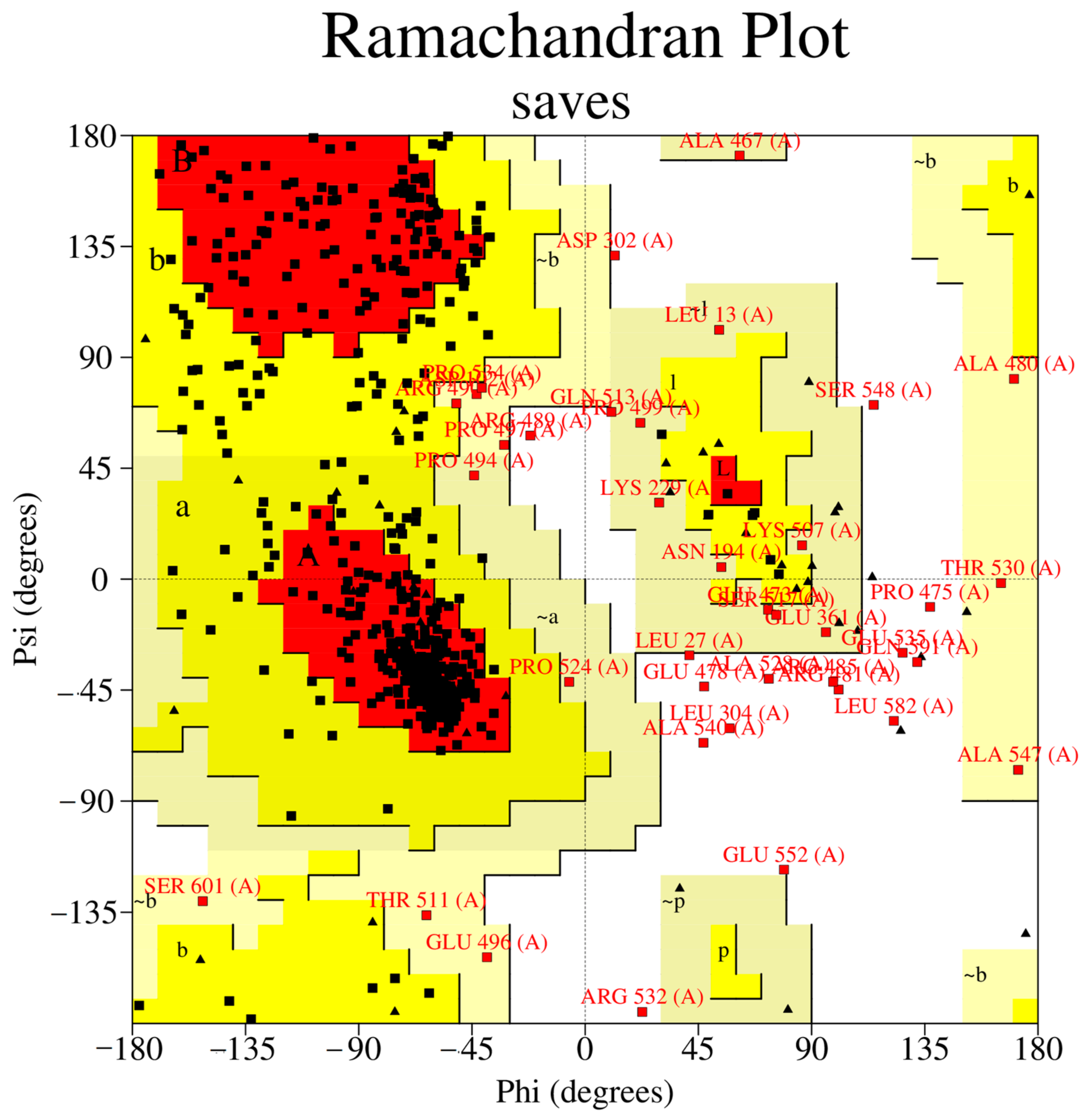
3.2.4. Protein Model Quality Assessment
3.3. Force Field Selection
3.4. Preparation of Screening Library
3.5. Virtual Screening against ITAS1
3.6. ADMET Prediction
3.7. Prediction of Biological Activity of Lead Compounds
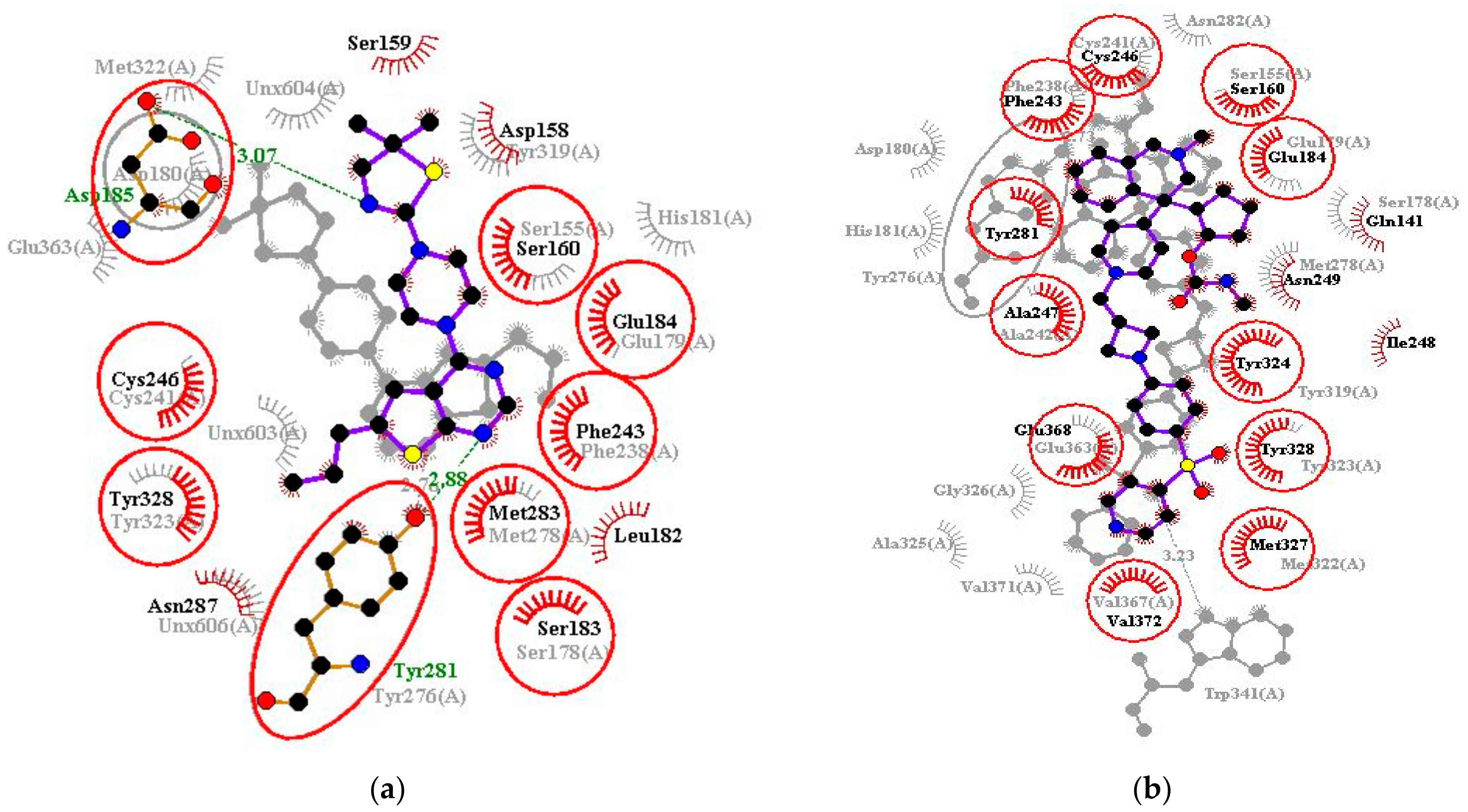
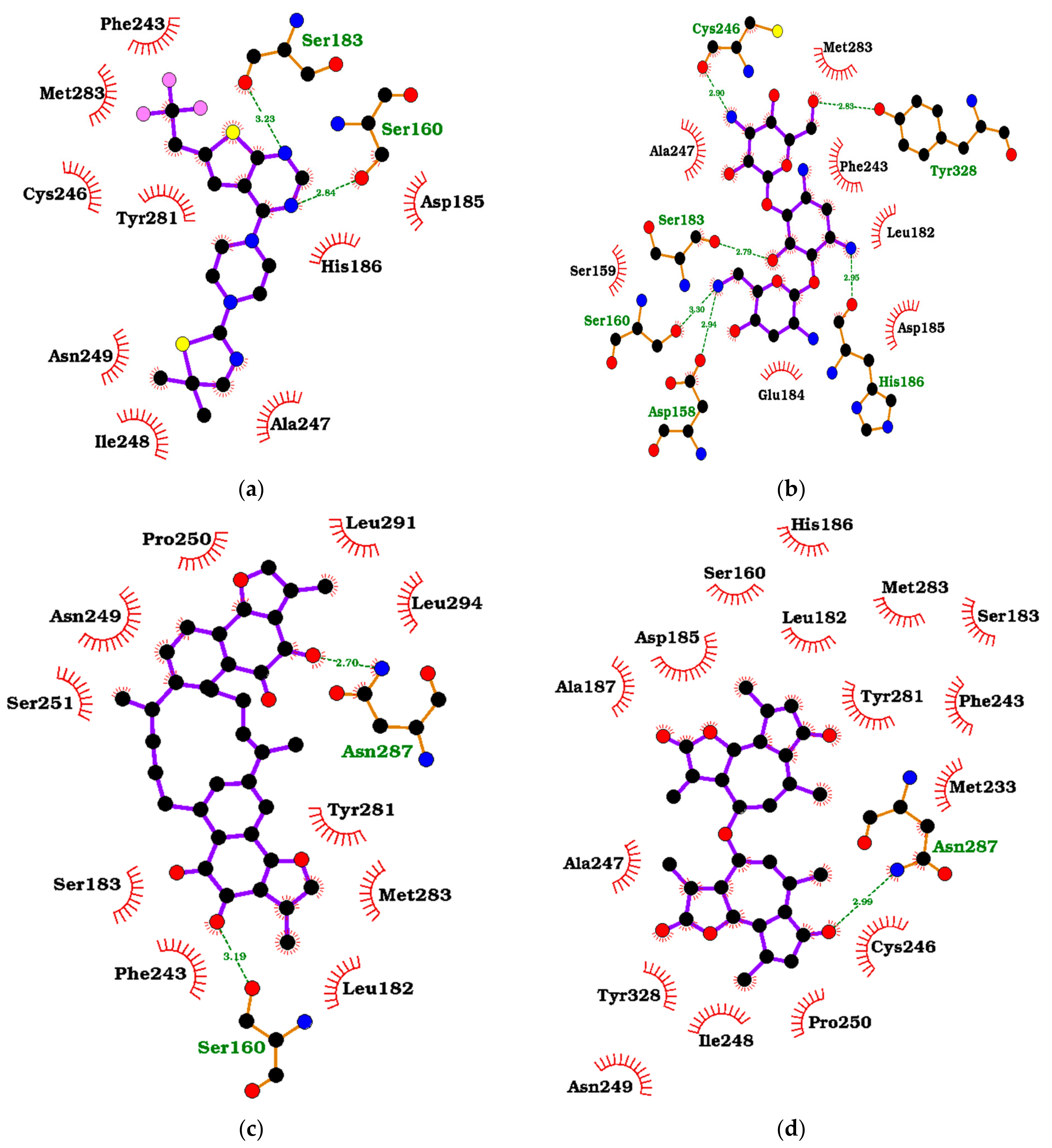
3.8. Visualization of the Protein–Ligand Interactions
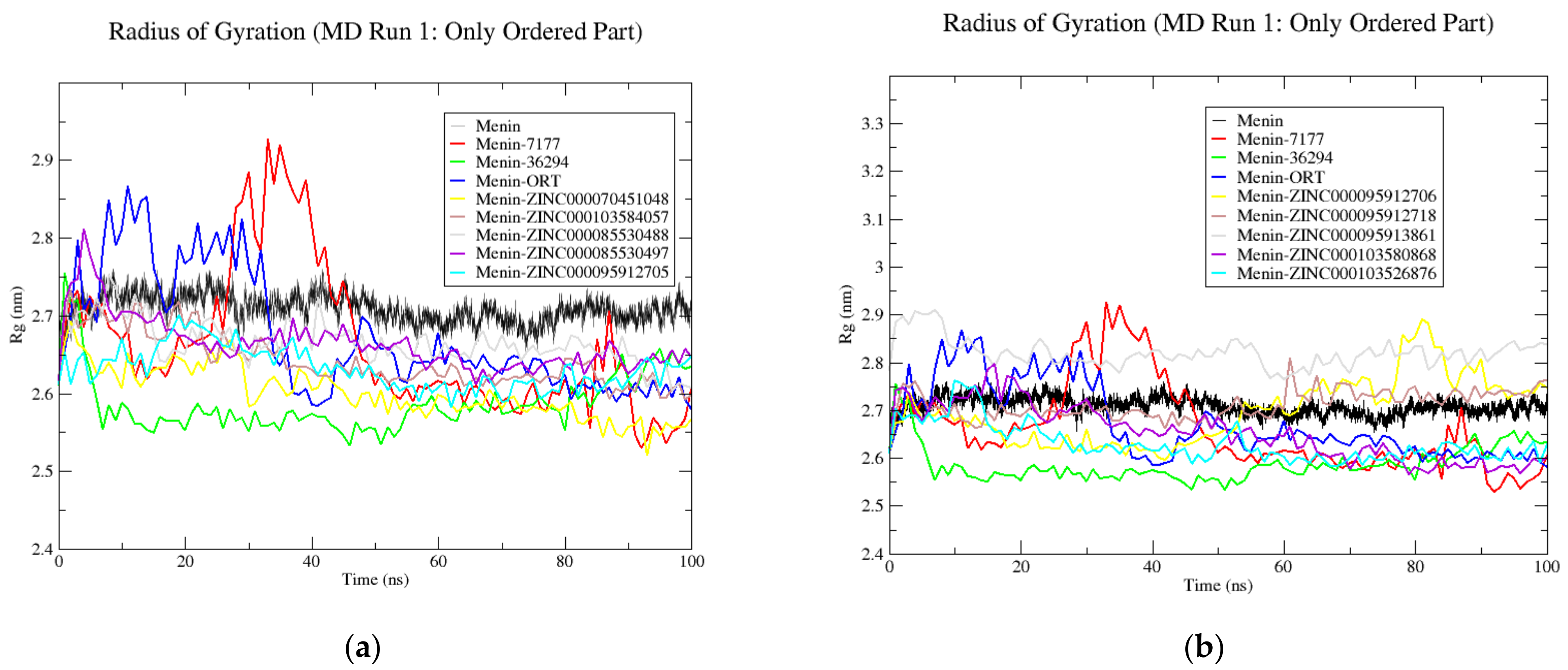
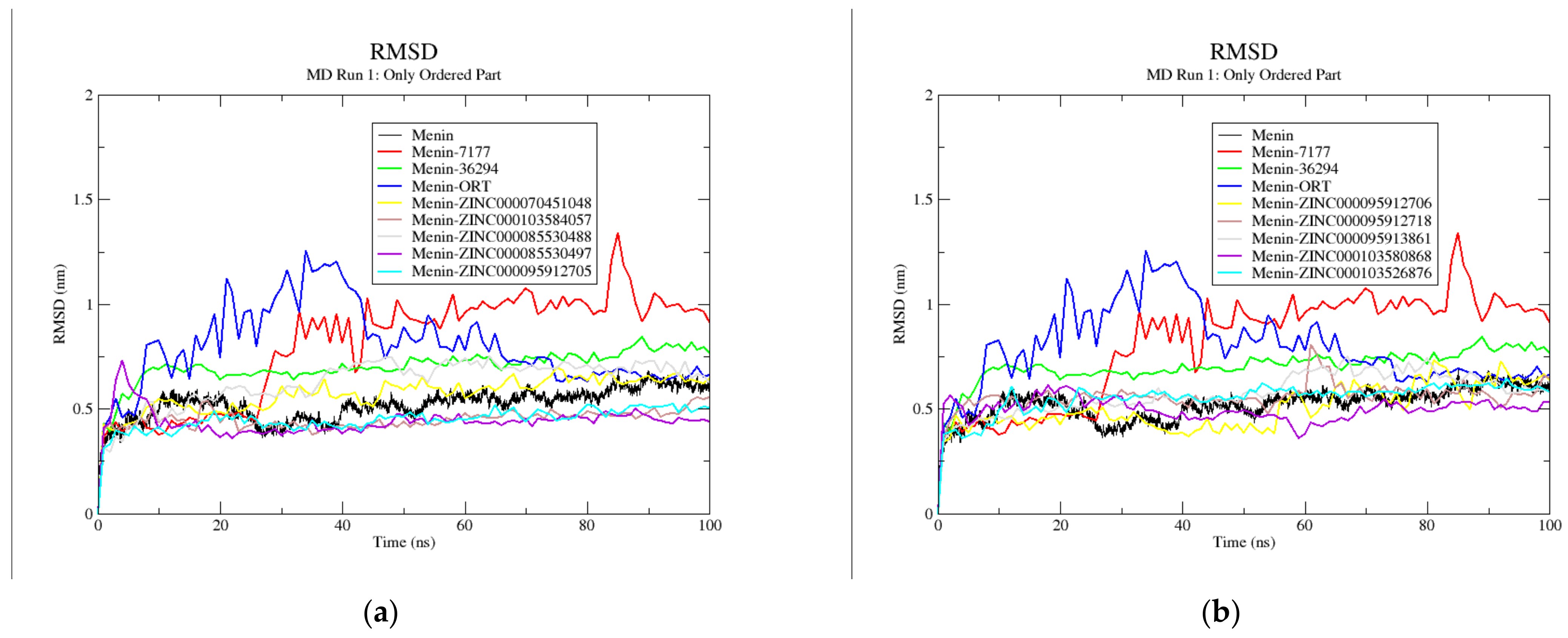
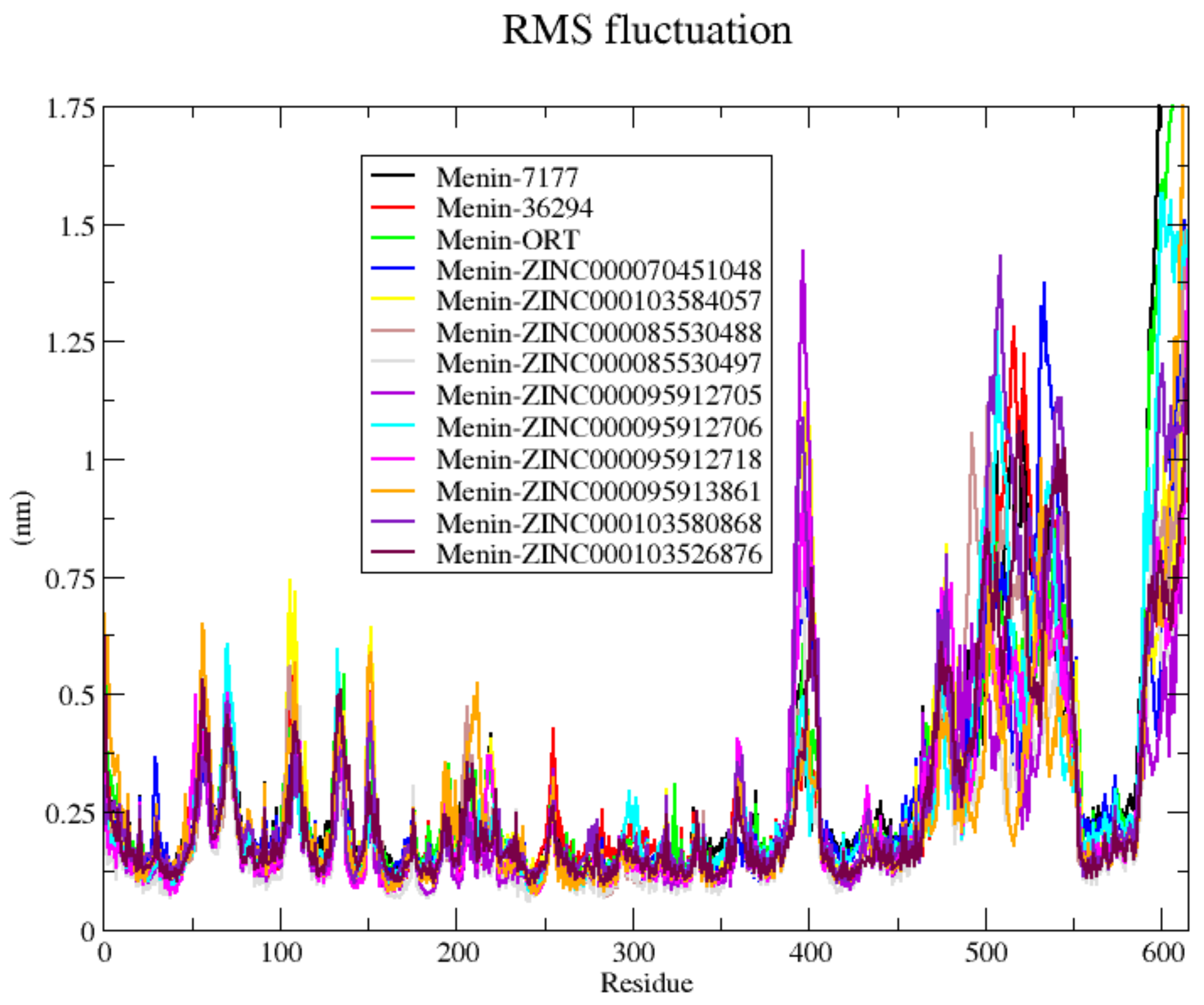
3.9. Molecular Dynamics Simulations
3.10. MM/PBSA Computations
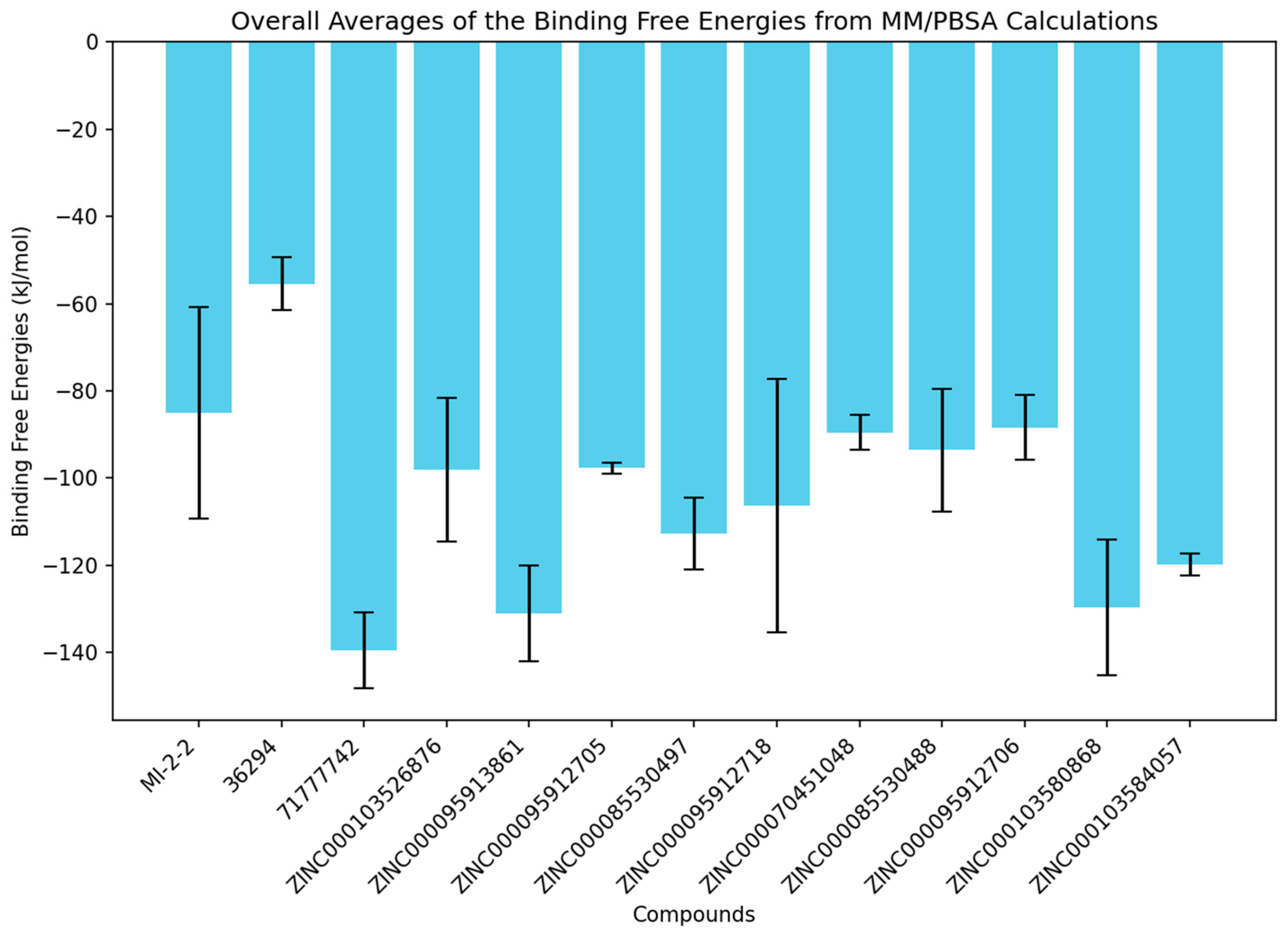
3.10.1. Binding Energies Involved in Menin–Ligand Binding
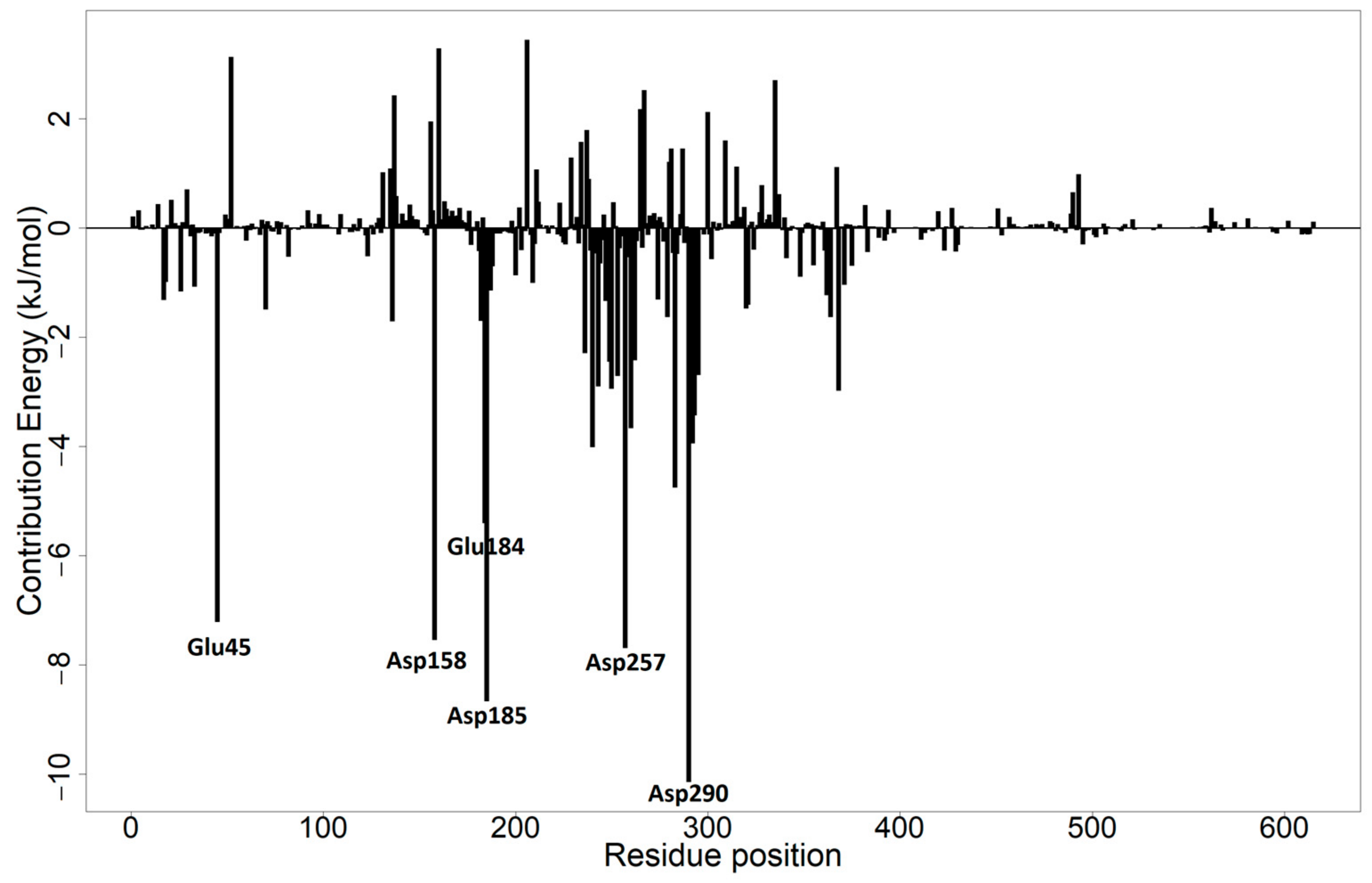
3.10.2. Per-Residue Energy Decomposition
3.11. Future Outlook and Implications
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Krivtsov, A.V.; Evans, K.; Gadrey, J.Y.; Eschle, B.K.; Hatton, C.; Uckelmann, H.J.; Ross, K.N.; Perner, F.; Olsen, S.N.; Pritchard, T.; et al. A Menin-MLL Inhibitor Induces Specific Chromatin Changes and Eradicates Disease in Models of MLL-Rearranged Leukemia. Cancer Cell 2019, 36, 660–673.e11. [Google Scholar] [CrossRef] [PubMed]
- Thiel, A.T.; Huang, J.; Lei, M.; Hua, X. Menin as a hub controlling mixed lineage leukemia. BioEssays 2012, 34, 771–780. [Google Scholar] [CrossRef] [PubMed]
- Lei, H.; Zhang, S.-Q.; Fan, S.; Bai, H.-R.; Zhao, H.-Y.; Mao, S.; Xin, M. Recent Progress of Small Molecule Menin–MLL Interaction Inhibitors as Therapeutic Agents for Acute Leukemia. J. Med. Chem. 2021, 64, 15519–15533. [Google Scholar] [CrossRef] [PubMed]
- Brown, P. Treatment of Infant Leukemias: Challenge and Promise. Hematol. Am. Soc. Hematol. Educ. Progr. 2013, 176, 596–600. [Google Scholar] [CrossRef]
- Winters, A.C.; Bernt, K.M. MLL-rearranged leukemias- An update on science and clinical approaches. Front. Pediatr. 2017, 5, 4. [Google Scholar] [CrossRef]
- McBride, A.; Houtmann, S.; Wilde, L.; Vigil, C.; Eischen, C.M.; Kasner, M.; Palmisiano, N. The role of inhibition of apoptosis in acute leukemias and myelodysplastic syndrome. Front. Oncol. 2019, 9, 192. [Google Scholar] [CrossRef]
- Klossowski, S.; Miao, H.; Kempinska, K.; Wu, T.; Purohit, T.; Kim, E.G.; Linhares, B.M.; Chen, D.; Jih, G.; Perkey, E.; et al. Menin inhibitor MI-3454 induces remission in MLL1-rearranged and NPM1-mutated models of leukemia. J. Clin. Investig. 2020, 130, 981–997. [Google Scholar] [CrossRef]
- Xu, Y.; Yue, L.; Wang, Y.; Xing, J.; Chen, Z.; Shi, Z.; Liu, R.; Liu, Y.-C.; Luo, X.; Jiang, H.; et al. Discovery of Novel Inhibitors Targeting the Menin-Mixed Lineage Leukemia Interface Using Pharmacophore- and Docking-Based Virtual Screening. J. Chem. Inf. Model. 2016, 56, 1847–1855. [Google Scholar] [CrossRef]
- McMahon, C.M. Menin Inhibitors: A Promising Class of Targeted Therapies for KMT2A-rearranged and NPM1-mutated Acute Leukemias. Hematologist 2022, 19. [Google Scholar] [CrossRef]
- Li, X.; Song, Y. Structure, function and inhibition of critical protein–protein interactions involving mixed lineage leukemia 1 and its fusion oncoproteins. J. Hematol. Oncol. 2021, 14, 56. [Google Scholar] [CrossRef]
- Shi, A.; Murai, M.J.; He, S.; Lund, G.; Hartley, T.; Purohit, T.; Reddy, G.; Chruszcz, M.; Grembecka, J.; Cierpicki, T. Structural insights into inhibition of the bivalent menin-MLL interaction by small molecules in leukemia. Blood 2012, 120, 4461–4469. [Google Scholar] [CrossRef]
- Gundry, M.C.; Goodell, M.A.; Brunetti, L. It’s All About MEis: Menin-MLL Inhibition Eradicates NPM1-Mutated and MLL-Rearranged Acute Leukemias in Mice. Cancer Cell 2020, 37, 267–269. [Google Scholar] [CrossRef]
- Jiang, M.; Xu, S.; Bai, M.; Zhang, A. The emerging role of MEIS1 in cell proliferation and differentiation. Am. J. Physiol. Cell Physiol. 2021, 320, C264–C269. [Google Scholar] [CrossRef]
- Wu, Y.; Doepner, M.; Hojnacki, T.; Feng, Z.; Katona, B.W.; He, X.; Ma, J.; Cao, Y.; Busino, L.; Zhou, F.; et al. Disruption of the menin-MLL interaction triggers menin protein degradation via ubiquitin-proteasome pathway. Am. J. Cancer Res. 2019, 9, 1682–1694. [Google Scholar]
- Swaminathan, M.; Bourgeois, W.; Armstrong, S.A.; Wang, E.S. Menin Inhibitors in Acute Myeloid Leukemia—What Does the Future Hold? Cancer J. 2022, 28, 62–66. [Google Scholar] [CrossRef]
- Chen, C.Y.-C. TCM Database@Taiwan: The world’s largest traditional Chinese medicine database for drug screening in silico. PLoS ONE 2011, 6, e15939. [Google Scholar] [CrossRef]
- Eccleston, A. Investigating menin inhibitor action in leukaemia. Nat. Rev. Drug Discov. 2023, 22, 352. [Google Scholar] [CrossRef]
- Libbrecht, C.; Riedel, S.S.; Haladyna, J.; Bernt, K.M. Menin Is a Therapeutic Target in MN1 High Leukemia. Blood 2018, 132, 758. [Google Scholar] [CrossRef]
- Dhingra, N. Computer-Aided Drug Design and Development: An Integrated Approach. In Drug Development Life Cycle; IntechOpen: London, UK, 2022; Volume 11, p. 13. ISBN 0000957720. [Google Scholar]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Filimonov, D.A.; Rudik, A.V.; Dmitriev, A.V.; Poroikov, V.V. Computer-Aided Estimation of Biological Activity Profiles of Drug-like Compounds Taking into Account Their Metabolism in Human Body. Int. J. Mol. Sci. 2020, 21, 7492. [Google Scholar] [CrossRef]
- Burley, S.K.; Bhikadiya, C.; Bi, C.; Bittrich, S.; Chen, L.; Crichlow, G.V.; Christie, C.H.; Dalenberg, K.; Di Costanzo, L.; Duarte, J.M.; et al. RCSB Protein Data Bank: Powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences. Nucleic Acids Res. 2021, 49, D437–D451. [Google Scholar] [CrossRef]
- Coudert, E.; Gehant, S.; de Castro, E.; Pozzato, M.; Baratin, D.; Neto, T.; Sigrist, C.J.A.; Redaschi, N.; Bridge, A. Annotation of biologically relevant ligands in UniProtKB using ChEBI. Bioinformatics 2023, 39, btac793. [Google Scholar] [CrossRef]
- Kuntal, B.K.; Aparoy, P.; Reddanna, P. EasyModeller: A graphical interface to MODELLER. BMC Res. Notes 2010, 3, 226. [Google Scholar] [CrossRef]
- Fiser, A.; Šali, A. Modeller: Generation and Refinement of Homology-Based Protein Structure Models. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2003; pp. 461–491. ISBN 9780121827779. [Google Scholar]
- Eswar, N.; Eramian, D.; Webb, B.; Shen, M.Y.; Sali, A. Protein structure modeling with MODELLER. Methods Mol. Biol. 2008, 426, 145–159. [Google Scholar] [CrossRef]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2014, 12, 7–8. [Google Scholar] [CrossRef]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef]
- Zheng, W.; Zhang, C.; Li, Y.; Pearce, R.; Bell, E.W.; Zhang, Y. Folding non-homologous proteins by coupling deep-learning contact maps with I-TASSER assembly simulations. Cell Rep. Methods 2021, 1, 100014. [Google Scholar] [CrossRef]
- Yang, J.; Roy, A.; Zhang, Y. Protein-ligand binding site recognition using complementary binding-specific substructure comparison and sequence profile alignment. Bioinformatics 2013, 29, 2588–2595. [Google Scholar] [CrossRef]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef]
- Bowie, J.; Luthy, R.; Eisenberg, D. A method to identify protein sequences that fold into a known three-dimensional structure. Science 1991, 253, 164–170. [Google Scholar] [CrossRef]
- Lüthy, R.; Bowie, J.U.; Eisenberg, D. Assessment of protein models with three-dimensional profiles. Nature 1992, 356, 83–85. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Sander, T.; Freyss, J.; Von Korff, M.; Rufener, C. DataWarrior: An open-source program for chemistry aware data visualization and analysis. J. Chem. Inf. Model. 2015, 55, 460–473. [Google Scholar] [CrossRef]
- Broni, E.; Striegel, A.; Ashley, C.; Sakyi, P.O.; Peracha, S.; Velazquez, M.; Bebla, K.; Sodhi, M.; Kwofie, S.K.; Ademokunwa, A.; et al. Molecular Docking and Dynamics Simulation Studies Predict Potential Anti-ADAR2 Inhibitors: Implications for the Treatment of Cancer, Neurological, Immunological and Infectious Diseases. Int. J. Mol. Sci. 2023, 24, 6795. [Google Scholar] [CrossRef]
- Kwofie, S.; Broni, E.; Yunus, F.; Nsoh, J.; Adoboe, D.; Miller, W.; Wilson, M. Molecular Docking Simulation Studies Identifies Potential Natural Product Derived-Antiwolbachial Compounds as Filaricides against Onchocerciasis. Biomedicines 2021, 9, 1682. [Google Scholar] [CrossRef]
- Broni, E.; Ashley, C.; Adams, J.; Manu, H.; Aikins, E.; Okom, M.; Miller, W.A.; Wilson, M.D.; Kwofie, S.K. Cheminformatics-Based Study Identifies Potential Ebola VP40 Inhibitors. Int. J. Mol. Sci. 2023, 24, 6298. [Google Scholar] [CrossRef]
- Broni, E.; Ashley, C.; Velazquez, M.; Khan, S.; Striegel, A.; Sakyi, P.O.; Peracha, S.; Bebla, K.; Sodhi, M.; Kwofie, S.K.; et al. In Silico Discovery of Potential Inhibitors Targeting the RNA Binding Loop of ADAR2 and 5-HT2CR from Traditional Chinese Natural Compounds. Int. J. Mol. Sci. 2023, 24, 12612. [Google Scholar] [CrossRef]
- Rappe, A.K.; Casewit, C.J.; Colwell, K.S.; Goddard, W.A.; Skiff, W.M. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc. 1992, 114, 10024–10035. [Google Scholar] [CrossRef]
- Artemova, S.; Jaillet, L.; Redon, S. Automatic molecular structure perception for the universal force field. J. Comput. Chem. 2016, 37, 1191–1205. [Google Scholar] [CrossRef]
- Yuan, S.; Chan, H.C.S.; Hu, Z. Using PyMOL as a platform for computational drug design. WIREs Comput. Mol. Sci. 2017, 7, e1298. [Google Scholar] [CrossRef]
- Kondapuram, S.K.; Sarvagalla, S.; Coumar, M.S. Docking-Based Virtual Screening Using PyRx Tool: Autophagy Target Vps34 as a Case Study. In Molecular Docking for Computer-Aided Drug Design; Coumar, M.S., Ed.; Academic Press: Cambridge, MA, USA, 2021; pp. 463–477. [Google Scholar] [CrossRef]
- Alves, M.; Froufe, H.; Costa, A.; Santos, A.; Oliveira, L.; Osório, S.; Abreu, R.; Pintado, M.; Ferreira, I. Docking Studies in Target Proteins Involved in Antibacterial Action Mechanisms: Extending the Knowledge on Standard Antibiotics to Antimicrobial Mushroom Compounds. Molecules 2014, 19, 1672–1684. [Google Scholar] [CrossRef]
- Kwofie, S.; Dankwa, B.; Odame, E.; Agamah, F.; Doe, L.; Teye, J.; Agyapong, O.; Miller, W.; Mosi, L.; Wilson, M. In Silico Screening of Isocitrate Lyase for Novel Anti-Buruli Ulcer Natural Products Originating from Africa. Molecules 2018, 23, 1550. [Google Scholar] [CrossRef]
- Jaundoo, R.; Bohmann, J.; Gutierrez, G.; Klimas, N.; Broderick, G.; Craddock, T. Using a Consensus Docking Approach to Predict Adverse Drug Reactions in Combination Drug Therapies for Gulf War Illness. Int. J. Mol. Sci. 2018, 19, 3355. [Google Scholar] [CrossRef]
- Kapale, S.S.; Mali, S.N.; Chaudhari, H.K. Molecular modelling studies for 4-oxo-1,4-dihydroquinoline-3-carboxamide derivatives as anticancer agents. Med. Drug Discov. 2019, 2, 100008. [Google Scholar] [CrossRef]
- Hu, J.; Liu, Z.; Yu, D.J.; Zhang, Y. LS-align: An atom-level, flexible ligand structural alignment algorithm for high-throughput virtual screening. Bioinformatics 2018, 34, 2209–2218. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.W.; Lindstrom, W.; Olson, A.J.; Belew, R.K. Analysis of HIV wild-type and mutant structures via in silico docking against diverse ligand libraries. J. Chem. Inf. Model. 2007, 47, 1258–1262. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Druzhilovskiy, D.S.; Rudik, A.V.; Filimonov, D.A.; Gloriozova, T.A.; Lagunin, A.A.; Dmitriev, A.V.; Pogodin, P.V.; Dubovskaya, V.I.; Ivanov, S.M.; Tarasova, O.A.; et al. Computational platform Way2Drug: From the prediction of biological activity to drug repurposing. Russ. Chem. Bull. 2018, 66, 1832–1841. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple Ligand-_Protein Interaction Diagrams for Drug Discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Raman, E.P.; MacKerell, A.D.J. Automation of the CHARMM General Force Field (CGenFF) II: Assignment of bonded parameters and partial atomic charges. J. Chem. Inf. Model. 2012, 52, 3155–3168. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Lynn, A. g_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Campanera, J.M.; Pouplana, R. MMPBSA Decomposition of the Binding Energy throughout a Molecular Dynamics Simulation of Amyloid-Beta (Aß10−35) Aggregation. Molecules 2010, 15, 2730–2748. [Google Scholar] [CrossRef]
- Wang, C.; Greene, D.; Xiao, L.; Qi, R.; Luo, R. Recent Developments and Applications of the MMPBSA Method. Front. Mol. Biosci. 2018, 4, 87. [Google Scholar] [CrossRef]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Development Core Team: Vienna, Austria, 2008; Volume 2, ISBN 3-900051-07-0. [Google Scholar]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server. In The Proteomics Protocols Handbook; Walker, J.M., Ed.; Humana Press: Totowa, NJ, USA, 2005; ISBN 978-1-58829-343-5. [Google Scholar]
- Borkin, D.; He, S.; Miao, H.; Kempinska, K.; Pollock, J.; Chase, J.; Purohit, T.; Malik, B.; Zhao, T.; Wang, J.; et al. Pharmacologic inhibition of the menin-MLL interaction blocks progression of MLL leukemia in vivo. Cancer Cell 2015, 27, 589–602. [Google Scholar] [CrossRef]
- Huang, J.; Gurung, B.; Wan, B.; Matkar, S.; Veniaminova, N.A.; Wan, K.; Merchant, J.L.; Hua, X.; Lei, M. The same pocket in menin binds both MLL and JUND but has opposite effects on transcription. Nature 2012, 482, 542–546. [Google Scholar] [CrossRef]
- Hoda, S.; Gupta, L.; Shankar, J.; Gupta, A.K.; Vijayaraghavan, P. cis-9-Hexadecenal, a Natural Compound Targeting Cell Wall Organization, Critical Growth Factor, and Virulence of Aspergillus fumigatus. ACS Omega 2020, 5, 10077–10088. [Google Scholar] [CrossRef]
- He, S.; Senter, T.J.; Pollock, J.; Han, C.; Upadhyay, S.K.; Purohit, T.; Gogliotti, R.D.; Lindsley, C.W.; Cierpicki, T.; Stauffer, S.R.; et al. High-Affinity Small-Molecule Inhibitors of the Menin-Mixed Lineage Leukemia (MLL) Interaction Closely Mimic a Natural Protein–Protein Interaction. J. Med. Chem. 2014, 57, 1543–1556. [Google Scholar] [CrossRef]
- Li, L.; Zhou, R.; Geng, H.; Yue, L.; Ye, F.; Xie, Y.; Liu, J.; Kong, X.; Jiang, H.; Huang, J.; et al. Discovery of two aminoglycoside antibiotics as inhibitors targeting the menin-mixed lineage leukaemia interface. Bioorganic Med. Chem. Lett. 2014, 24, 2090–2093. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef]
- Ramírez, D.; Caballero, J. Is It Reliable to Take the Molecular Docking Top Scoring Position as the Best Solution without Considering Available Structural Data? Molecules 2018, 23, 1038. [Google Scholar] [CrossRef]
- Houston, D.R.; Walkinshaw, M.D. Consensus docking: Improving the reliability of docking in a virtual screening context. J. Chem. Inf. Model. 2013, 53, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Biancaniello, C.; D’argenio, A.; Giordano, D.; Dotolo, S.; Scafuri, B.; Marabotti, A.; D’acierno, A.; Tagliaferri, R.; Facchiano, A. Investigating the Effects of Amino Acid Variations in Human Menin. Molecules 2022, 27, 1747. [Google Scholar] [CrossRef]
- Benet, L.Z.; Hosey, C.M.; Ursu, O.; Oprea, T.I. BDDCS, the Rule of 5 and Drugability. Adv. Drug Deliv. Rev. 2016, 101, 89–98. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Cragg, G.M.; Pezzuto, J.M. Natural Products as a Vital Source for the Discovery of Cancer Chemotherapeutic and Chemopreventive Agents. Med. Princ. Pract. 2016, 25, 41–59. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef]
- Jamkhande, P.G.; Pathan, S.K.; Wadher, S.J. In silico PASS analysis and determination of antimycobacterial, antifungal, and antioxidant efficacies of maslinic acid in an extract rich in pentacyclic triterpenoids. Int. J. Mycobacteriology 2016, 5, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Parasuraman, S. Prediction of activity spectra for substances. J. Pharmacol. Pharmacother. 2011, 2, 52–53. [Google Scholar] [CrossRef] [PubMed]
- Eldridge, S.; Davis, M. Antineoplastic Agents. In Comprehensive Toxicology, 3rd ed.; McQueen, C.A., Ed.; Elsevier Science: Amsterdam, The Netherlands, 2018; Volume 3, pp. 219–232. [Google Scholar] [CrossRef]
- Dagli, M.L.Z. Connexins/Gap Junction Based Agents in Cancer. In Comprehensive Pharmacology; Elsevier: Amsterdam, The Netherlands, 2022; Volume 6, pp. 419–437. ISBN 9780128204726. [Google Scholar]
- Dorai, T.; Aggarwal, B.B. Role of chemopreventive agents in cancer therapy. Cancer Lett. 2004, 215, 129–140. [Google Scholar] [CrossRef]
- Kotecha, R.; Takami, A.; Espinoza, J.L. Dietary phytochemicals and cancer chemoprevention: A review of the clinical evidence. Oncotarget 2016, 7, 52517–52529. [Google Scholar] [CrossRef]
- Langner, E.; Rzeski, W. Dietary derived compounds in cancer chemoprevention. Contemp. Oncol. 2012, 16, 394–400. [Google Scholar] [CrossRef]
- Kagan, A.B.; Moses, B.S.; Mott, B.T.; Rai, G.; Anders, N.M.; Rudek, M.A.; Civin, C.I. A Novel 2-Carbon-Linked Dimeric Artemisinin With Potent Antileukemic Activity and Favorable Pharmacology. Front. Oncol. 2022, 11, 790037. [Google Scholar] [CrossRef]
- Herschlag, D.; Pinney, M.M. Hydrogen Bonds: Simple after All? Biochemistry 2018, 57, 3338–3352. [Google Scholar] [CrossRef]
- McRee, D.E. Computational Techniques. In Practical Protein Crystallography, 2nd ed.; Elsevier: San Diego, CA, USA, 1999; p. 91–cp1. ISBN 978-0-12-486052-0. [Google Scholar]
- Lobanov, M.Y.; Bogatyreva, N.S.; Galzitskaya, O.V. Radius of gyration as an indicator of protein structure compactness. Mol. Biol. 2008, 42, 623–628. [Google Scholar] [CrossRef]
- Cheng, X.; Ivanov, I. Molecular Dynamics. Computational Toxicology: Volume I. In Methods in Molecular Biology; Clifton, N.J., Ed.; Springer: Berlin/Heidelberg, Germany, 2012; Volume 929, pp. 243–285. ISBN 9781627030496. [Google Scholar] [CrossRef]
- Deng, N.; Zhang, P.; Cieplak, P.; Lai, L. Elucidating the Energetics of Entropically Driven Protein–Ligand Association: Calculations of Absolute Binding Free Energy and Entropy. J. Phys. Chem. B 2011, 115, 11902–11910. [Google Scholar] [CrossRef]
- Tian, M.; Li, H.; Yan, X.; Gu, J.; Zheng, P.; Luo, S.; Zhangsun, D.; Chen, Q.; Ouyang, Q. Application of per-Residue Energy Decomposition to Design Peptide Inhibitors of PSD95 GK Domain. Front. Mol. Biosci. 2022, 9, 848353. [Google Scholar] [CrossRef]
- Kwofie, S.; Dankwa, B.; Enninful, K.; Adobor, C.; Broni, E.; Ntiamoah, A.; Wilson, M. Molecular Docking and Dynamics Simulation Studies Predict Munc18b as a Target of Mycolactone: A Plausible Mechanism for Granule Exocytosis Impairment in Buruli Ulcer Pathogenesis. Toxins 2019, 11, 181. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Binding Energy (kcal/mol) | Interacting Residues | ||
|---|---|---|---|---|
| ITAS1 | Best Cluster | Hydrogen Bonds [Bond Length (Å)] | Hydrophobic Bonds | |
| MI-2-2 | −7.8 | −7.5 | Ser160 (2.84) and Ser183 (3.23) | Asp185, His186, Phe243, Cys246, Ala247, Ile248, Asn249, Tyr281, and Met283 |
| 36294 | −6.7 | −6.7 | Asp158 (2.94), Ser160 (3.3), Ser183 (2.79), His186 (2.95), Cys246 (2.9), and Tyr328 (2.83) | Ser159, Leu182, Glu184, Asp185, Phe243, Ala247, and Met283 |
| 71777742 | −6.5 | −6.9 | - | Gln141, Asp158, Ser183, Phe243, Cys246, Ala247, Tyr281, and Met283 |
| ZINC000103526876 | −11.0 | −9.8 | Ser160 (2.92) | Gln141, Asp158, Leu182, Glu184, Asp185, His186, Ala187, His204, Phe243, Cys246, Ala247, Tyr281, Met283, and Glu364 |
| ZINC000095913861 | −10.7 | −10.1 | Ser160 (3.19) and Asn287 (2.7) | Leu182, Ser183, Phe243, Asn249, Pro250, Ser251, Tyr281, Met283, Leu291, and Leu294 |
| ZINC000095912705 | −10.6 | −8.1 | Asn287 (2.99) | Ser160, Leu182, Ser183, Asp185, His186, Ala187, Met233, Phe243, Cys246, Ala247, Ile248, Asn249, Pro250, Tyr281, Met283, and Tyr328 |
| ZINC000085530497 | −10.2 | −9.5 | - | Asp158, Ser160, Phe243, Cys246, Ala247, Ile248, Asn249, Gln265, Tyr281, Met283, and Asn287 |
| ZINC000095912718 | −10.2 | −9.5 | - | Ser160, Phe243, Ala247, Cys246, Pro250, Gln265, Tyr281, Met283, Asn287, Asp290, and Tyr328 |
| ZINC000070451048 | −9.9 | −8.1 | Cys246 (3.02), Ile248 (2.9) and Tyr328 (3.14) | Leu182, Ser183, Glu184, Asp185, His186, Phe243, Ala247, Tyr281, Asn287, Met283, and Asp290 |
| ZINC000085530488 | −9.9 | −8.9 | Ser160 (3.15) and Tyr328 (2.7) | Gln141, Asp158, Asp185, Phe243, Cys246, Ala247, Tyr281, Met283, Asn287, and Tyr324 |
| ZINC000095912706 | −9.9 | −8.1 | Ser160 (3.13 and 3.19), Asn249 (2.97) and er251 (2.93) | Leu182, Ser183, Asp185, His186, Ala187, Phe243, Cys246, Ala247, Ile248, Pro250, Tyr281, Met283, and Asn287 |
| ZINC000103580868 | −9.9 | −9.5 | - | Ser160, Glu184, Phe243, Cys246, Ala247, Ile248, Asn249, Pro250, Ser251, Asp257, Leu262, Gln265, Tyr281, Met283, Asn287, Leu291, and Tyr324 |
| ZINC000103584057 | −9.9 | −8.7 | Arg335 (2.94) | Phe243, Cys246, Tyr281, Met283, Asn287, Asp290, Tyr324, Tyr328, and Glu368 |
| System/Complex | Run | RMSD | Rg | RMSF | H-Bonds | |||
|---|---|---|---|---|---|---|---|---|
| Complete | Ordered | Complete | Ordered | Complete | Ordered | |||
| Menin | 1 | 0.66 ± 0.11 | 0.52 ± 0.07 | 2.94 ± 0.02 | 2.71 ± 0.02 | - | - | - |
| 2 | 0.47 ± 0.08 | 0.45 ± 0.07 | 2.79 ± 0.03 | 2.64 ± 0.03 | - | - | - | |
| 3 | 0.49 ± 0.08 | 0.45 ± 0.06 | 2.84 ± 0.03 | 2.66 ± 0.03 | - | - | - | |
| Avg | 0.54 ± 0.09 | 0.47 ± 0.03 | 2.86 ± 0.06 | 2.67 ± 0.03 | - | - | - | |
| Menin–MI-2-2 | 1 | 0.8 ± 0.18 | 0.79 ± 0.2 | 2.87 ± 0.08 | 2.67 ± 0.08 | 0.32 ± 0.34 | 0.29 ± 0.34 | 0.61 ± 0.54 |
| 2 | 0.79 ± 0.16 | 0.72 ± 0.18 | 2.84 ± 0.05 | 2.63 ± 0.06 | 0.32 ± 0.31 | 0.26 ± 0.31 | 1.11 ± 0.76 | |
| 3 | 0.57 ± 0.07 | 0.53 ± 0.07 | 2.8 ± 0.03 | 2.6 ± 0.04 | 0.26 ± 0.22 | 0.19 ± 0.17 | 0.16 ± 0.37 | |
| Avg | 0.72 ± 0.1 | 0.68 ± 0.11 | 2.84 ± 0.03 | 2.63 ± 0.03 | 0.3 ± 0.03 | 0.25 ± 0.04 | 0.63 ± 0.39 | |
| Menin–36294 | 1 | 0.75 ± 0.13 | 0.7 ± 0.1 | 2.84 ± 0.06 | 2.59 ± 0.04 | 0.27 ± 0.22 | 0.2 ± 0.14 | 2.9 ± 1.53 |
| 2 * | 0.56 ± 0.09 * | 0.46 ± 0.09 * | 2.82 ± 0.04 * | 2.62 ± 0.03 * | 0.26 ± 0.2 * | 0.2 ± 0.16 * | 0.95 ± 1.4 * | |
| 3 | 0.55 ± 0.07 | 0.46 ± 0.05 | 2.83 ± 0.04 | 2.65 ± 0.03 | 0.24 ± 0.2 | 0.19 ± 0.17 | 2.82 ± 1.33 | |
| Avg | 0.65 ± 0.1 | 0.58 ± 0.12 | 2.84 ± 0.01 | 2.62 ± 0.03 | 0.255 ± 0.02 | 0.195 ± 0.01 | 2.86 ± 0.04 | |
| Menin–71777742 | 1 | 0.8 ± 0.23 | 0.8 ± 0.26 | 2.86 ± 0.07 | 2.66 ± 0.09 | 0.35 ± 0.44 | 0.33 ± 0.44 | 0.11 ± 0.31 |
| 2 * | 0.65 ± 0.1 * | 0.59 ± 0.1 * | 2.77 ± 0.03 * | 2.61 ± 0.04 * | 0.25 ± 0.25 * | 0.21 ± 0.24 * | 0.15 ± 0.36 * | |
| 3 | 0.72 ± 0.1 | 0.72 ± 0.1 | 2.74 ± 0.04 | 2.57 ± 0.03 | 0.22 ± 0.19 | 0.18 ± 0.18 | 0.32 ± 0.53 | |
| Avg | 0.76 ± 0.04 | 0.76 ± 0.04 | 2.8 ± 0.06 | 2.62 ± 0.05 | 0.29 ± 0.07 | 0.26 ± 0.08 | 0.22 ± 0.11 | |
| Menin–ZINC000103526876 | 1 | 0.71 ± 0.12 | 0.55 ± 0.08 | 2.85 ± 0.04 | 2.63 ± 0.04 | 0.27 ± 0.22 | 0.19 ± 0.17 | 1.56 ± 0.76 |
| 2 | 0.58 ± 0.11 | 0.47 ± 0.09 | 2.84 ± 0.03 | 2.65 ± 0.04 | 0.3 ± 0.23 | 0.22 ± 0.2 | 1.75 ± 0.91 | |
| 3 | 0.59 ± 0.07 | 0.49 ± 0.07 | 2.85 ± 0.05 | 2.64 ± 0.05 | 0.29 ± 0.25 | 0.22 ± 0.23 | 2.51 ± 0.73 | |
| Avg | 0.63 ± 0.06 | 0.5 ± 0.03 | 2.85 | 2.64 ± 0.01 | 0.29 ± 0.01 | 0.21 ± 0.01 | 1.94 ± 0.41 | |
| Menin–ZINC000095913861 | 1 | 0.6 ± 0.1 | 0.57 ± 0.1 | 2.82 ± 0.03 | 2.65 ± 0.03 | 0.25 ± 0.22 | 0.23 ± 0.22 | 1.1 ± 0.72 |
| 2 | 0.78 ± 0.29 | 0.81 ± 0.35 | 2.81 ± 0.03 | 2.61 ± 0.05 | 0.35 ± 0.47 | 0.35 ± 0.5 | 0.59 ± 0.72 | |
| 3 | 0.68 ± 0.09 | 0.55 ± 0.07 | 2.86 ± 0.05 | 2.64 ± 0.03 | 0.26 ± 0.23 | 0.19 ± 0.18 | 0.91 ± 0.63 | |
| Avg | 0.69 ± 0.07 | 0.64 ± 0.12 | 2.83 ± 0.02 | 2.63 ± 0.02 | 0.29 ± 0.04 | 0.26 ± 0.07 | 0.87 ± 0.21 | |
| Menin–ZINC000095912705 | 1 | 0.54 ± 0.09 | 0.45 ± 0.06 | 2.89 ± 0.04 | 2.63 ± 0.03 | 0.24 ± 0.2 | 0.18 ± 0.16 | 1.51 ± 0.57 |
| 2 | 0.58 ± 0.09 | 0.52 ± 0.08 | 2.9 ± 0.02 | 2.69 ± 0.02 | 0.23 ± 0.18 | 0.18 ± 0.16 | 1.13 ± 0.36 | |
| 3 | 0.74 ± 0.14 | 0.58 ± 0.08 | 2.8 ± 0.02 | 2.58 ± 0.02 | 0.26 ± 0.24 | 0.18 ± 0.15 | 1.09 ± 0.28 | |
| Avg | 0.62 ± 0.09 | 0.52 ± 0.05 | 2.86 ± 0.04 | 2.63 ± 0.04 | 0.24 ± 0.01 | 0.18 ± 0 | 1.24 ± 0.19 | |
| Menin–ZINC000085530497 | 1 | 0.5 ± 0.07 | 0.44 ± 0.07 | 2.84 ± 0.03 | 2.66 ± 0.03 | 0.21 ± 0.17 | 0.17 ± 0.15 | 0.24 ± 0.45 |
| 2 | 0.83 ± 0.17 | 0.72 ± 0.18 | 2.92 ± 0.05 | 2.62 ± 0.05 | 0.31 ± 0.32 | 0.25 ± 0.28 | 0.8 ± 0.7 | |
| 3 | 0.75 ± 0.15 | 0.46 ± 0.08 | 2.9 ± 0.03 | 2.63 ± 0.03 | 0.26 ± 0.24 | 0.18 ± 0.18 | 1.51 ± 0.61 | |
| Avg | 0.69 ± 0.14 | 0.54 ± 0.13 | 2.89 ± 0.03 | 2.64 ± 0.02 | 0.26 ± 0.04 | 0.2 ± 0.04 | 0.85 ± 0.52 | |
| Menin–ZINC000095912718 | 1 | 0.65 ± 0.1 | 0.56 ± 0.08 | 2.9 ± 0.03 | 2.71 ± 0.03 | 0.26 ± 0.2 | 0.19 ± 0.17 | 0.81 ± 0.69 |
| 2 | 0.64 ± 0.15 | 0.63 ± 0.15 | 2.82 ± 0.03 | 2.62 ± 0.04 | 0.26 ± 0.28 | 0.22 ± 0.26 | 0.97 ± 0.26 | |
| 3 | 0.64 ± 0.15 | 0.5 ± 0.08 | 2.96 ± 0.06 | 2.73 ± 0.04 | 0.31 ± 0.24 | 0.21 ± 0.18 | 1.43 ± 0.96 | |
| Avg | 0.64 | 0.56 ± 0.05 | 2.89 | 2.69 ± 0.05 | 0.28 ± 0.02 | 0.21 ± 0.01 | 1.07 ± 0.26 | |
| Menin–ZINC000070451048 | 1 | 0.67 ± 0.12 | 0.57 ± 0.09 | 2.75 ± 0.04 | 2.6 ± 0.03 | 0.29 ± 0.24 | 0.2 ± 0.2 | 1.15 ± 0.99 |
| 2 * | 0.64 ± 0.12 * | 0.52 ± 0.15 * | 2.8 ± 0.06 * | 2.65 ± 0.05 * | 0.29 ± 0.27 * | 0.24 ± 0.24 * | 0.72 ± 0.73 * | |
| 3 | 0.7 ± 0.09 | 0.55 ± 0.07 | 2.86 ± 0.04 | 2.7 ± 0.03 | 0.28 ± 0.24 | 0.21 ± 0.2 | 1.11 ± 0.85 | |
| Avg | 0.69 ± 0.02 | 0.56 ± 0.01 | 2.8 ± 0.06 | 2.65 ± 0.05 | 0.29 ± 0.01 | 0.21 ± 0.01 | 1.13 ± 0.02 | |
| Menin–ZINC000085530488 | 1 | 0.87 ± 0.13 | 0.63 ± 0.12 | 2.9 ± 0.03 | 2.67 ± 0.03 | 0.27 ± 0.24 | 0.19 ± 0.2 | 0.15 ± 0.41 |
| 2 | 0.53 ± 0.1 | 0.5 ± 0.11 | 2.81 ± 0.05 | 2.66 ± 0.04 | 0.25 ± 0.24 | 0.2 ± 0.23 | 1.03 ± 0.78 | |
| 3 | 0.63 ± 0.1 | 0.47 ± 0.06 | 2.93 ± 0.04 | 2.67 ± 0.02 | 0.26 ± 0.22 | 0.18 ± 0.16 | 1.01 ± 0.92 | |
| Avg | 0.68 ± 0.14 | 0.53 ± 0.07 | 2.88 ± 0.05 | 2.67 | 0.26 ± 0.01 | 0.19 ± 0.01 | 0.73 ± 0.41 | |
| Menin–ZINC000095912706 | 1 | 0.58 ± 0.12 | 0.5 ± 0.11 | 2.86 ± 0.06 | 2.7 ± 0.07 | 0.29 ± 0.28 | 0.24 ± 0.25 | 1.27 ± 0.49 |
| 2 | 0.61 ± 0.08 | 0.53 ± 0.07 | 2.81 ± 0.04 | 2.64 ± 0.04 | 0.23 ± 0.18 | 0.19 ± 0.15 | 1.78 ± 0.84 | |
| 3 | 0.47 ± 0.05 | 0.41 ± 0.05 | 2.82 ± 0.03 | 2.67 ± 0.03 | 0.22 ± 0.18 | 0.18 ± 0.16 | 1.47 ± 0.57 | |
| Avg | 0.55 ± 0.06 | 0.48 ± 0.05 | 2.83 ± 0.02 | 2.67 ± 0.02 | 0.25 ± 0.03 | 0.2 ± 0.03 | 1.51 ± 0.21 | |
| Menin–ZINC000103580868 | 1 | 0.66 ± 0.11 | 0.49 ± 0.07 | 2.83 ± 0.06 | 2.65 ± 0.06 | 0.3 ± 0.28 | 0.21 ± 0.2 | 0.17 ± 0.37 |
| 2 | 0.69 ± 0.11 | 0.7 ± 0.12 | 2.79 ± 0.04 | 2.58 ± 0.04 | 0.27 ± 0.22 | 0.22 ± 0.19 | 0.01 ± 0.1 | |
| 3 | 0.52 ± 0.07 | 0.44 ± 0.05 | 2.85 ± 0.04 | 2.67 ± 0.03 | 0.24 ± 0.17 | 0.18 ± 0.12 | 0.02 ± 0.14 | |
| Avg | 0.62 ± 0.07 | 0.54 ± 0.11 | 2.82 ± 0.02 | 2.63 ± 0.04 | 0.27 ± 0.02 | 0.2 ± 0.02 | 0.07 ± 0.07 | |
| Menin–ZINC000103584057 | 1 | 0.56 ± 0.09 | 0.45 ± 0.06 | 2.84 ± 0.04 | 2.64 ± 0.03 | 0.28 ± 0.21 | 0.21 ± 0.19 | 0.2 ± 0.42 |
| 2 | 0.56 ± 0.08 | 0.5 ± 0.08 | 2.79 ± 0.04 | 2.62 ± 0.03 | 0.25 ± 0.2 | 0.2 ± 0.17 | 0.19 ± 0.39 | |
| 3 | 0.54 ± 0.1 | 0.45 ± 0.08 | 2.85 ± 0.03 | 2.62 ± 0.03 | 0.25 ± 0.19 | 0.19 ± 0.14 | 0.66 ± 0.63 | |
| Avg | 0.55 ± 0.01 | 0.47 ± 0.02 | 2.83 ± 0.03 | 2.63 ± 0.01 | 0.26 ± 0.01 | 0.2 ± 0.01 | 0.35 ± 0.22 | |
| Compound | MD Run | vdW | Electrostatic | Polar Solvation | SASA | Binding Energy |
|---|---|---|---|---|---|---|
| MI−2−2 | 1 | −131.5 ± 1.2 | −16.2 ± 1.3 | 82.4 ± 2 | −16.8 ± 0.1 | −82.1 ± 2 |
| 2 | −123.5 ± 1.7 | −20.7 ± 1.7 | 103.3 ± 3.9 | −15.8 ± 0.2 | −56.8 ± 2.9 | |
| 3 | −157 ± 1.3 | −2.9 ± 0.6 | 62.2 ± 1.3 | −18.5 ± 0.1 | −116.2 ± 1.5 | |
| Avg | −137.3 ± 14.3 | −13.3 ± 7.6 | 82.6 ± 16.7 | −17 ± 1.1 | −85 ± 24.3 | |
| 36294 | 1 | −108.3 ± 1.5 | −53.6 ± 2.5 | 129.1 ± 2.7 | −16.3 ± 0.1 | −49.3 ± 1.7 |
| 2 * | −36.5 ± 3.9 * | −19.4 ± 2.8 * | 50.5 ± 7.5 * | −6.1 ± 0.6 * | −11.9 ± 5.9 * | |
| 3 | −126 ± 1.6 | −61 ± 2.3 | 143.3 ± 3.4 | −17.7 ± 0.1 | −61.4 ± 2.1 | |
| Avg | −117.2 ± 8.9 | −57.3 ± 3.7 | 136.2 ± 7.1 | −17 ± 0.7 | −55.4 ± 6.1 | |
| 71777742 | 1 | −169.6 ± 1.8 | −12.9 ± 0.8 | 54.4 ± 1.6 | −20 ± 0.2 | −148.1 ± 1.7 |
| 2 * | −75.6 ± 4.6 * | −5.5 ± 0.8 * | 46.1 ± 5.3 * | −9.8 ± 0.6 * | −45 ± 6.8 * | |
| 3 | −168.9 ± 2.7 | −20.6 ± 1 | 77.7 ± 2.1 | −19.1 ± 0.2 | −130.7 ± 2.1 | |
| Avg | −169.3 ± 0.4 | −16.8 ± 3.9 | 66.1 ± 11.7 | −19.55 ± 0.5 | −139.4 ± 8.7 | |
| ZINC000103526876 | 1 | −131.4 ± 1.5 | −37.4 ± 1.8 | 106.3 ± 3.6 | −18.1 ± 0.2 | −80.8 ± 2.7 |
| 2 | −173.7 ± 2.5 | −34.1 ± 2 | 110.1 ± 3.3 | −22.4 ± 0.2 | −120.1 ± 2.5 | |
| 3 | −149.3 ± 1.3 | −25 ± 1.6 | 99.4 ± 2.2 | −18.8 ± 0.1 | −93.5 ± 1.8 | |
| Avg | −151.5 ± 17.3 | −32.2 ± 5.3 | 105.3 ± 4.4 | −19.7 ± 1.9 | −98.1 ± 16.4 | |
| ZINC000095913861 | 1 | −172.8 ± 1.1 | −53.3 ± 1.3 | 109.3 ± 2 | −21.2 ± 0.1 | −138 ± 1.7 |
| 2 | −163.4 ± 2.6 | −27.6 ± 2.2 | 95.4 ± 4.1 | −19.7 ± 0.3 | −115.5 ± 2.3 | |
| 3 | −170.3 ± 1.4 | −37.8 ± 1.1 | 89 ± 1.3 | −20.9 ± 0.1 | −139.8 ± 1.7 | |
| Avg | −168.8 ± 4 | −39.6 ± 10.6 | 97.9 ± 8.5 | −20.6 ± 0.6 | −131.1 ± 11 | |
| ZINC000095912705 | 1 | −149.9 ± 1.2 | −21.1 ± 1 | 90.8 ± 1.8 | −19.1 ± 0.1 | −99.3 ± 1.5 |
| 2 | −134 ± 1.2 | −22.5 ± 1.1 | 77.8 ± 1.7 | −17.8 ± 0.1 | −96.5 ± 1.5 | |
| 3 | −151 ± 1.4 | −18.4 ± 1 | 91.4 ± 2 | −19.3 ± 0.1 | −97.3 ± 1.6 | |
| Avg | −145 ± 7.8 | −20.7 ± 1.7 | 86.7 ± 6.3 | −18.7 ± 0.7 | −97.7 ± 1.2 | |
| ZINC000085530497 | 1 | −171 ± 1 | −2 ± 1 | 80 ± 1.9 | −20.3 ± 0.1 | −113.3 ± 1.5 |
| 2 | −177.1 ± 1.9 | −28.4 ± 1.8 | 103.1 ± 2.4 | −20.1 ± 0.1 | −122.4 ± 2.2 | |
| 3 | −140.7 ± 1.3 | −46.3 ± 1.2 | 102.3± 2.4 | −17.5 ± 0.1 | −102.3 ± 2 | |
| Avg | −162.9 ± 15.9 | −25.6 ± 18.2 | 95.1 ± 10.7 | −19.3 ± 1.2 | −112.7 ± 8.2 | |
| ZINC000095912718 | 1 | −163.1 ± 1.8 | −19 ± 1.3 | 131.4 ± 2.3 | −21.4 ± 0.2 | −72.1 ± 1.7 |
| 2 | −146.1 ± 1.6 | −51.1 ± 1.1 | 72.5 ± 1.4 | −18.5 ± 0.2 | −143.2 ± 2.1 | |
| 3 | −147.2 ± 1.1 | −59.9 ± 1.3 | 122.6 ± 1.8 | −18.9 ± 0.1 | −103.4 ± 1.8 | |
| Avg | −152.1 ± 7.7 | −43.4 ± 17.6 | 108.8 ± 25.9 | −19.6 ± 1.3 | −106.3 ± 29.1 | |
| ZINC000070451048 | 1 | −123.1 ± 1.8 | −123 ± 1.7 | 68.3 ± 2.6 | −16.3 ± 0.2 | −85.4 ± 1.9 |
| 2 * | −95.4 ± 4.8 * | −28.4 ± 2.2 * | 85.4 ± 5.5 * | −13.3 ± 0.6 * | −51.6 ± 4.4 * | |
| 3 | −165.9 ± 2.2 | −18.5 ± 1.7 | 110.7 ± 3.4 | −19.8 ± 0.2 | −93.5 ± 1.6 | |
| Avg | −144.5 ± 21.4 | −70.8 ± 52.3 | 89.5 ± 21.2 | −18.1 ± 1.8 | −89.5 ± 4.1 | |
| ZINC000085530488 | 1 | −154.9 ± 1 | −34.2 ± 1.7 | 106.8 ± 2.3 | −19.4 ± 0.1 | −101.6 ± 1.8 |
| 2 | −173.9 ± 1.3 | −82.1 ± 1.2 | 170.4 ± 1.4 | −19.8 ± 0.1 | −105.3 ± 1.5 | |
| 3 | −147.8 ± 1.4 | −29.1 ± 1.8 | 120.9 ± 2.8 | −18 ± 0.1 | −74 ± 2.3 | |
| Avg | −158.9 ± 11 | −48.4 ± 23.9 | 132.7 ± 27.3 | −19.1 ± 0.8 | −93.6 ± 14 | |
| ZINC000095912706 | 1 | −152.8 ± 1.2 | −40.6 ± 1.2 | 115.3 ± 2 | −19.1 ± 0.1 | −97.3 ± 1.5 |
| 2 | −163.8 ± 1.4 | −31.9 ± 1.3 | 127.3 ± 2.3 | −20.5 ± 0.1 | −88.9 ± 1.9 | |
| 3 | −165 ± 1.5 | −30.4 ± 1.5 | 137.2 ± 3.8 | −21 ± 0.2 | −79 ± 2.1 | |
| Avg | −160.5 ± 5.5 | −34.3 ± 4.5 | 126.6 ± 9 | −20.2 ± 0.8 | −88.4 ± 7.4 | |
| ZINC000103580868 | 1 | −172.4 ± 1.9 | −8.6 ± 0.7 | 94.3 ± 3.4 | −21.6 ± 0.2 | −108.3 ± 3.2 |
| 2 | −177.9 ± 1.6 | −1.8 ± 0.4 | 56.3 ± 1.8 | −21.1 ± 0.2 | −144.6 ± 2 | |
| 3 | −153.6 ± 2.2 | −2.2 ± 0.6 | 39.6 ± 2.3 | −19.7 ± 0.2 | −135.9 ± 2.8 | |
| Avg | −168 ± 10.4 | −4.2 ± 3.1 | 63.4 ± 22.9 | −20.8 ± 0.8 | −129.6 ± 15.5 | |
| ZINC000103584057 | 1 | −128.6 ± 0.9 | −1.8 ± 0.9 | 22.3 ± 2 | −15.2 ± 0.1 | −123.3 ± 1.7 |
| 2 | −165.5 ± 1.5 | −4.3 ± 0.9 | 70.6 ± 1.6 | −18.2 ± 0.1 | −117.4 ± 1.9 | |
| 3 | −175.6 ± 1.9 | −9.8 ± 1.2 | 85.8 ± 1.7 | −19.4 ± 0.1 | −119 ± 1.7 | |
| Avg | −156.6 ± 20.2 | −5.3 ± 3.3 | 59.6 ± 27.1 | −17.6 ± 1.8 | −119.9 ± 2.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arthur, M.N.; Bebla, K.; Broni, E.; Ashley, C.; Velazquez, M.; Hua, X.; Radhakrishnan, R.; Kwofie, S.K.; Miller, W.A., III. Design of Inhibitors That Target the Menin–Mixed-Lineage Leukemia Interaction. Computation 2024, 12, 3. https://doi.org/10.3390/computation12010003
Arthur MN, Bebla K, Broni E, Ashley C, Velazquez M, Hua X, Radhakrishnan R, Kwofie SK, Miller WA III. Design of Inhibitors That Target the Menin–Mixed-Lineage Leukemia Interaction. Computation. 2024; 12(1):3. https://doi.org/10.3390/computation12010003
Chicago/Turabian StyleArthur, Moses N., Kristeen Bebla, Emmanuel Broni, Carolyn Ashley, Miriam Velazquez, Xianin Hua, Ravi Radhakrishnan, Samuel K. Kwofie, and Whelton A. Miller, III. 2024. "Design of Inhibitors That Target the Menin–Mixed-Lineage Leukemia Interaction" Computation 12, no. 1: 3. https://doi.org/10.3390/computation12010003