
In Silico Identification of Natural Products and World-Approved Drugs Targeting the KEAP1/NRF2 Pathway Endowed with Potential Antioxidant Profile

 , ,
, ,  and
and
Abstract
:1. Introduction
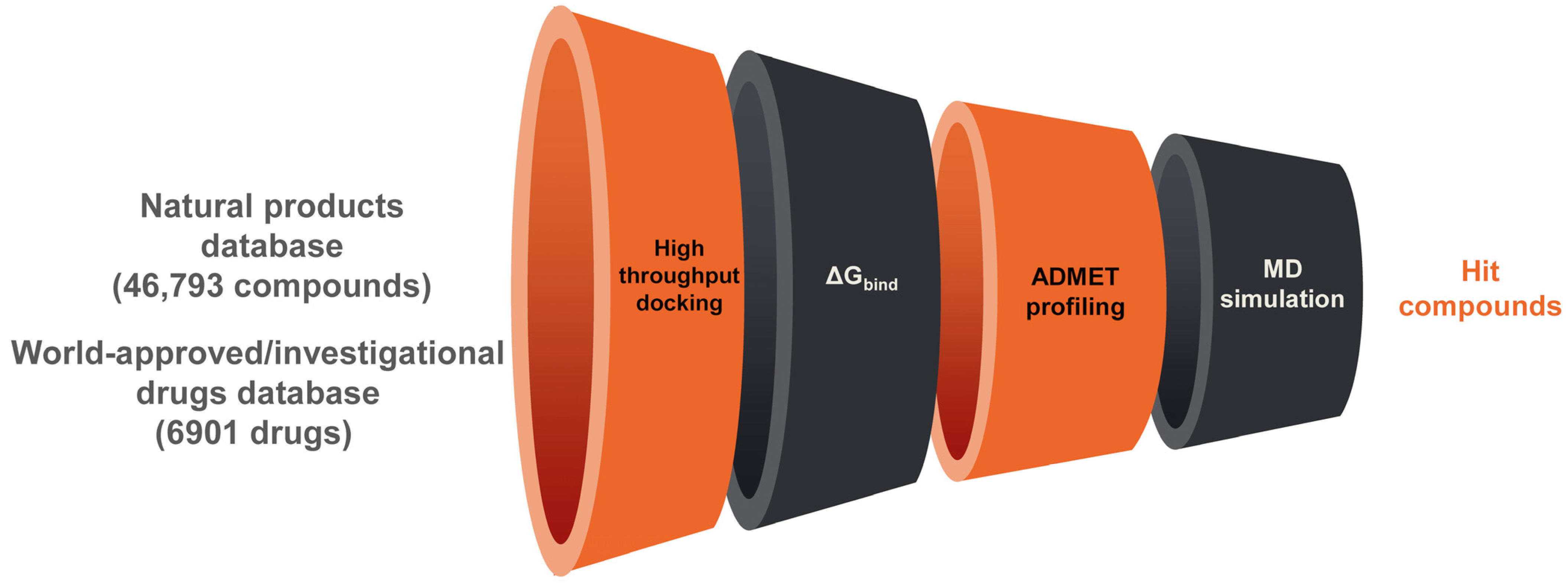
2. Materials and Methods
2.1. Databases and Protein Preparation
2.2. High-Throughput Docking
2.3. ADMET Profiling
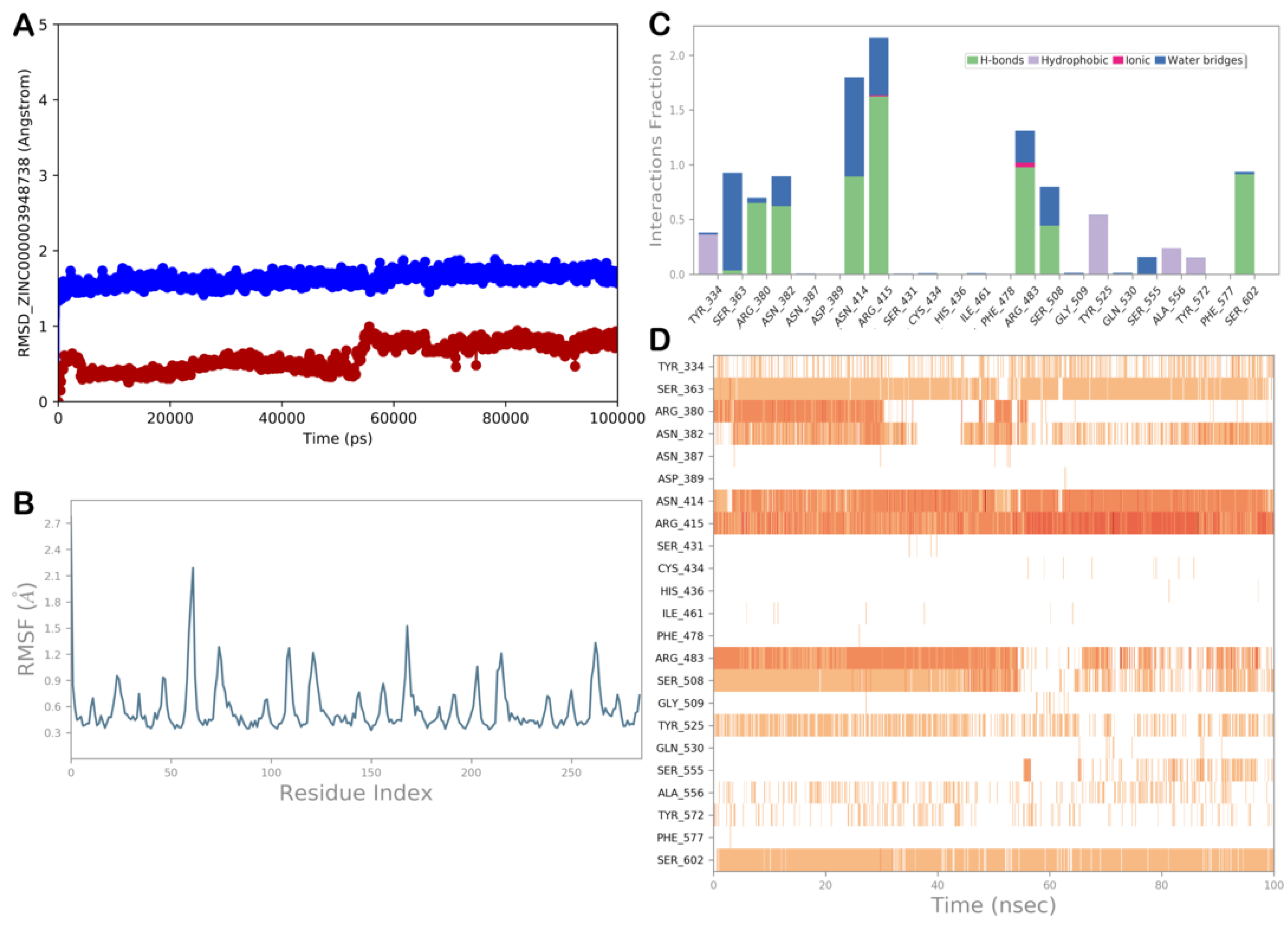
2.4. Molecular Dynamics Simulation
2.5. Covalent Docking
3. Results and Discussion
3.1. Natural Products Database Screening
3.1.1. Potential Natural Products Hit Molecules Targeting the NRF2 Binding Site on KEAP1 Protein
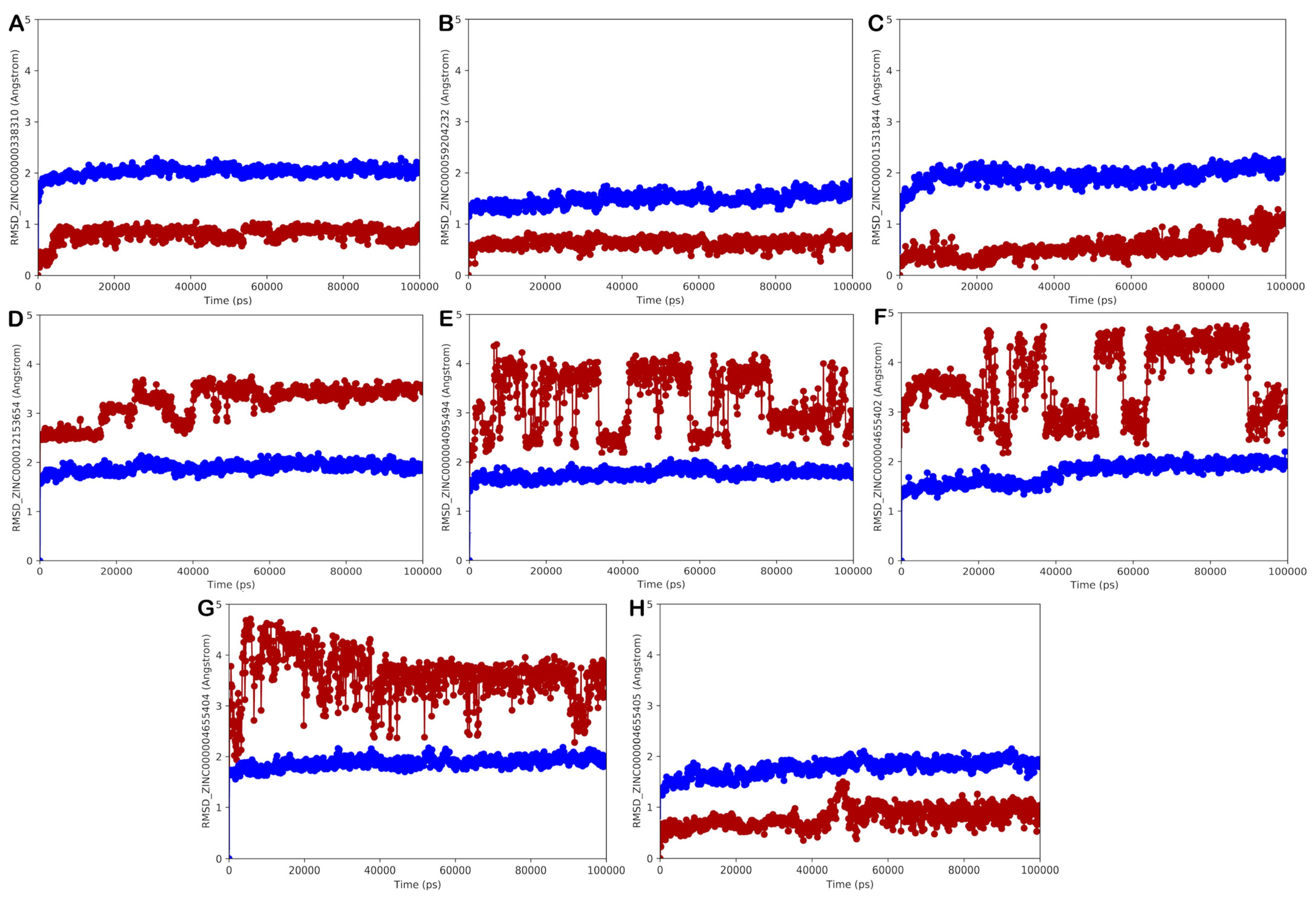
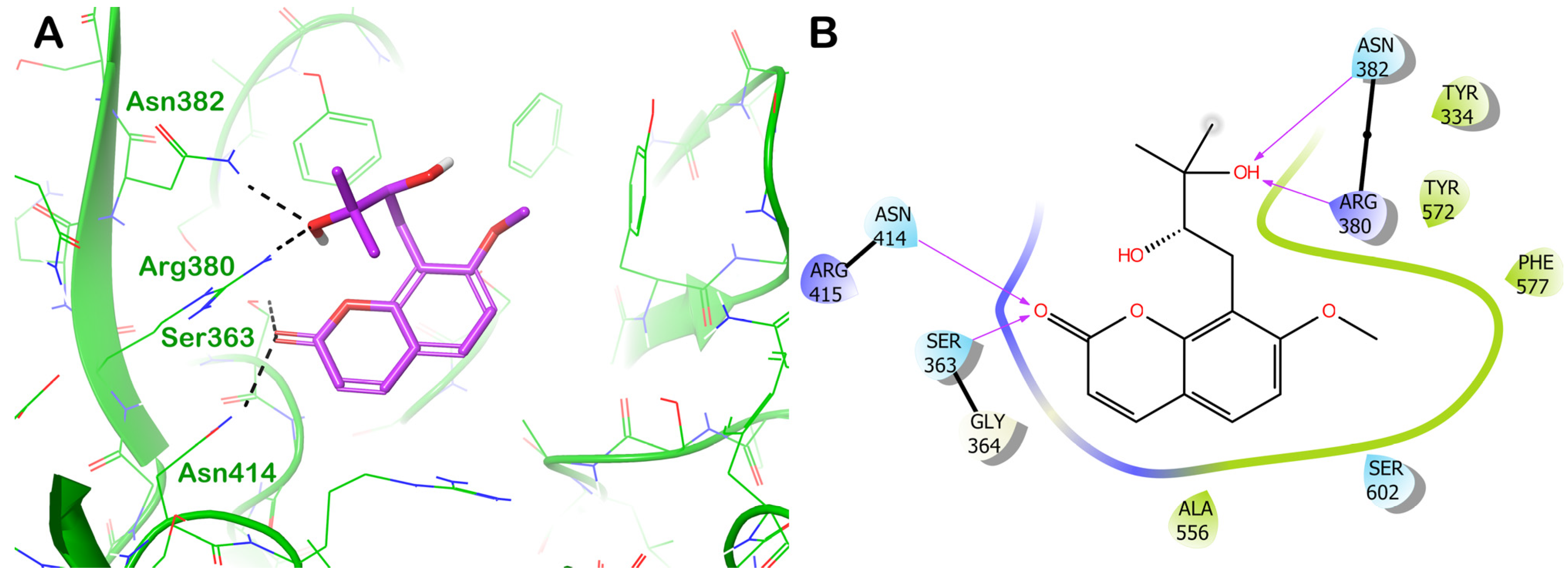
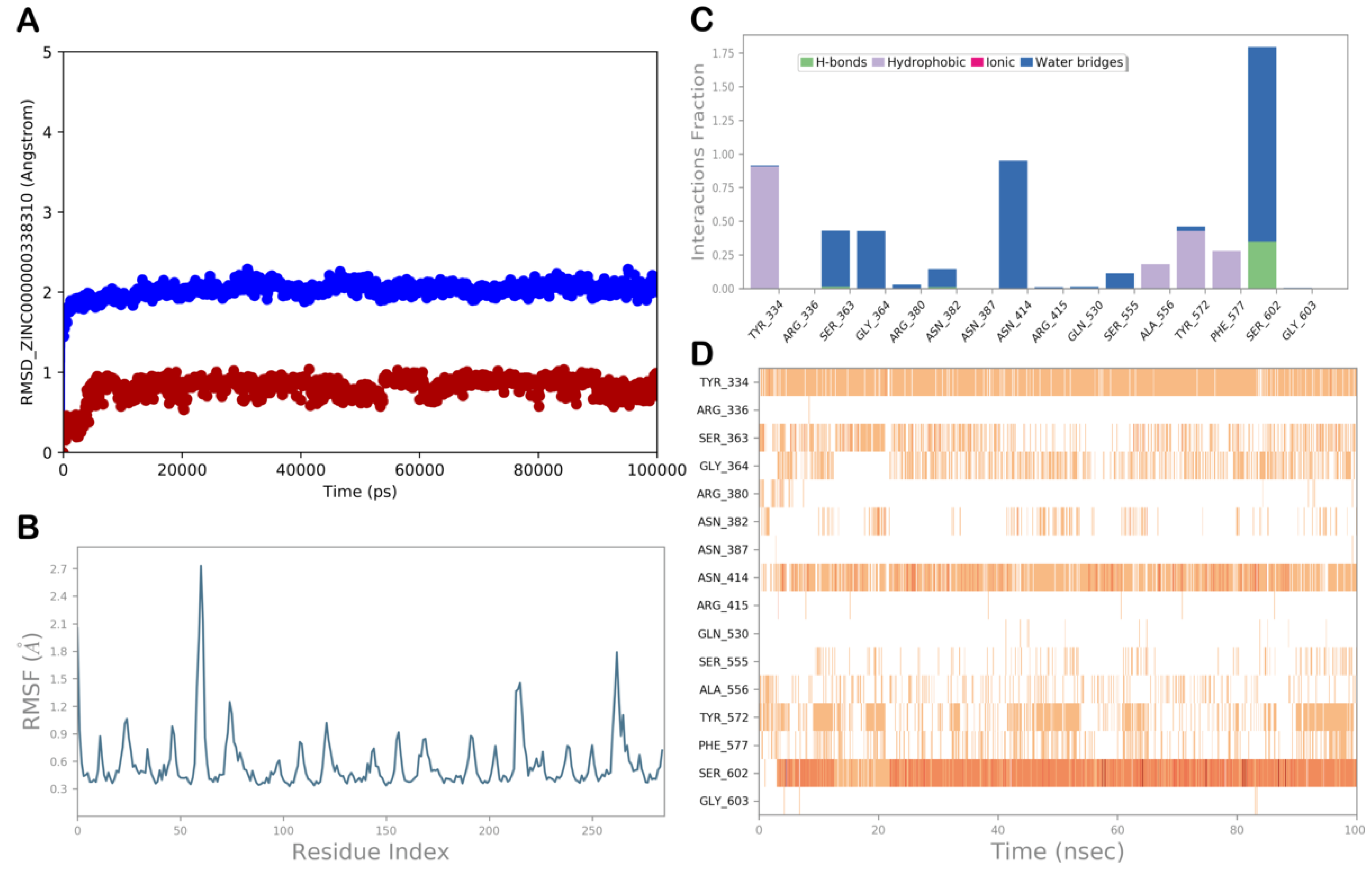
ZINC000000338310 (Meranzin Hydrate)
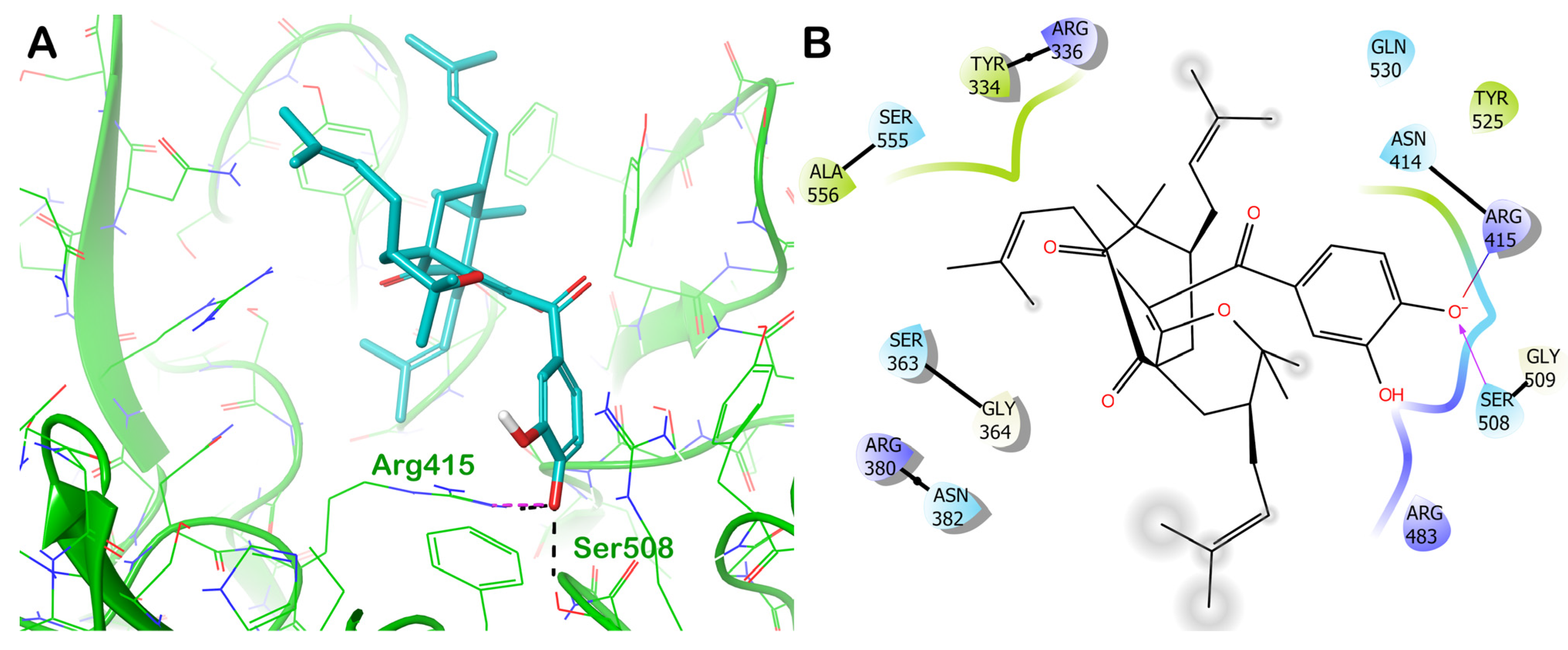
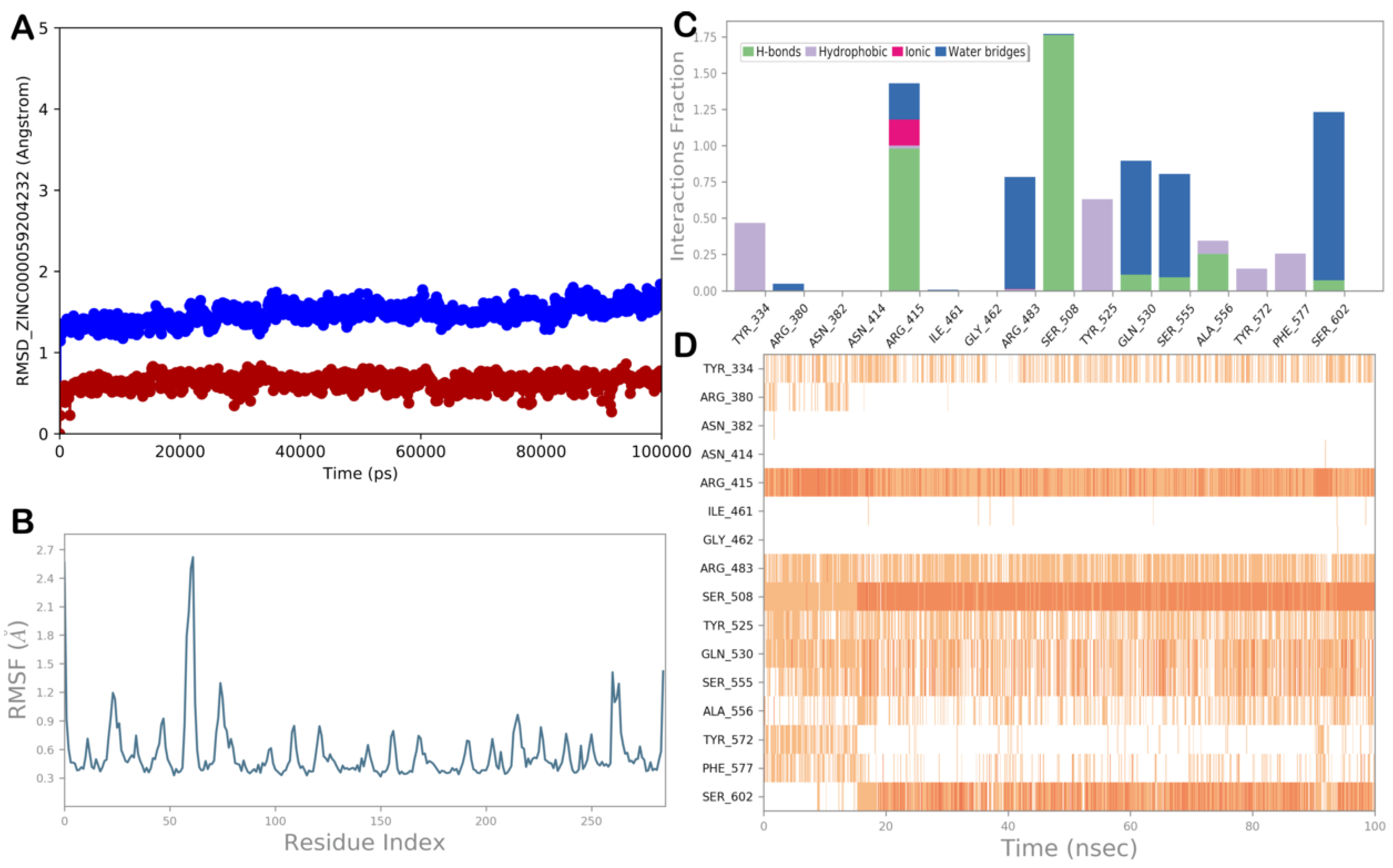
ZINC000059204232 (Isoxanthochymol)
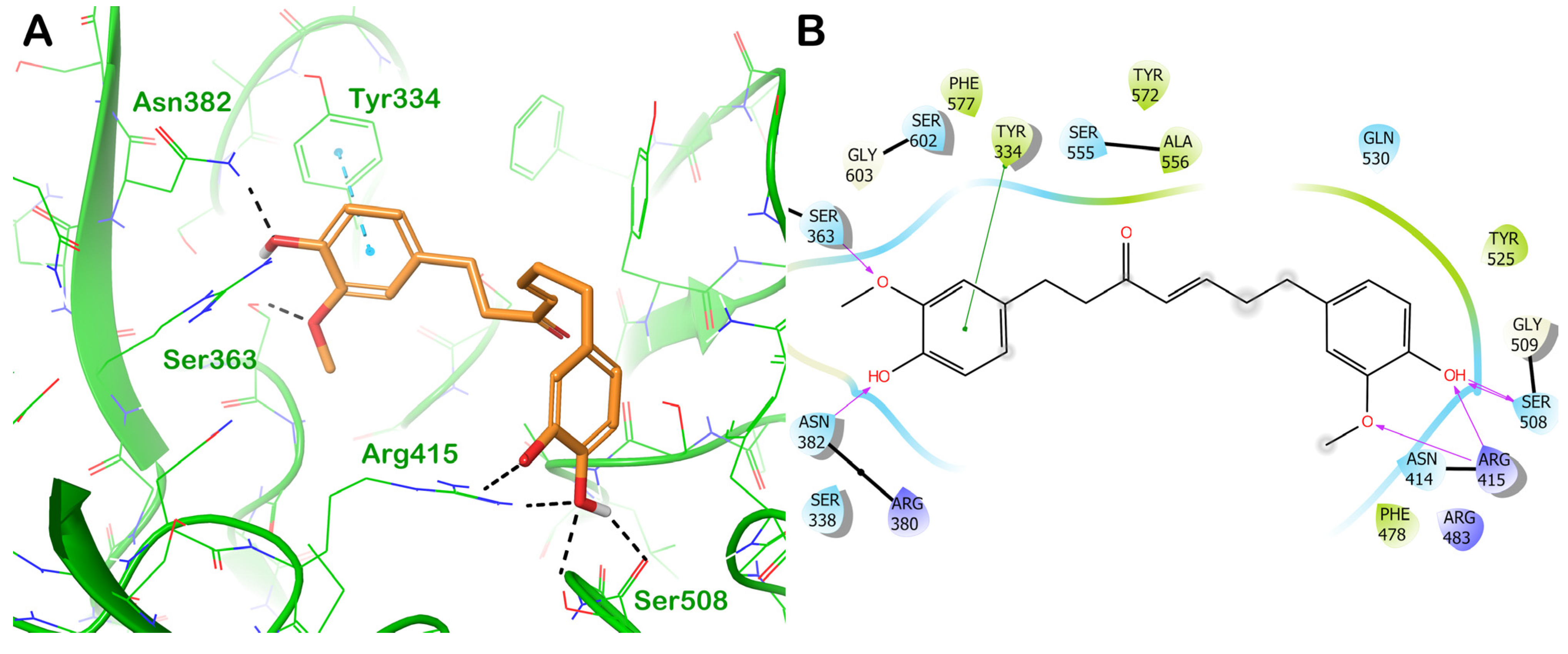
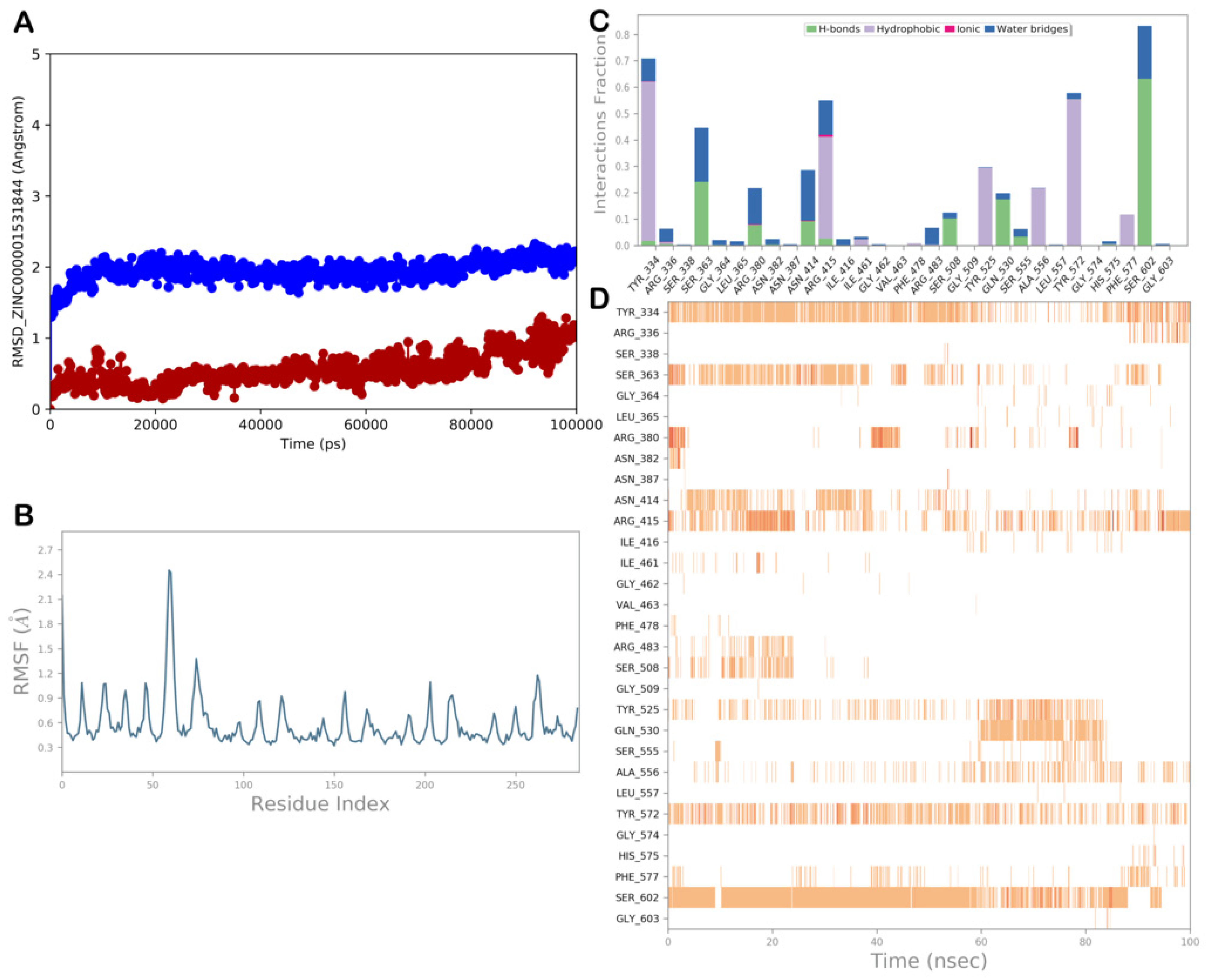
ZINC000001531844 (Gingerenone A)
3.2. Approved and Investigational Drugs Database Screening
3.2.1. Potential Approved and Investigational Drug Hits Targeting the NRF2 Binding Site on KEAP1 Protein
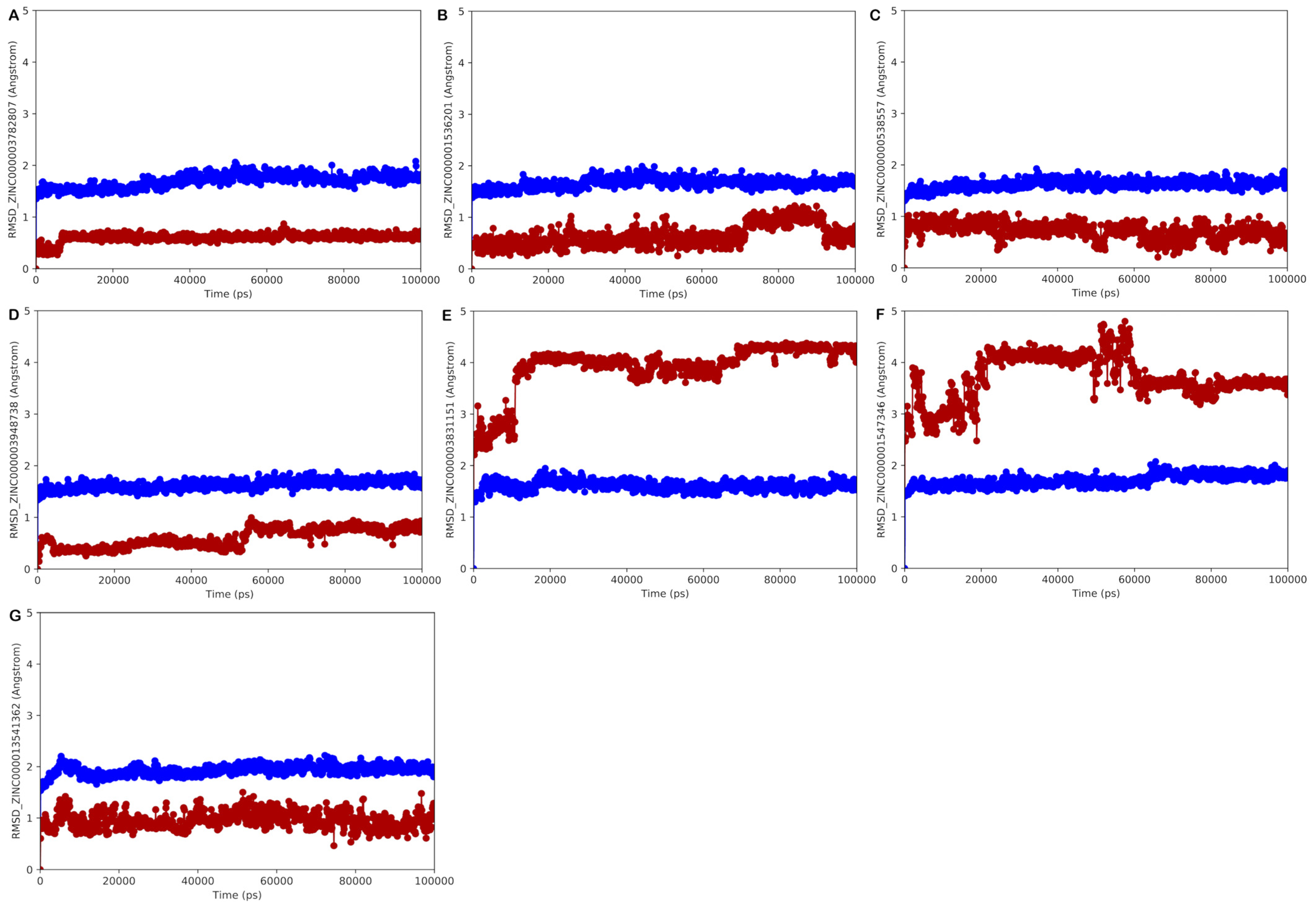
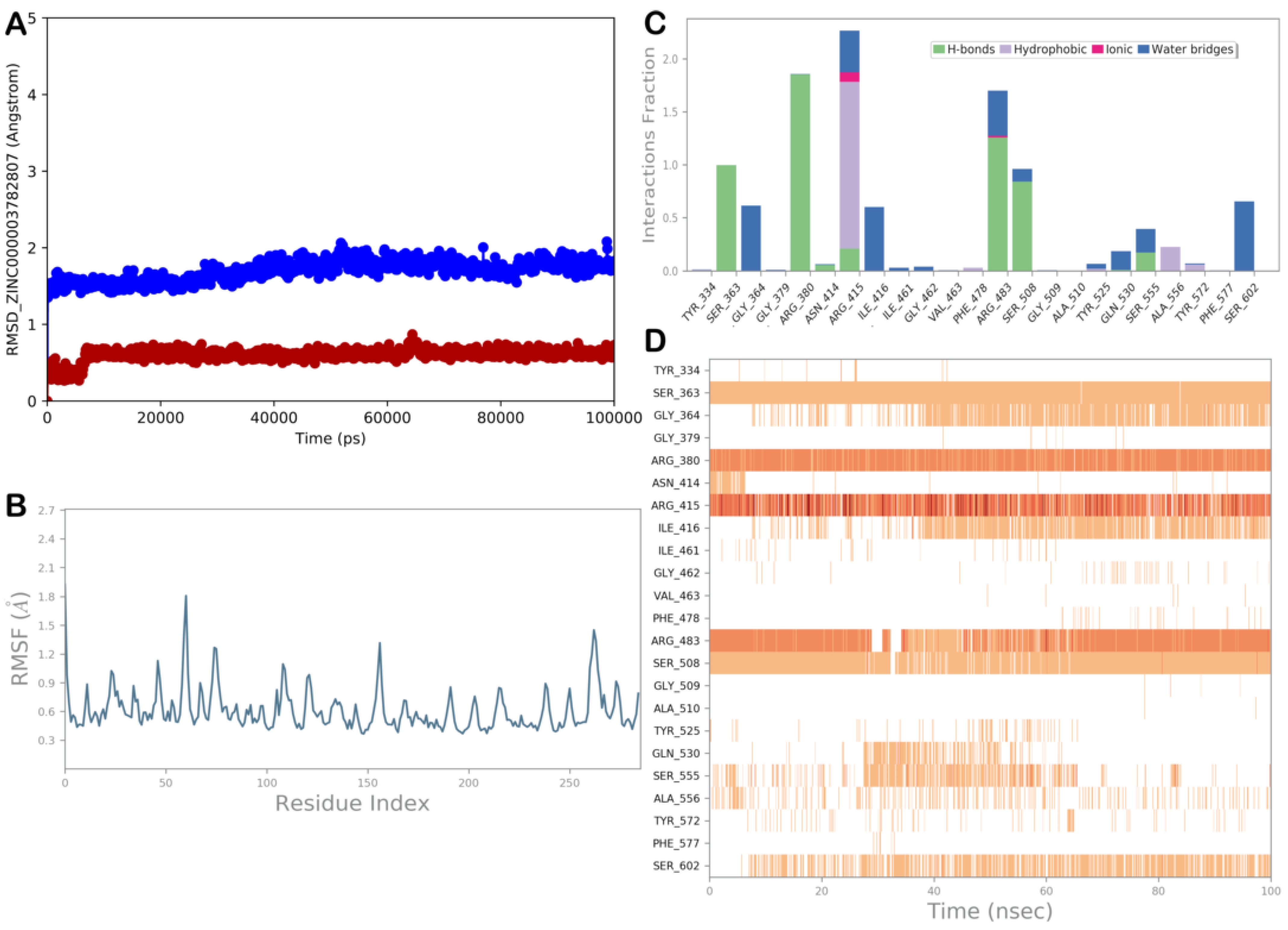
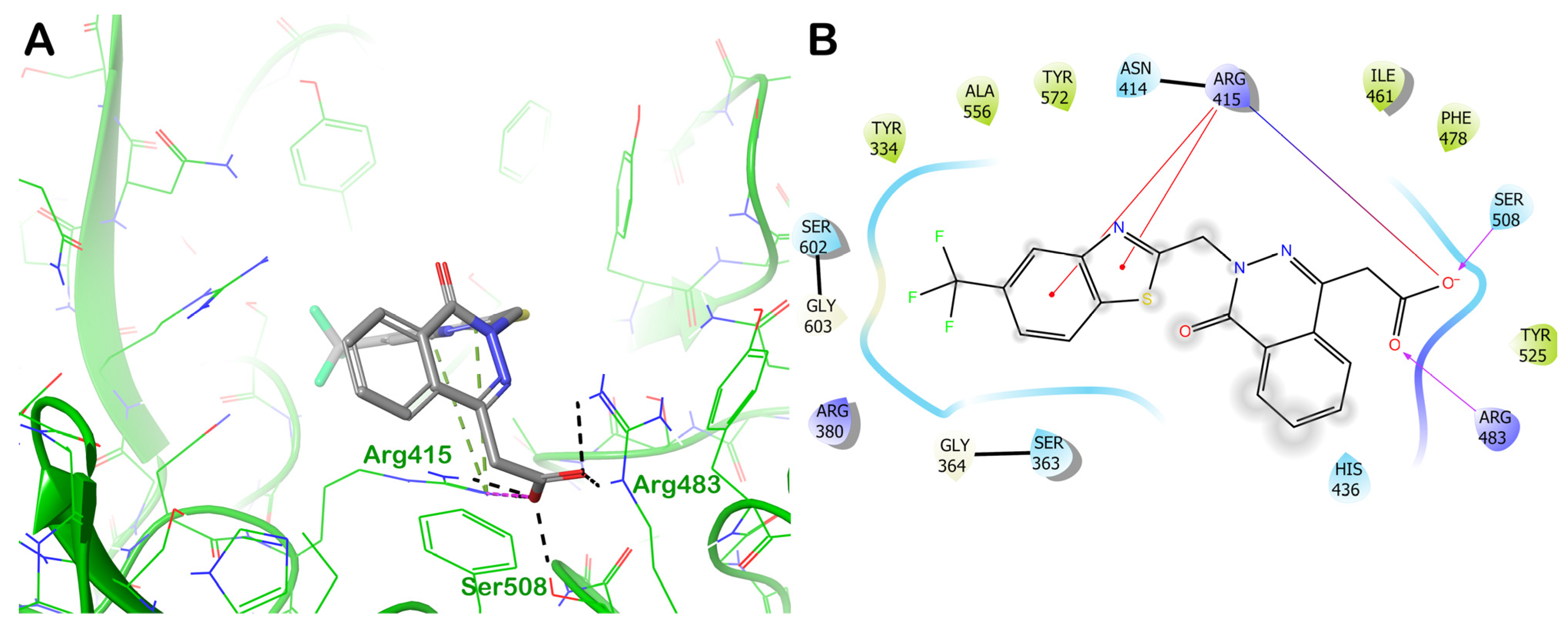
ZINC000003782807 (Nedocromil)
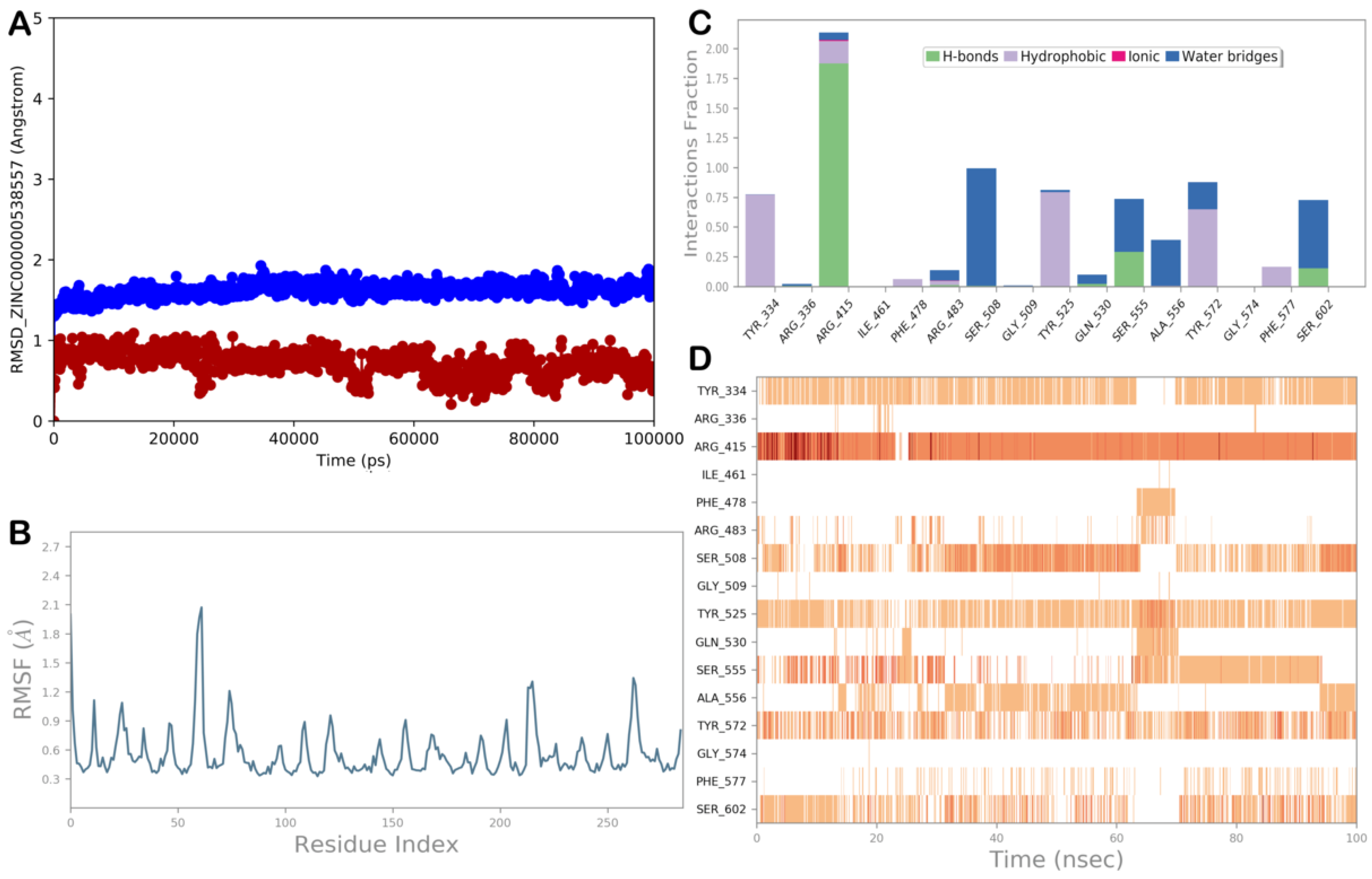
ZINC000000538557 (Zopolrestat)
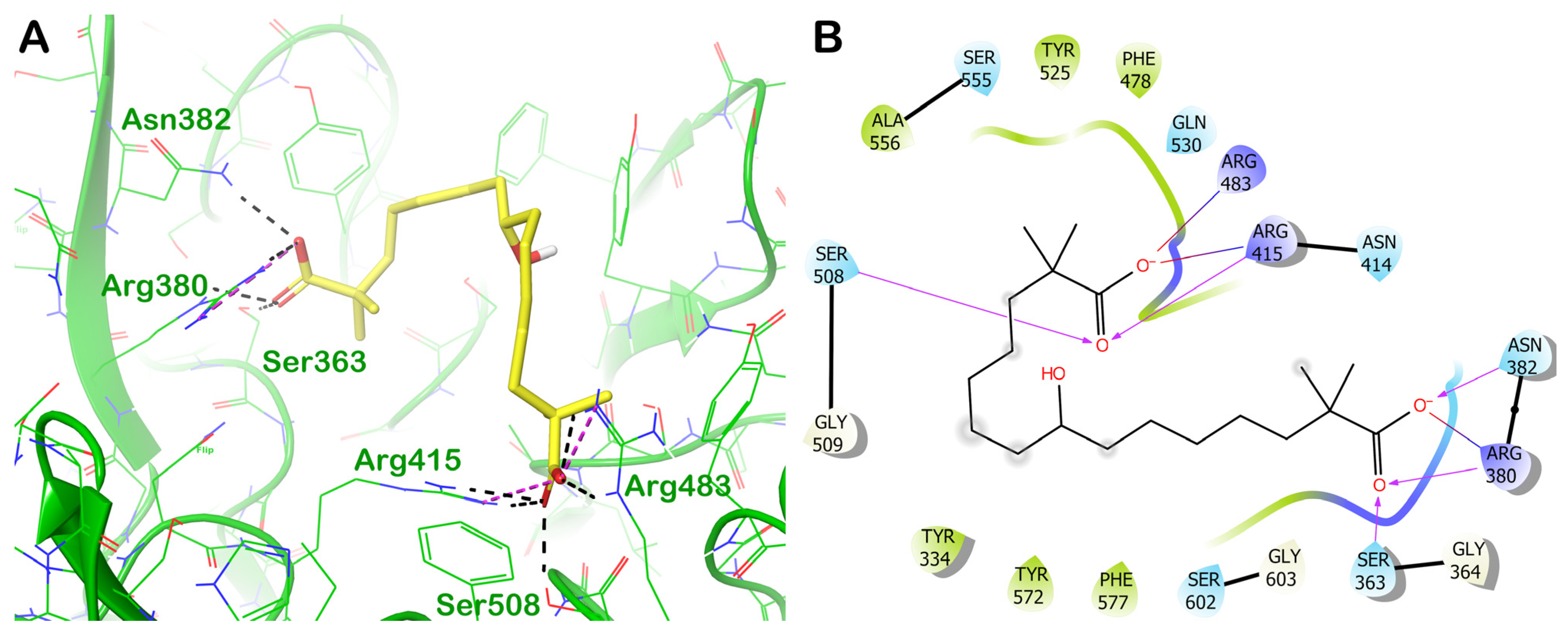
ZINC000003948738 (Bempedoic Acid)
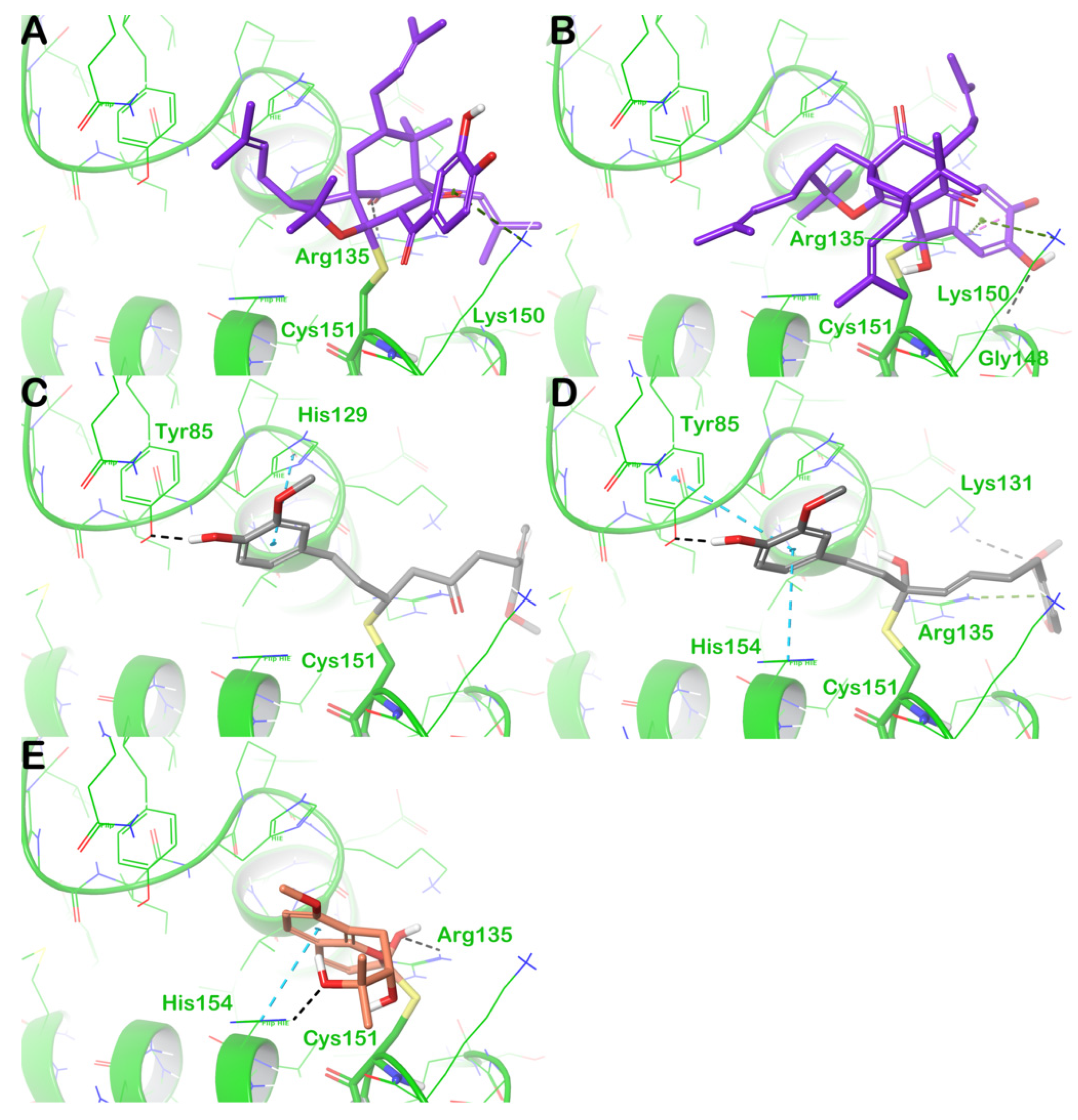
3.3. Covalent Docking Studies
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef] [PubMed]
- Durackova, Z. Some current insights into oxidative stress. Physiol. Res. 2010, 59, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Ray, P.D.; Huang, B.W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Xu, L.; Porter, N.A. Free radical lipid peroxidation: Mechanisms and analysis. Chem. Rev. 2011, 111, 5944–5972. [Google Scholar] [CrossRef] [PubMed]
- Birnboim, H.C. DNA strand breaks in human leukocytes induced by superoxide anion, hydrogen peroxide and tumor promoters are repaired slowly compared to breaks induced by ionizing radiation. Carcinogenesis 1986, 7, 1511–1517. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Reactive species and antioxidants. Redox biology is a fundamental theme of aerobic life. Plant Physiol. 2006, 141, 312–322. [Google Scholar] [CrossRef]
- Davies, M.J. The oxidative environment and protein damage. Biochim. Biophys. Acta 2005, 1703, 93–109. [Google Scholar] [CrossRef]
- Chang, K.C.; Liu, P.F.; Chang, C.H.; Lin, Y.C.; Chen, Y.J.; Shu, C.W. The interplay of autophagy and oxidative stress in the pathogenesis and therapy of retinal degenerative diseases. Cell Biosci. 2022, 12, 1. [Google Scholar] [CrossRef]
- Barrera, G. Oxidative stress and lipid peroxidation products in cancer progression and therapy. ISRN Oncol. 2012, 2012, 137289. [Google Scholar] [CrossRef]
- Ishikawa, K.; Takenaga, K.; Akimoto, M.; Koshikawa, N.; Yamaguchi, A.; Imanishi, H.; Nakada, K.; Honma, Y.; Hayashi, J. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science 2008, 320, 661–664. [Google Scholar] [CrossRef] [PubMed]
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Oxidative stress and cancer: Have we moved forward? Biochem. J. 2007, 401, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Izakovic, M.; Mazur, M.; Rhodes, C.J.; Telser, J. Role of oxygen radicals in DNA damage and cancer incidence. Mol. Cell. Biochem. 2004, 266, 37–56. [Google Scholar] [CrossRef]
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Oxidative stress and heart failure. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H2181–H2190. [Google Scholar] [CrossRef] [PubMed]
- Ramond, A.; Godin-Ribuot, D.; Ribuot, C.; Totoson, P.; Koritchneva, I.; Cachot, S.; Levy, P.; Joyeux-Faure, M. Oxidative stress mediates cardiac infarction aggravation induced by intermittent hypoxia. Fundam. Clin. Pharmacol. 2013, 27, 252–261. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Fernandez, S.; Gurpegui, M.; Diaz-Atienza, F.; Perez-Costillas, L.; Gerstenberg, M.; Correll, C.U. Oxidative stress and antioxidant parameters in patients with major depressive disorder compared to healthy controls before and after antidepressant treatment: Results from a meta-analysis. J. Clin. Psychiatry 2015, 76, 1658–1667. [Google Scholar] [CrossRef]
- Batty, M.; Bennett, M.R.; Yu, E. The Role of Oxidative Stress in Atherosclerosis. Cells 2022, 11, 3843. [Google Scholar] [CrossRef]
- Kennedy, G.; Spence, V.A.; McLaren, M.; Hill, A.; Underwood, C.; Belch, J.J. Oxidative stress levels are raised in chronic fatigue syndrome and are associated with clinical symptoms. Free Radic. Biol. Med. 2005, 39, 584–589. [Google Scholar] [CrossRef]
- Aly, D.G.; Shahin, R.S. Oxidative stress in lichen planus. Acta Dermatovenerol. Alp. Pannonica Adriat. 2010, 19, 3–11. [Google Scholar] [PubMed]
- Omata, N.; Tsukahara, H.; Ito, S.; Ohshima, Y.; Yasutomi, M.; Yamada, A.; Jiang, M.; Hiraoka, M.; Nambu, M.; Deguchi, Y.; et al. Increased oxidative stress in childhood atopic dermatitis. Life Sci. 2001, 69, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Arican, O.; Kurutas, E.B. Oxidative stress in the blood of patients with active localized vitiligo. Acta Dermatovenerol. Alp. Pannonica Adriat. 2008, 17, 12–16. [Google Scholar] [PubMed]
- Dobrica, E.C.; Cozma, M.A.; Gaman, M.A.; Voiculescu, V.M.; Gaman, A.M. The Involvement of Oxidative Stress in Psoriasis: A Systematic Review. Antioxidants 2022, 11, 282. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef] [PubMed]
- Hwang, O. Role of oxidative stress in Parkinson’s disease. Exp. Neurobiol. 2013, 22, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.J.; Zhang, X.; Chen, W.W. Role of oxidative stress in Alzheimer’s disease. Biomed. Rep. 2016, 4, 519–522. [Google Scholar] [CrossRef] [PubMed]
- Moreira, D.C.; Oliveira, M.F.; Liz-Guimaraes, L.; Diniz-Rojas, N.; Campos, E.G.; Hermes-Lima, M. Current Trends and Research Challenges Regarding “Preparation for Oxidative Stress”. Front. Physiol. 2017, 8, 702. [Google Scholar] [CrossRef]
- Willcox, J.K.; Ash, S.L.; Catignani, G.L. Antioxidants and prevention of chronic disease. Crit. Rev. Food Sci. Nutr. 2004, 44, 275–295. [Google Scholar] [CrossRef]
- Diplock, A.T. Antioxidants and disease prevention. Mol. Aspects Med. 1994, 15, 293–376. [Google Scholar] [CrossRef]
- Tardif, J.C. Antioxidants: The good, the bad and the ugly. Can. J. Cardiol. 2006, 22 (Suppl. B), 61B–65B. [Google Scholar] [CrossRef] [PubMed]
- Casas, A.I.; Nogales, C.; Mucke, H.A.M.; Petraina, A.; Cuadrado, A.; Rojo, A.I.; Ghezzi, P.; Jaquet, V.; Augsburger, F.; Dufrasne, F.; et al. On the Clinical Pharmacology of Reactive Oxygen Species. Pharmacol. Rev. 2020, 72, 801–828. [Google Scholar] [CrossRef] [PubMed]
- Silvestro, S.; Mazzon, E. Nrf2 Activation: Involvement in Central Nervous System Traumatic Injuries. A Promising Therapeutic Target of Natural Compounds. Int. J. Mol. Sci. 2022, 24, 199. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A.; Manda, G.; Hassan, A.; Alcaraz, M.J.; Barbas, C.; Daiber, A.; Ghezzi, P.; Leon, R.; Lopez, M.G.; Oliva, B.; et al. Transcription Factor NRF2 as a Therapeutic Target for Chronic Diseases: A Systems Medicine Approach. Pharmacol. Rev. 2018, 70, 348–383. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef] [PubMed]
- Canning, P.; Sorrell, F.J.; Bullock, A.N. Structural basis of Keap1 interactions with Nrf2. Free Radic. Biol. Med. 2015, 88, 101–107. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Ru, X.; Wen, T. NRF2, a Transcription Factor for Stress Response and Beyond. Int. J. Mol. Sci. 2020, 21, 4777. [Google Scholar] [CrossRef]
- Nguyen, T.; Sherratt, P.J.; Pickett, C.B. Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu. Rev. Pharmacol. Toxicol. 2003, 43, 233–260. [Google Scholar] [CrossRef]
- Saha, S.; Buttari, B.; Panieri, E.; Profumo, E.; Saso, L. An Overview of Nrf2 Signaling Pathway and Its Role in Inflammation. Molecules 2020, 25, 5474. [Google Scholar] [CrossRef]
- Sporn, M.B.; Liby, K.T. NRF2 and cancer: The good, the bad and the importance of context. Nat. Rev. Cancer 2012, 12, 564–571. [Google Scholar] [CrossRef]
- Wang, M.X.; Zhao, J.; Zhang, H.; Li, K.; Niu, L.Z.; Wang, Y.P.; Zheng, Y.J. Potential Protective and Therapeutic Roles of the Nrf2 Pathway in Ocular Diseases: An Update. Oxidative Med. Cell. Longev. 2020, 2020, 9410952. [Google Scholar] [CrossRef] [PubMed]
- Gan, L.; Johnson, J.A. Oxidative damage and the Nrf2-ARE pathway in neurodegenerative diseases. Biochim. Biophys. Acta 2014, 1842, 1208–1218. [Google Scholar] [CrossRef] [PubMed]
- Zgorzynska, E.; Dziedzic, B.; Walczewska, A. An Overview of the Nrf2/ARE Pathway and Its Role in Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 9592. [Google Scholar] [CrossRef] [PubMed]
- Ooi, B.K.; Goh, B.H.; Yap, W.H. Oxidative Stress in Cardiovascular Diseases: Involvement of Nrf2 Antioxidant Redox Signaling in Macrophage Foam Cells Formation. Int. J. Mol. Sci. 2017, 18, 2336. [Google Scholar] [CrossRef]
- Da Costa, R.M.; Rodrigues, D.; Pereira, C.A.; Silva, J.F.; Alves, J.V.; Lobato, N.S.; Tostes, R.C. Nrf2 as a Potential Mediator of Cardiovascular Risk in Metabolic Diseases. Front. Pharmacol. 2019, 10, 382. [Google Scholar] [CrossRef]
- Flori, L.; Brogi, S.; Sirous, H.; Calderone, V. Disruption of Irisin Dimerization by FDA-Approved Drugs: A Computational Repurposing Approach for the Potential Treatment of Lipodystrophy Syndromes. Int. J. Mol. Sci. 2023, 24, 7578. [Google Scholar] [CrossRef]
- MacroModel Schrödinger, LLC, New York, NY, USA. 2020. Available online: https://www.schrodinger.com/products/macromodel (accessed on 11 May 2023).
- LigPrep Schrödinger, LLC, New York, NY, USA. 2020. Available online: https://www.schrodinger.com/products/ligprep (accessed on 11 May 2023).
- Maestro Schrödinger, LLC, New York, NY, USA. 2020. Available online: https://www.schrodinger.com/products/maestro (accessed on 11 April 2023).
- Battah, B.; Chemi, G.; Butini, S.; Campiani, G.; Brogi, S.; Delogu, G.; Gemma, S. A Repurposing Approach for Uncovering the Anti-Tubercular Activity of FDA-Approved Drugs with Potential Multi-Targeting Profiles. Molecules 2019, 24, 4373. [Google Scholar] [CrossRef]
- Brogi, S.; Ramunno, A.; Savi, L.; Chemi, G.; Alfano, G.; Pecorelli, A.; Pambianchi, E.; Galatello, P.; Compagnoni, G.; Focher, F.; et al. First dual AK/GSK-3beta inhibitors endowed with antioxidant properties as multifunctional, potential neuroprotective agents. Eur. J. Med. Chem. 2017, 138, 438–457. [Google Scholar] [CrossRef]
- Brogi, S.; Corelli, F.; Di Marzo, V.; Ligresti, A.; Mugnaini, C.; Pasquini, S.; Tafi, A. Three-dimensional quantitative structure-selectivity relationships analysis guided rational design of a highly selective ligand for the cannabinoid receptor 2. Eur. J. Med. Chem. 2011, 46, 547–555. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Vallone, A.; D’Alessandro, S.; Brogi, S.; Brindisi, M.; Chemi, G.; Alfano, G.; Lamponi, S.; Lee, S.G.; Jez, J.M.; Koolen, K.J.M.; et al. Antimalarial agents against both sexual and asexual parasites stages: Structure-activity relationships and biological studies of the Malaria Box compound 1-[5-(4-bromo-2-chlorophenyl)furan-2-yl]-N-[(piperidin-4-yl)methyl]methanamine (MMV019918) and analogues. Eur. J. Med. Chem. 2018, 150, 698–718. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, E.R.; Brogi, S.; Lucon-Junior, J.F.; Campiani, G.; Gemma, S.; Maquiaveli, C.D.C. Dietary polyphenols rutin, taxifolin and quercetin related compounds target Leishmania amazonensis arginase. Food Funct. 2019, 10, 3172–3180. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Gao, Q.; Lu, C.; Wang, X.W.; Zhang, J.W.; Song, Y.; Xue, Y.L. Molecular dynamics simulation and steered molecular dynamics simulation on irisin dimers. J. Mol. Model. 2018, 24, 95. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, M.A.; Chinnam, N.; Ohashi, T.; Shah, R.S.; Erickson, H.P. The structure of irisin reveals a novel intersubunit beta-sheet fibronectin type III (FNIII) dimer: Implications for receptor activation. J. Biol. Chem. 2013, 288, 33738–33744. [Google Scholar] [CrossRef] [PubMed]
- Prime Schrödinger, LLC, New York, NY, USA. 2020. Available online: https://newsite.schrodinger.com/platform/products/prime (accessed on 11 May 2023).
- Brogi, S.; Rossi, S.; Ibba, R.; Butini, S.; Calderone, V.; Campiani, G.; Gemma, S. In Silico Analysis of Peptide-Based Derivatives Containing Bifunctional Warheads Engaging Prime and Non-Prime Subsites to Covalent Binding SARS-CoV-2 Main Protease (Mpro). Computation 2022, 10, 69. [Google Scholar] [CrossRef]
- Brindisi, M.; Gemma, S.; Kunjir, S.; Di Cerbo, L.; Brogi, S.; Parapini, S.; D’Alessandro, S.; Taramelli, D.; Habluetzel, A.; Tapanelli, S.; et al. Synthetic spirocyclic endoperoxides: New antimalarial scaffolds. Medchemcomm 2015, 6, 357–362. [Google Scholar] [CrossRef]
- Ciccone, L.; Piragine, E.; Brogi, S.; Camodeca, C.; Fucci, R.; Calderone, V.; Nencetti, S.; Martelli, A.; Orlandini, E. Resveratrol-like Compounds as SIRT1 Activators. Int J Mol Sci 2022, 23, 15105. [Google Scholar] [CrossRef]
- D’Alessandro, S.; Alfano, G.; Di Cerbo, L.; Brogi, S.; Chemi, G.; Relitti, N.; Brindisi, M.; Lamponi, S.; Novellino, E.; Campiani, G.; et al. Bridged bicyclic 2,3-dioxabicyclo[3.3.1]nonanes as antiplasmodial agents: Synthesis, structure-activity relationships and studies on their biomimetic reaction with Fe(II). Bioorganic Chem. 2019, 89, 103020. [Google Scholar] [CrossRef]
- QikProp Schrödinger, L., New York, NY, USA. 2020. Available online: https://www.schrodinger.com/products/qikprop (accessed on 11 May 2023).
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Sirous, H.; Chemi, G.; Gemma, S.; Butini, S.; Debyser, Z.; Christ, F.; Saghaie, L.; Brogi, S.; Fassihi, A.; Campiani, G.; et al. Identification of Novel 3-Hydroxy-pyran-4-One Derivatives as Potent HIV-1 Integrase Inhibitors Using in silico Structure-Based Combinatorial Library Design Approach. Front. Chem. 2019, 7, 574. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, D.D.; Friesner, R.A.; Berne, B.J. A Multiple-Time-Step Molecular Dynamics Algorithm for Macromolecules. J. Phys. Chem. 1994, 98, 6885–6892. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef] [PubMed]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Brogi, S.; Fiorillo, A.; Chemi, G.; Butini, S.; Lalle, M.; Ilari, A.; Gemma, S.; Campiani, G. Structural characterization of Giardia duodenalis thioredoxin reductase (gTrxR) and computational analysis of its interaction with NBDHEX. Eur. J. Med. Chem. 2017, 135, 479–490. [Google Scholar] [CrossRef]
- Giovani, S.; Penzo, M.; Brogi, S.; Brindisi, M.; Gemma, S.; Novellino, E.; Savini, L.; Blackman, M.J.; Campiani, G.; Butini, S. Rational design of the first difluorostatone-based PfSUB1 inhibitors. Bioorganic Med. Chem. Lett. 2014, 24, 3582–3586. [Google Scholar] [CrossRef]
- Zhu, K.; Borrelli, K.W.; Greenwood, J.R.; Day, T.; Abel, R.; Farid, R.S.; Harder, E. Docking covalent inhibitors: A parameter free approach to pose prediction and scoring. J. Chem. Inf. Model. 2014, 54, 1932–1940. [Google Scholar] [CrossRef]
- Liu, Y.L.; Xu, J.J.; Han, L.R.; Liu, X.F.; Lin, M.H.; Wang, Y.; Xiao, Z.; Huang, Y.K.; Ren, P.; Huang, X. Meranzin Hydrate Improves Depression-Like Behaviors and Hypomotility via Ghrelin and Neurocircuitry. Chin. J. Integr. Med. 2023, 29, 490–499. [Google Scholar] [CrossRef]
- Huang, X.; Guo, Y.; Huang, W.H.; Zhang, W.; Tan, Z.R.; Peng, J.B.; Wang, Y.C.; Hu, D.L.; Ouyang, D.S.; Xiao, J.; et al. Searching the cytochrome p450 enzymes for the metabolism of meranzin hydrate: A prospective antidepressant originating from Chaihu-Shugan-San. PLoS ONE 2014, 9, e113819. [Google Scholar] [CrossRef]
- Li, L.; Yu, A.L.; Wang, Z.L.; Chen, K.; Zheng, W.; Zhou, J.J.; Xie, Q.; Yan, H.B.; Ren, P.; Huang, X. Chaihu-Shugan-San and absorbed meranzin hydrate induce anti-atherosclerosis and behavioral improvements in high-fat diet ApoE(-/-) mice via anti-inflammatory and BDNF-TrkB pathway. Biomed. Pharmacother. 2019, 115, 108893. [Google Scholar] [CrossRef] [PubMed]
- Rosselli, S.; Maggio, A.; Bellone, G.; Formisano, C.; Basile, A.; Cicala, C.; Alfieri, A.; Mascolo, N.; Bruno, M. Antibacterial and anticoagulant activities of coumarins isolated from the flowers of Magydaris tomentosa. Planta Med. 2007, 73, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Shorbagi, M.; Fayek, N.M.; Shao, P.; Farag, M.A. Citrus reticulata Blanco (the common mandarin) fruit: An updated review of its bioactive, extraction types, food quality, therapeutic merits, and bio-waste valorization practices to maximize its economic value. Food Biosci. 2022, 47, 101699. [Google Scholar] [CrossRef]
- Chattopadhyay, S.K.; Kumar, S. Identification and quantification of two biologically active polyisoprenylated benzophenones xanthochymol and isoxanthochymol in Garcinia species using liquid chromatography-tandem mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2006, 844, 67–83. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Chattopadhyay, S.K.; Darokar, M.P.; Garg, A.; Khanuja, S.P. Cytotoxic activities of xanthochymol and isoxanthochymol substantiated by LC-MS/MS. Planta Med. 2007, 73, 1452–1456. [Google Scholar] [CrossRef] [PubMed]
- Lannang, A.M.; Louh, G.N.; Lontsi, D.; Specht, S.; Sarite, S.R.; Florke, U.; Hussain, H.; Hoerauf, A.; Krohn, K. Antimalarial compounds from the root bark of Garcinia polyantha Olv. J. Antibiot. 2008, 61, 518–523. [Google Scholar] [CrossRef] [PubMed]
- Pasaribu, Y.P.; Fadlan, A.; Fatmawati, S.; Ersam, T. Biological Activity Evaluation and In Silico Studies of Polyprenylated Benzophenones from Garcinia celebica. Biomedicines 2021, 9, 1654. [Google Scholar] [CrossRef]
- Moaddel, R.; Rossi, M.; Rodriguez, S.; Munk, R.; Khadeer, M.; Abdelmohsen, K.; Gorospe, M.; Ferrucci, L. Identification of gingerenone A as a novel senolytic compound. PLoS ONE 2022, 17, e0266135. [Google Scholar] [CrossRef]
- Suk, S.; Kwon, G.T.; Lee, E.; Jang, W.J.; Yang, H.; Kim, J.H.; Thimmegowda, N.R.; Chung, M.Y.; Kwon, J.Y.; Yang, S.; et al. Gingerenone A, a polyphenol present in ginger, suppresses obesity and adipose tissue inflammation in high-fat diet-fed mice. Mol. Nutr. Food Res. 2017, 61, 1700139. [Google Scholar] [CrossRef]
- Yu, T.J.; Tang, J.Y.; Shiau, J.P.; Hou, M.F.; Yen, C.H.; Ou-Yang, F.; Chen, C.Y.; Chang, H.W. Gingerenone A Induces Antiproliferation and Senescence of Breast Cancer Cells. Antioxidants 2022, 11, 587. [Google Scholar] [CrossRef]
- Rampogu, S.; Baek, A.; Gajula, R.G.; Zeb, A.; Bavi, R.S.; Kumar, R.; Kim, Y.; Kwon, Y.J.; Lee, K.W. Ginger (Zingiber officinale) phytochemicals-gingerenone-A and shogaol inhibit SaHPPK: Molecular docking, molecular dynamics simulations and in vitro approaches. Ann. Clin. Microbiol. Antimicrob. 2018, 17, 16. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhu, S.; Chen, Z.; Liu, Y.; Pei, C.; Huang, H.; Hou, S.; Ning, W.; Liang, J. Gingerenone A Alleviates Ferroptosis in Secondary Liver Injury in Colitis Mice via Activating Nrf2-Gpx4 Signaling Pathway. J. Agric. Food Chem. 2022, 70, 12525–12534. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.W.; Chun, K.S.; Kim, D.H.; Kim, S.J.; Kim, S.H.; Cho, N.C.; Na, H.K.; Surh, Y.J. Curcumin induces stabilization of Nrf2 protein through Keap1 cysteine modification. Biochem Pharmacol 2020, 173, 113820. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Lu, J.Y.; Wu, X.; Summer, S.; Whoriskey, J.; Saris, C.; Reagan, J.D. G-protein-coupled receptor 35 is a target of the asthma drugs cromolyn disodium and nedocromil sodium. Pharmacology 2010, 86, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Malik, R.A. Can diabetic neuropathy be prevented by angiotensin-converting enzyme inhibitors? Ann. Med. 2000, 32, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Goyal, A.; Changez, M.I.K.; Tariq, M.D.; Mushtaq, F.; Shamim, U.; Sohail, A.H.; Mahalwar, G. Efficacy and Outcomes of Bempedoic Acid Versus Placebo in Patients with Statin-Intolerance: A Pilot Systematic Review and Meta-analysis of Randomized Controlled Trials. Curr. Probl. Cardiol. 2023, in press. [Google Scholar] [CrossRef] [PubMed]
- De Filippo, O.; D’Ascenzo, F.; Iannaccone, M.; Bertaina, M.; Leone, A.; Borzillo, I.; Ravetti, E.; Solano, A.; Pagliassotto, I.; Nebiolo, M.; et al. Safety and efficacy of bempedoic acid: A systematic review and meta-analysis of randomised controlled trials. Cardiovasc. Diabetol. 2023, 22, 324. [Google Scholar] [CrossRef]
- Trotta, V.; Lee, W.H.; Loo, C.Y.; Young, P.M.; Traini, D.; Scalia, S. Co-spray dried resveratrol and budesonide inhalation formulation for reducing inflammation and oxidative stress in rat alveolar macrophages. Eur. J. Pharm. Sci. 2016, 86, 20–28. [Google Scholar] [CrossRef]
- Schichlein, K.D.; Smith, G.J.; Jaspers, I. Protective effects of inhaled antioxidants against air pollution-induced pathological responses. Respir. Res. 2023, 24, 187. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cpd | GlideScore (SP) (kcal/mol) | ΔGbind (kcal/mol) | SASA a | QPlogP b | QPlogS c | QPPCaco d | QPPMDCK e | %HOA f |
|---|---|---|---|---|---|---|---|---|
| ZINC000000338310 (meranzin hydrate) | −8.254 | −45.94 | 490 | 2.02 | −2.39 | 1505 | 770 | 95 |
| ZINC000059204232 (isoxanthochymol) | −8.698 | −41.96 | 930 | 6.575 | −8.35 | 424 | 197 | 86 |
| ZINC000001531844 (gingerenone A) | −8.426 | −45.23 | 679 | 3.685 | −4.92 | 359 | 165 | 94 |
| ZINC000012153654 (olivil) | −8.223 | −36.62 | 621 | 2.150 | −3.48 | 308 | 139 | 84 |
| ZINC000004095494 (leukotriene A4) | −8.808 | −44.70 | 665 | 5.22 | −4.67 | 289 | 165 | 88 |
| ZINC000004655402 (5,6-epoxy-8,11,14-eicosatrienoic acid) | −8.197 | −42.27 | 665 | 5.12 | −4.66 | 287 | 164 | 87 |
| ZINC000004655404 (13′-carboxy-γ-tocopherol) | −8.546 | −40.11 | 654 | 5.01 | −4.44 | 277 | 157 | 87 |
| ZINC000004655405 (8,9-epoxyeicosatrienoic acid) | −8.409 | −42.78 | 656 | 4.939 | −4.49 | 254 | 143 | 100 |
| Cpd | GlideScore (SP) (kcal/mol) | ΔGbind (kcal/mol) | LD50 a mg/kg | Therapeutic Indications |
|---|---|---|---|---|
| ZINC000003782807 (nedocromil) | −8.243 | −54.91 | 980 | Approved anti-asthma medication. It is used prophylactically in asthma including allergy-related asthma. Ophthalmic nedocromil is used to treat itchy eyes caused by allergies. |
| ZINC000001536201 (sacubitrilat) | −8.089 | −49.77 | 2000 | Active form of sacubitril, and it belongs to the class of therapeutics called angiotensin receptor neprilysin inhibitors. This drug improves endothelial cell function, and it is recommended for treating cardiovascular disorders. |
| ZINC000000538557 (zopolrestat) | −7.148 | −47.53 | 1034 | Zopolrestat is a potent inhibitor of aldose reductase, and it is approved for the treatment of diabetic complications such as diabetic cardiovascular autonomic neuropathy or diabetic neuropathy. |
| ZINC000003948738 (bempedoic acid) | −6.792 | −50.96 | >1000 | Bempedoic acid is a prescription-only, once-daily oral tablet used to lower low-density lipoprotein cholesterol (LDL-C) levels in the blood. It is used for the treatment of hypercholesterolemia. |
| ZINC000003831151 (montelukast) | −6.695 | −41.48 | 1552 | FDA-approved drug for treating chronic asthma and prophylaxis and prevention of exercise-induced bronchoconstriction. It is also approved to relieve seasonal and perennial allergic rhinitis symptoms. |
| ZINC000001547346 (solabegron) | −6.658 | −54.89 | 1036 | Solabegron is a selective adrenergic β-3 adrenoceptor agonist, and it was developed for the treatment of overactive bladder and irritable bowel syndrome. |
| ZINC000013541362 (dinoprost) | −6.531 | −51.32 | 1170 | Dinoprost is a medication used to induce a second trimester abortion and is used in evacuating the uterus in cases of fetal death. It has been investigated for the treatment of headaches. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brogi, S.; Guarino, I.; Flori, L.; Sirous, H.; Calderone, V. In Silico Identification of Natural Products and World-Approved Drugs Targeting the KEAP1/NRF2 Pathway Endowed with Potential Antioxidant Profile. Computation 2023, 11, 255. https://doi.org/10.3390/computation11120255
Brogi S, Guarino I, Flori L, Sirous H, Calderone V. In Silico Identification of Natural Products and World-Approved Drugs Targeting the KEAP1/NRF2 Pathway Endowed with Potential Antioxidant Profile. Computation. 2023; 11(12):255. https://doi.org/10.3390/computation11120255
Chicago/Turabian StyleBrogi, Simone, Ilaria Guarino, Lorenzo Flori, Hajar Sirous, and Vincenzo Calderone. 2023. "In Silico Identification of Natural Products and World-Approved Drugs Targeting the KEAP1/NRF2 Pathway Endowed with Potential Antioxidant Profile" Computation 11, no. 12: 255. https://doi.org/10.3390/computation11120255