

Spectrum of Genetic Variants in the Dystrophin Gene: A Single Centre Retrospective Analysis of 750 Duchenne and Becker Patients from Southern Italy
 ,
,  , ,
, ,
Abstract
:1. Introduction
1.1. Clinical Pictures of Dystrophinopathies
1.1.1. Rapidly Evolving Dystrophinopathy
1.1.2. Benign Evolving Dystrophinopathy
1.1.3. X-Linked Dilated Cardiomyopathy
1.1.4. Carrier’s Dystrophinopathy
1.1.5. Treatment of Dystrophinopathies
2. Patients and Methods
2.1. Patients
2.2. Genomic Analysis
3. Results
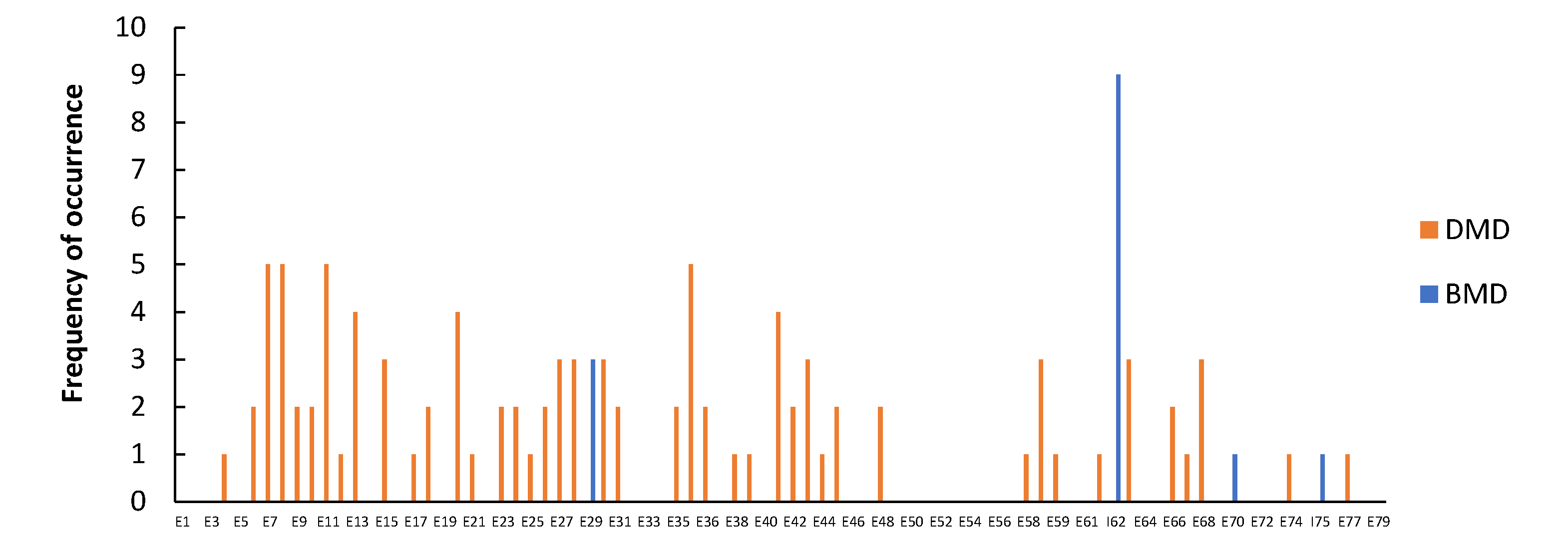
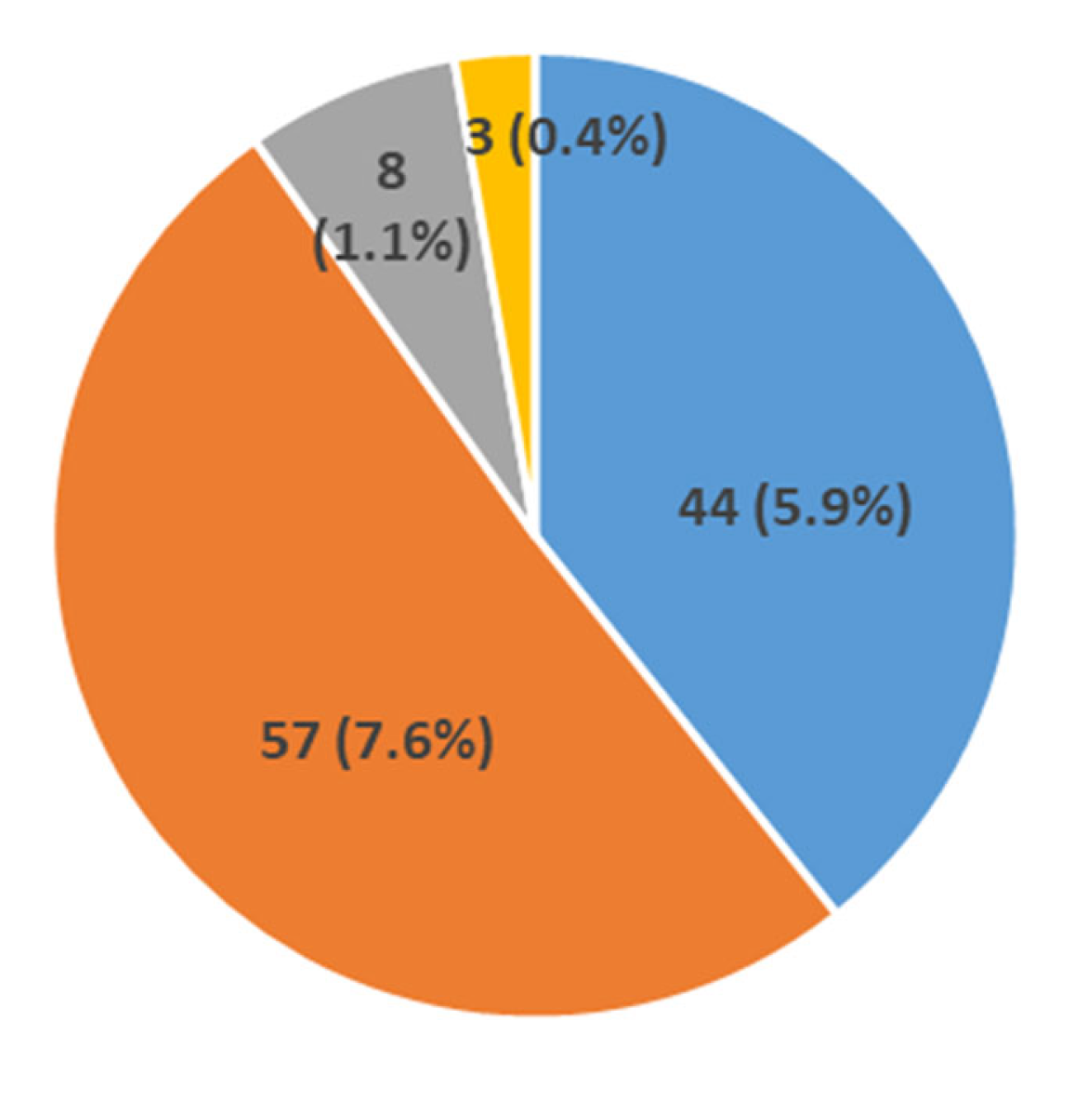
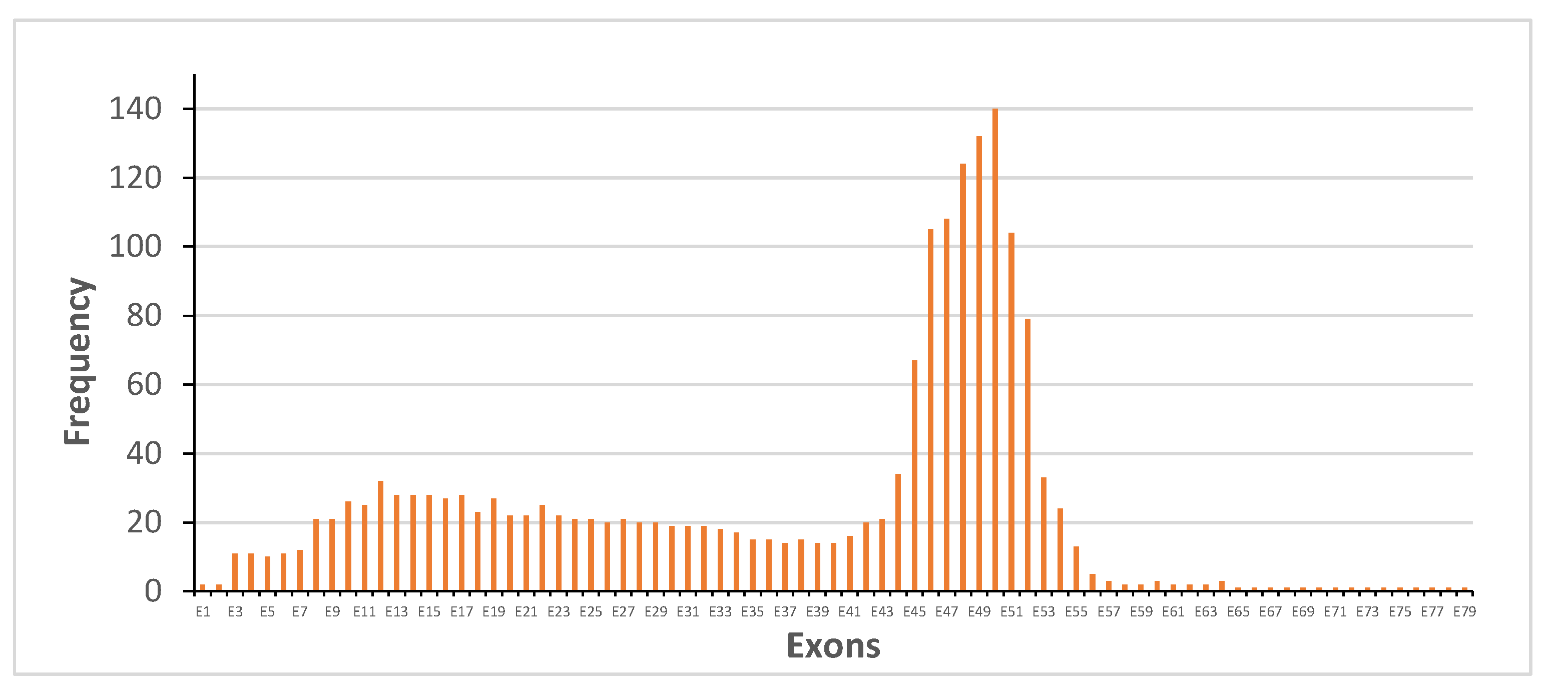
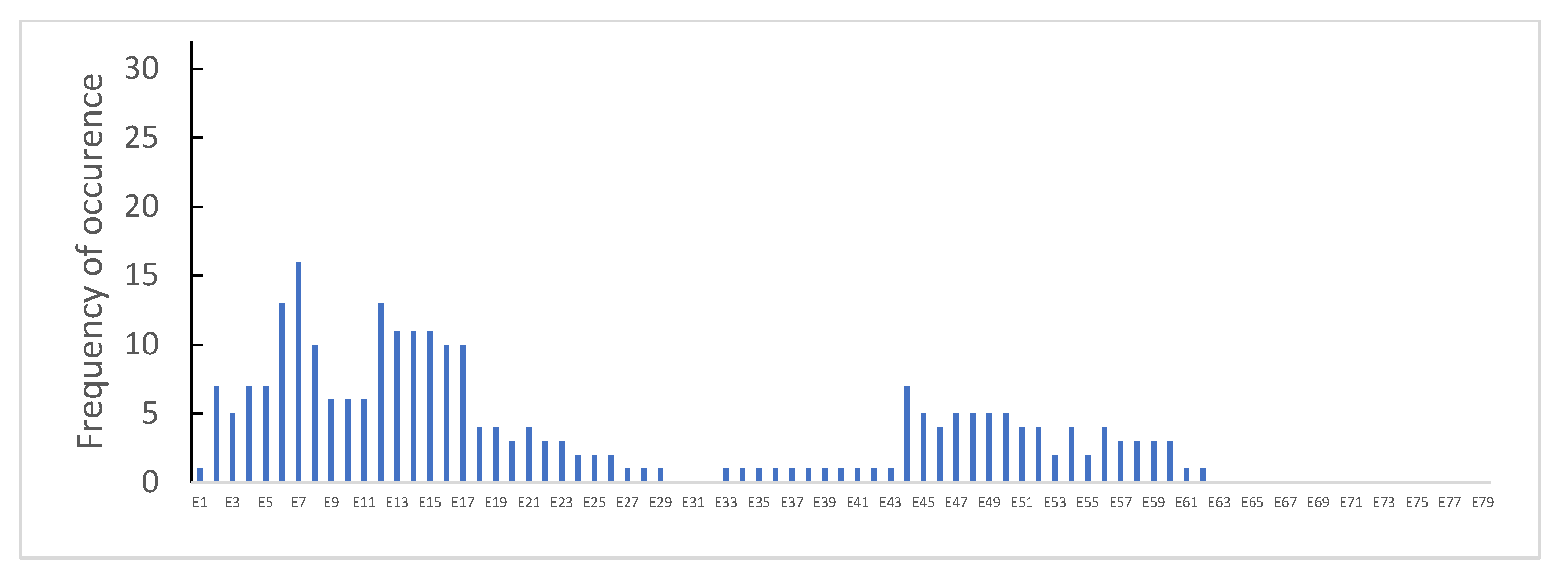
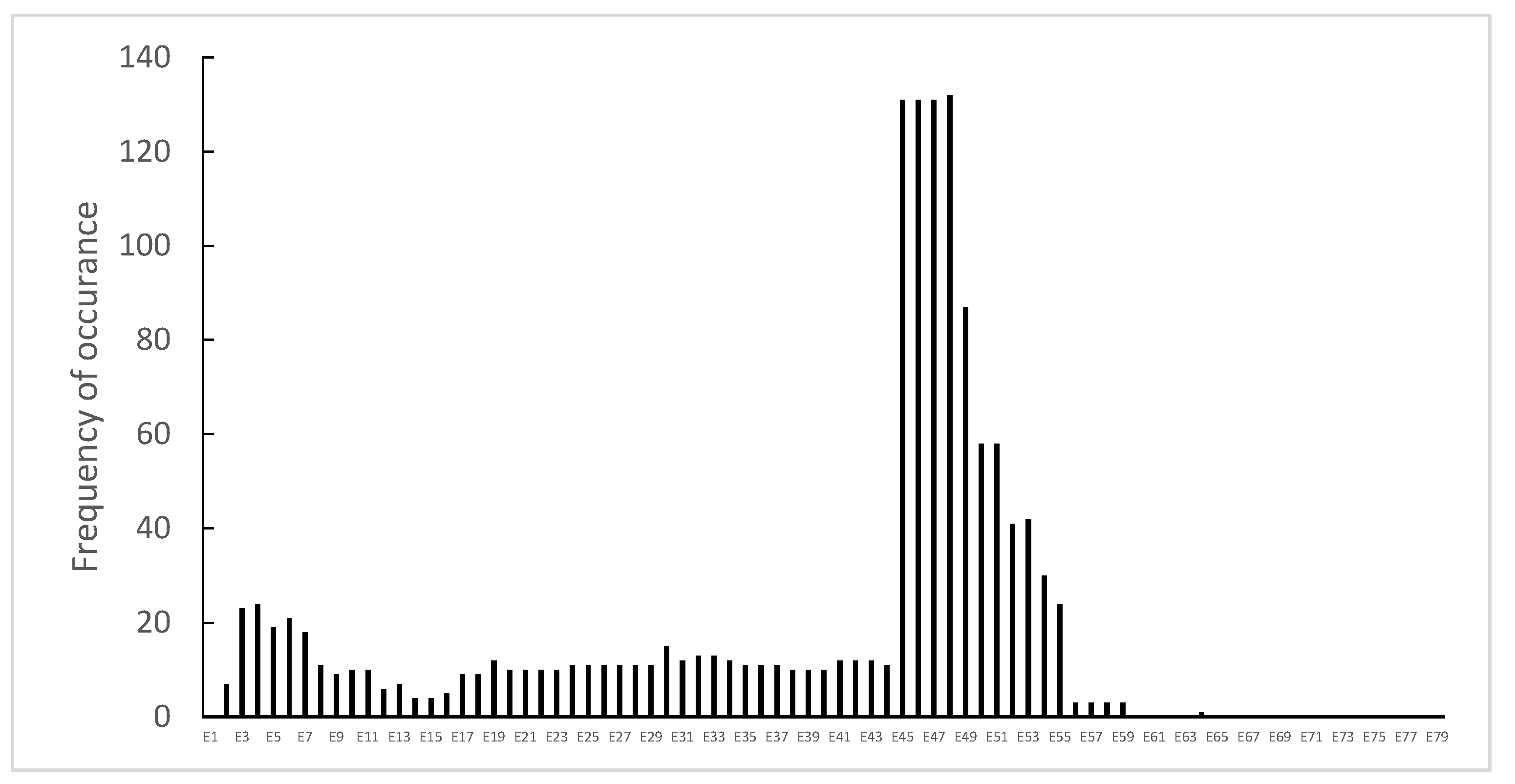
3.1. Distribution of Dystrophin Gene Pathogenic Variants in the Study Population
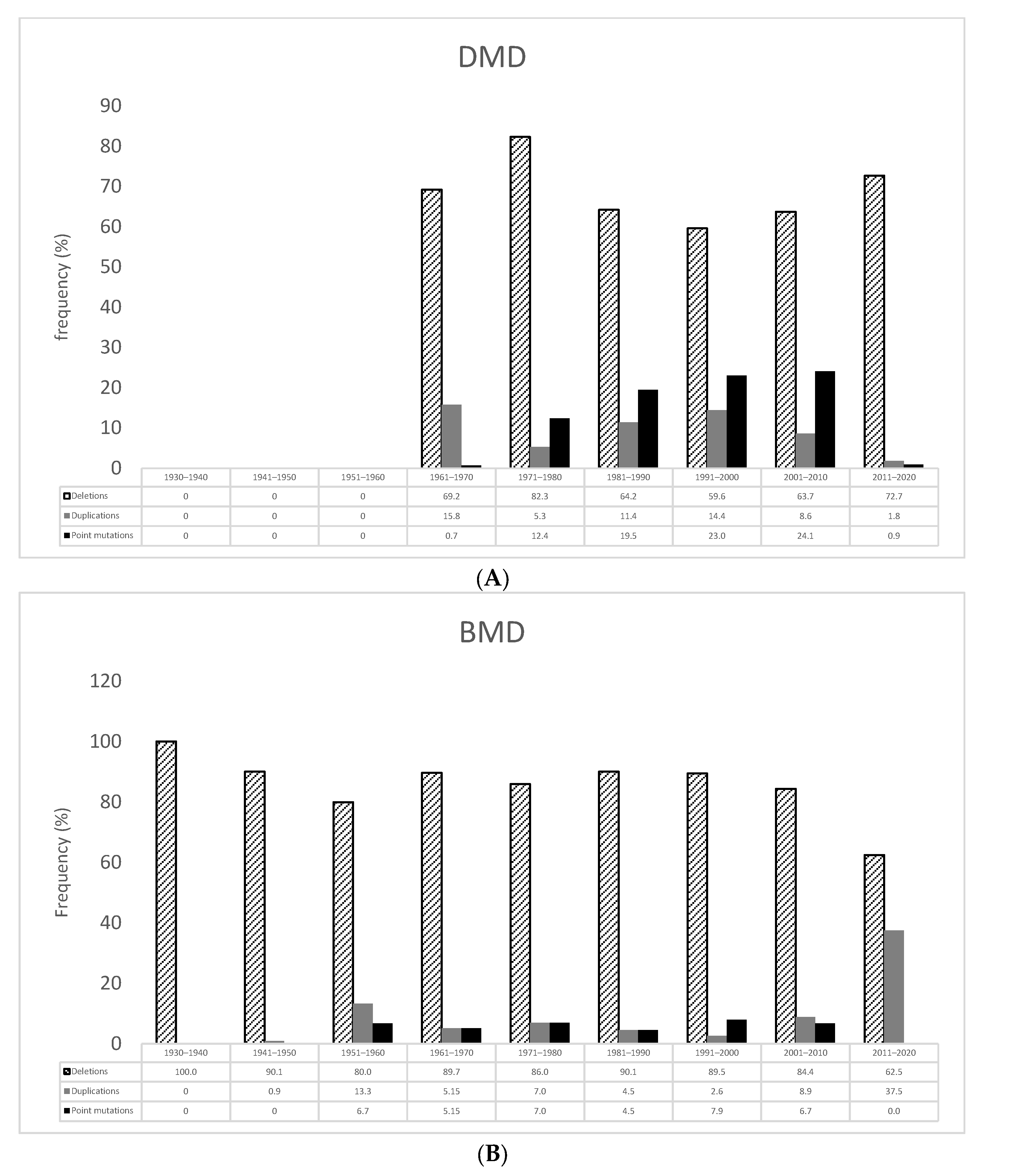
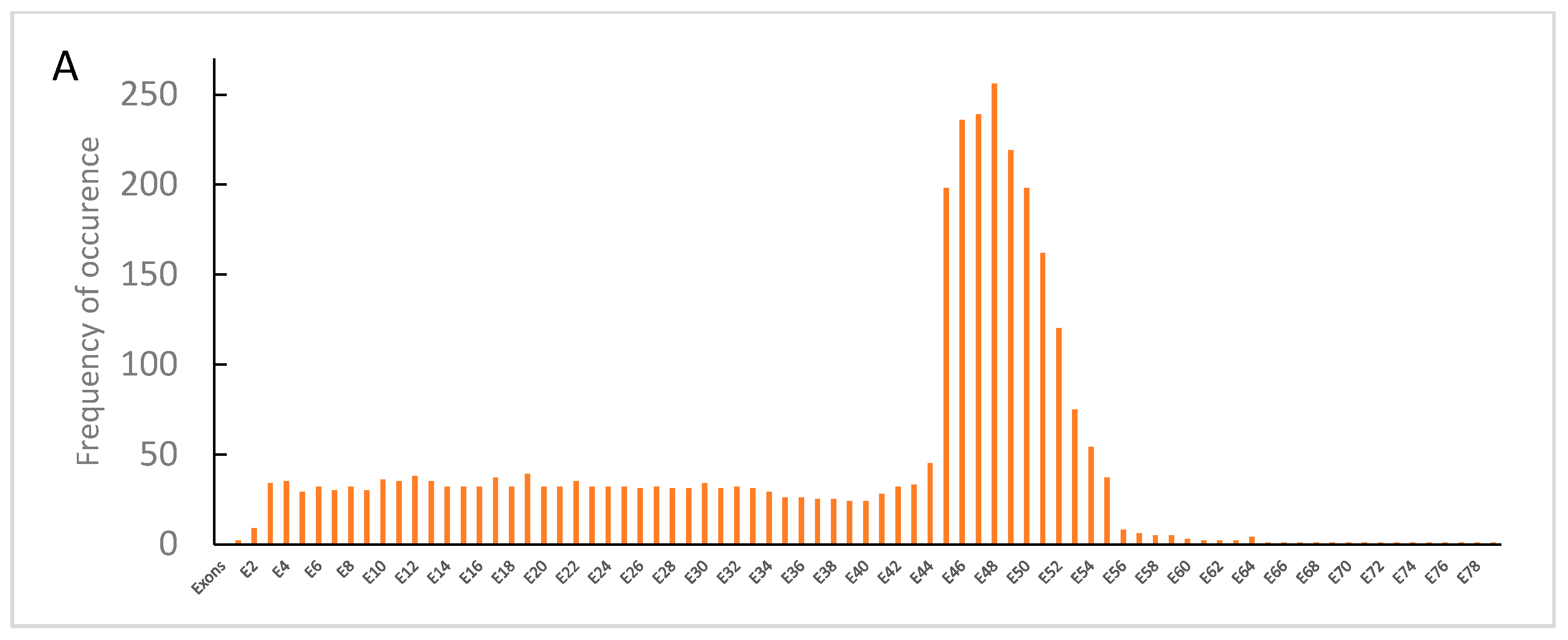
3.2. Dystrophin Gene Variant Analysis in Duchenne Population
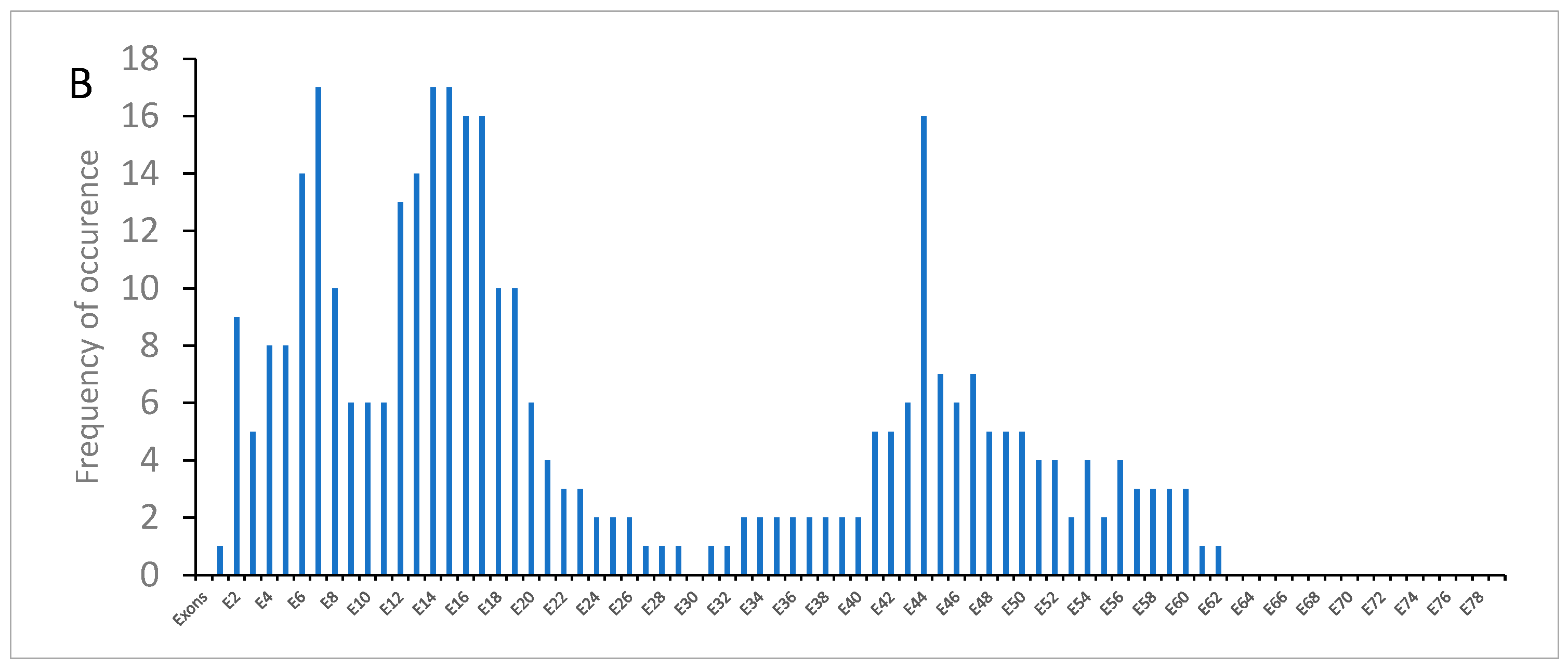
3.3. Dystrophin Gene Variant Analysis in the Becker Population
4. Discussion
- -
- selection bias, as some studies analyzed subjects from different institutions [79];
- -
- -
- failure to sequence all patients negative for deletions or duplications, due to the cost problem [93];
- -
- -
- -
- -
- higher rate of inbreeding in some countries, such as the Middle East and Turkey, leading to a higher incidence of autosomal recessive diseases;
- -
- -
- the geographic size of North America, which limits the access to diagnostic procedures;
- -
- the large difference in urban and rural medical care;
- -
- the lack of knowledge in primary care providers about DMD, resulting in less CK and transaminase testing;
- -
- insurance coverage issues, especially for genetic testing procedures.
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Emery, A.E.H. The muscular dystrophies. Lancet 2002, 359, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Mercuri, E.; Bönnemann, C.G.; Muntoni, F. Muscular dystrophies. Lancet 2019, 394, 2025–2038. [Google Scholar] [CrossRef] [PubMed]
- Brandsema, J.F.; Darras, B.T. Dystrophinopathies. Semin. Neurol. 2015, 35, 369–384. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, E.P.; Brown, R.H., Jr.; Kunkel, L.M. Dystrophin: The protein product of the Duchenne muscular dystrophy locus. Cell 1987, 51, 919–928. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, E.P.; Kunkel, L.M. Dystrophin abnormalities in Duchenne/Becker muscular dystrophy. Neuron 1989, 2, 1019–1029. [Google Scholar] [CrossRef]
- Koeks, Z.; Bladen, C.L.; Salgado, D.; van Zwet, E.; Pogoryelova, O.; McMacken, G.; Monges, S.; Foncuberta, M.E.; Kekou, K.; Kosma, K.; et al. Clinical Outcomes in Duchenne Muscular Dystrophy: A Study of 5345 Patients from the TREAT-NMD DMD Global Database. J. Neuromuscul. Dis. 2017, 4, 293–306. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, E.P. Causes of clinical variability in Duchenne and Becker muscular dystrophies and implications for exon skipping therapies. Acta Myol. 2020, 39, 179–186. [Google Scholar] [CrossRef]
- Leturcq, F.; Kaplan, J.-C. Bases moléculaires des dystrophinopathies [Molecular bases of dystrophinopathies]. J. Soc. Biol. 2005, 199, 5–11. (In French) [Google Scholar] [CrossRef]
- Blake, D.J.; Weir, A.; Newey, S.E.; Davies, K.E.; Spaulding, H.R.; Quindry, T.; Hammer, K.; Quindry, J.C.; Selsby, J.T.; Amirouche, A.; et al. Function and Genetics of Dystrophin and Dystrophin-Related Proteins in Muscle. Physiol. Rev. 2002, 82, 291–329. [Google Scholar] [CrossRef] [Green Version]
- Haldane, J.B. The rate of spontaneous mutation of a human gene. J. Genet. 2004, 83, 235–244. [Google Scholar] [CrossRef]
- Duchenne, G.B.A. Recherches sur la paralysie musculaire pseudohypertrophique ou paralysie myo-sclerosique. Arch. Gen. Med. 1868, 11, 5–25. [Google Scholar]
- Conte, G.; Gioia, L. Scrofola del sistema muscolare. Ann. Clin. Osp. Incurabili Napoli 1836, 2, 66–79. [Google Scholar]
- Thangarajh, M. The Dystrophinopathies. Continuum 2019, 25, 1619–1639. [Google Scholar] [CrossRef] [PubMed]
- Nigro, G.; Comi, L.I.; Politano, L.; Bain, R.J. The incidence and evolution of cardiomyopathy in Duchenne muscular dystrophy. Int. J. Cardiol. 1990, 26, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Nigro, G.; Comi, L.I.; Politano, L.; Nigro, G. Cardiomyopathies associated with Muscular Dystrophies. In Engel & Franzini-Armstrong: Myology; McGraw-Hill: New York, NY, USA, 2004; pp. 1239–1256. [Google Scholar]
- LoMauro, A.; Romei, M.; Gandossini, S.; Pascuzzo, R.; Vantini, S.; D’Angelo, M.G.; Aliverti, A. Evolution of respiratory function in Duchenne muscular dystrophy from childhood to adulthood. Eur. Respir. J. 2018, 51, 1701418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banihani, R.; Smile, S.; Yoon, G.; Dupuis, A.; Mosleh, M.; Snider, A.; McAdam, L. Cognitive and Neurobehavioral Profile in Boys With Duchenne Muscular Dystrophy. J. Child Neurol. 2015, 30, 1472–1482. [Google Scholar] [CrossRef] [PubMed]
- Kiessling, W.R.; Beckmann, R. Serum levels of myoglobin and creatine kinase in duchenne muscular dystrophy. Klin. Wochenschr. 1981, 59, 347–348. [Google Scholar] [CrossRef]
- Becker, P.E.; Kiener, F. A new X-chromosomal muscular dystrophy. Arch. Psychiatr. Nervenkr Z Gesamte Neurol. Psychiatry 1955, 193, 427–448. [Google Scholar] [CrossRef]
- Bushby, K.M.; Gardner-Medwin, D. The clinical, genetic and dystrophin characteristics of Becker muscular dystrophy. I. Natural history. J. Neurol. 1993, 240, 98–104. [Google Scholar] [CrossRef]
- Bushby, K.M.; Gardner-Medwin, D.; Nicholson, L.V.; Johnson, M.A.; Haggerty, I.D.; Cleghorn, N.J.; Harris, J.B.; Bhattacharyal, S.S. The clinical, genetic and dystrophin characteristics of Becker muscular dystrophy. II. Correlation of phenotype with genetic and protein abnormalities. J. Neurol. 1993, 240, 105–112. [Google Scholar] [CrossRef]
- Miyazaki, D.; Yoshida, K.; Fukushima, K.; Nakamura, A.; Suzuki, K.; Sato, T.; Takeda, S.; Ikeda, S.-I. Characterization of deletion breakpoints in patients with dystrophinopathy carrying a deletion of exons 45–55 of the Duchenne muscular dystrophy (DMD) gene. J. Hum. Genet. 2009, 54, 127–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taglia, A.; Petillo, R.; D’Ambrosio, P.; Picillo, E.; Torella, A.; Orsini, C.; Ergoli, M.; Scutifero, M.; Passamano, L.; Palladino, A.; et al. Clinical features of patients with dystrophinopathy sharing the 45–55 exon deletion of DMD gene. Acta Myol. 2015, 34, 9–13. [Google Scholar] [PubMed]
- Nigro, G.; Comi, L.I.; Politano, L.; Limongelli, F.M.; Nigro, V.; De Rimini, M.L.; Giugliano, M.A.; Petretta, V.R.; Passamano, L.; Restucci, B.; et al. Evaluation of the cardiomyopathy in becker muscular dystrophy. Muscle Nerve 1995, 18, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Arbustini, E.; Diegoli, M.; Morbini, P.; Bello, B.D.; Banchieri, N.; Pilotto, A.; Magani, F.; Grasso, M.; Narula, J.; Gavazzi, A.; et al. Prevalence and characteristics of the dystrophin defects in adult male patients with dilated cardiomyopathy. J. Am. Coll. Cardiol. 2000, 35, 1760–1768. [Google Scholar] [CrossRef] [Green Version]
- Nicolas, A.; Raguénès-Nicol, C.; Ben Yaou, R.; Ameziane-Le Hir, S.; Chéron, A.; Vié, V.; Claustres, M.; Leturcq, F.; Delalande, O.; Hubert, J.F.; et al. French network of clinical reference centres for neuromuscular diseases (CORNEMUS). Becker muscular dystrophy severity is linked to the structure of dystrophin. Hum. Mol. Genet. 2015, 24, 1267–1279. [Google Scholar] [CrossRef]
- Nigro, G.; Politano, L.; Nigro, V.; Petretta, V.R.; Comi, L.I. Mutation of dystrophin gene and cardiomyopathy. Neuromuscul. Disord. 1994, 4, 371–379. [Google Scholar] [CrossRef]
- Kaspar, R.W.; Allen, H.D.; Ray, W.C.; Alvarez, C.E.; Kissel, J.T.; Pestronk, A.; Weiss, R.B.; Flanigan, K.M.; Mendell, J.R.; Montanaro, F. Analysis of Dystrophin Deletion Mutations Predicts Age of Cardiomyopathy Onset in Becker Muscular Dystrophy. Circ. Cardiovasc. Genet. 2009, 2, 544–551. [Google Scholar] [CrossRef]
- Papa, A.A.; D’Ambrosio, P.; Petillo, R.; Palladino, A.; Politano, L. Heart transplantation in patients with dystrophinopathic cardiomyopathy: Review of the literature and personal series. Intractable Rare Dis. Res. 2017, 6, 95–101. [Google Scholar] [CrossRef] [Green Version]
- Ascencio-Lemus, M.G.; Barge-Caballero, E.; Paniagua-Martín, M.J.; Barge-Caballero, G.; Couto-Mallón, D.; Crespo-Leiro, M.G. Is Becker Dystrophinopathy a Contraindication to Heart Transplant? Experience in a Single Institution. Rev. Esp. Cardiol. 2019, 72, 584–585. [Google Scholar] [CrossRef]
- Towbin, J.A.; Hejtmancik, J.F.; Brink, P.; Gelb, B.; Zhu, X.M.; Chamberlain, J.S.; McCabe, E.R.; Swift, M. X-linked dilated cardiomyopathy. Molecular genetic evidence of linkage to the Duchenne muscular dystrophy (dystrophin) gene at the Xp21 locus. Circulation 1993, 87, 1854–1865. [Google Scholar] [CrossRef] [Green Version]
- Muntoni, F.; Cau, M.; Ganau, A.; Congiu, R.; Arvedi, G.; Mateddu, A.; Marrosu, M.G.; Cianchetti, C.; Realdi, G.; Cao, A.; et al. Brief report: Deletion of the Dystrophin Muscle-Promoter Region Associated with X-Linked Dilated Cardiomyopathy. N. Engl. J. Med. 1993, 329, 921–925. [Google Scholar] [CrossRef] [PubMed]
- Ferlini, A.; Sewry, C.; Melis, M.A.; Mateddu, A.; Muntoni, F. X-linked dilated cardiomyopathy and the dystrophin gene. Neuromuscul. Disord. 1999, 9, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Xie, Y.; Bhandari, V.; Chen, G.; Dang, Y.; Liao, H.; Zhang, J.; Lan, D. Clinical and genetic characteristics of female dystrophinopathy carriers. Mol. Med. Rep. 2019, 19, 3035–3044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norman, A.; Harper, P. A survey of manifesting carriers of Duchenne and Becker muscular dystrophy in Wales. Clin. Genet. 1989, 36, 31–37. [Google Scholar] [CrossRef]
- Seemann, N.; Selby, K.; McAdam, L.; Biggar, D.; Kolski, H.; Goobie, S.; Yoon, G.; Campbell, C.; Canadian Pediatric Neuro-muscular Group. Symptomatic dystrophinopathies in female children. Neuromuscul. Disord. 2011, 21, 172–177. [Google Scholar] [CrossRef]
- Politano, L.; Nigro, V.; Nigro, G.; Petretta, V.R.; Passamano, L.; Papparella, S.; Di Somma, S.; I Comi, L. Development of cardiomyopathy in female carriers of Duchenne and Becker muscular dystrophies. JAMA 1996, 275, 1335–1338. [Google Scholar] [CrossRef]
- Hoogerwaard, E.M.; Bakker, E.; Ippel, P.F.; Oosterwijk, J.C.; Majoor-Krakauer, D.F.; Leschot, N.J.; Van Essen, A.J.; Brunner, H.G.; van der Wouw, P.A.; Wilde, A.A.; et al. Signs and symptoms of Duchenne muscular dystrophy and Becker muscular dystrophy among carriers in the Netherlands: A cohort study. Lancet 1999, 353, 2116–2119. [Google Scholar] [CrossRef]
- Grain, L.; Cortina-Borja, M.; Forfar, C.; Hilton-Jones, D.; Hopkin, J.; Burch, M. Cardiac abnormalities and skeletal muscle weakness in carriers of Duchenne and Becker muscular dystrophies and controls. Neuromuscul. Disord. 2001, 11, 186–191. [Google Scholar] [CrossRef]
- Elangkovan, N.; Dickson, G. Gene Therapy for Duchenne Muscular Dystrophy. J. Neuromuscul. Dis. 2021, 8 (Suppl. S2), S303–S316. [Google Scholar] [CrossRef]
- Takeda, S.; Clemens, P.R.; Hoffman, E.P. Exon-Skipping in Duchenne Muscular Dystrophy. J. Neuromuscul. Dis. 2021, 8 (Suppl. S2), S343–S358. [Google Scholar] [CrossRef]
- Landfeldt, E.; Sejersen, T.; Tulinius, M. A mini-review and implementation model for using ataluren to treat nonsense muta-tion Duchenne muscular dystrophy. Acta Paediatr. 2019, 108, 224–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beggs, A.H.; Koenig, M.; Boyce, F.M.; Kunkel, L.M. Detection of 98% of DMD/BMD gene deletions by polymerase chain reaction. Hum. Genet. 1990, 86, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, J.S.; Farwell, N.J.; Chamberlain, J.R.; Cox, G.A.; Caskey, C.T. PCR analysis of dystrophin gene mutation and expression. J. Cell. Biochem. 1991, 46, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Dunnen, J.T.D.; Beggs, A.H. Multiplex PCR for Identifying DMD Gene Deletions. Curr. Protoc. Hum. Genet. 2006, 49, 9.3.1–9.3.22. [Google Scholar] [CrossRef]
- Lalic, T.; Vossen, R.H.; Coffa, J.; Schouten, J.P.; Guc-Scekic, M.; Radivojevic, D.; Djurisic, M.; Breuning, M.H.; White, S.J.; den Dunnen, J.T. Deletion and duplication screening in the DMD gene using MLPA. Eur. J. Hum. Genet. 2005, 13, 1231–1234. [Google Scholar] [CrossRef] [PubMed]
- Okizuka, Y.; Takeshima, Y.; Awano, H.; Zhang, Z.; Yagi, M.; Matsuo, M. Small Mutations Detected by Multiplex Ligation–Dependent Probe Amplification of the Dystrophin Gene. Genet. Test. Mol. Biomark. 2009, 13, 427–431. [Google Scholar] [CrossRef]
- Schwartz, M.; Dunø, M. Improved Molecular Diagnosis of Dystrophin Gene Mutations Using the Multiplex Ligation-Dependent Probe Amplification Method. Genet. Test. 2004, 8, 361–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slatko, B.E.; Gardner, A.F.; Ausubel, F.M. Overview of Next Generation Sequencing technologies (and bioinformatics) in cancer. Mol. Biol. 2018, 122, e59. [Google Scholar] [CrossRef]
- Volk, A.E.; Kubisch, C. The rapid evolution of molecular genetic diagnostics in neuromuscular diseases. Curr. Opin. Neurol. 2017, 30, 523–528. [Google Scholar] [CrossRef]
- Zhang, K.; Yang, X.; Lin, G.; Han, Y.; Li, J. Molecular genetic testing and diagnosis strategies for dystrophinopathies in the era of next generation sequencing. Clin. Chim. Acta 2019, 491, 66–73. [Google Scholar] [CrossRef]
- Lim, B.C.; Lee, S.; Shin, J.-Y.; Kim, J.-I.; Hwang, H.; Kim, K.J.; Hwang, Y.S.; Seo, J.-S.; Chae, J.H. Genetic diagnosis of Duchenne and Becker muscular dystrophy using next-generation sequencing technology: Comprehensive mutational search in a single platform. J. Med. Genet. 2011, 48, 731–736. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; Ginjaar, I.B.; Bushby, K. The importance of genetic diagnosis for Duchenne muscular dystrophy. J. Med. Genet. 2016, 53, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Nigro, V.; Politano, L.; Nigro, G.; Romano, S.C.; Molinari, A.M.; Puca, G.A. Detection of a nonsense mutation in the dystrophin gene by multiple SSCP. Hum. Mol. Genet. 1992, 1, 517–520. [Google Scholar] [CrossRef] [PubMed]
- Nigro, V.; Nigro, G.; Esposito, M.G.; Comi, L.I.; Molinari, A.M.; Puca, G.A.; Politano, L. Novel small mutations along the DMD/BMD gene associated with different phenotypes. Hum. Mol. Genet. 1994, 3, 1907–1908. [Google Scholar] [CrossRef] [PubMed]
- Trimarco, A.; Torella, A.; Piluso, G.; Maria Ventriglia, V.; Politano, L.; Nigro, V. Log-PCR: A New Tool for Immediate and Cost-Effective Diagnosis of up to 85% of Dystrophin Gene Mutations. Clin. Chem. 2008, 54, 973–981. [Google Scholar] [CrossRef] [Green Version]
- Torella, A.; Trimarco, A.; Blanco, F.D.V.; Cuomo, A.; Aurino, S.; Piluso, G.; Minetti, C.; Politano, L.; Nigro, V. One Hundred Twenty-One Dystrophin Point Mutations Detected from Stored DNA Samples by Combinatorial Denaturing High-Performance Liquid Chromatography. J. Mol. Diagn. 2010, 12, 65–73. [Google Scholar] [CrossRef]
- Torella, A.; Zanobio, M.; Zeuli, R.; Del Vecchio Blanco, F.; Savarese, M.; Giugliano, T.; Garofalo, A.; Piluso, G.; Politano, L.; Nigro, V. The position of nonsense mutations can predict the phenotype severity: A survey on the DMD gene. PLoS ONE 2020, 15, e0237803. [Google Scholar] [CrossRef]
- Savarese, M.; Qureshi, T.; Torella, A.; Laine, P.; Giugliano, T.; Jonson, P.H.; Johari, M.; Paulin, L.; Piluso, G.; Auvinen, P.; et al. Identification and Characterization of Splicing Defects by Single-Molecule Real-Time Sequencing Technology (PacBio). J. Neuromuscul. Dis. 2020, 7, 477–481. [Google Scholar] [CrossRef]
- Onore, M.E.; Torella, A.; Musacchia, F.; D’Ambrosio, P.; Zanobio, M.; Blanco, F.D.V.; Piluso, G.; Nigro, V. Linked-Read Whole Genome Sequencing Solves a Double DMD Gene Rearrangement. Genes 2021, 12, 133. [Google Scholar] [CrossRef] [PubMed]
- Mora, M.; Angelini, C.; Bignami, F.; Bodin, A.-M.; Crimi, M.; Di Donato, J.H.; Felice, A.; Jaeger, C.; Karcagi, V.; LeCam, Y.; et al. The EuroBioBank Network: 10 years of hands-on experience of collaborative, transnational biobanking for rare diseases. Eur. J. Hum. Genet. 2014, 23, 1116–1123. [Google Scholar] [CrossRef]
- Filocamo, M.; Baldo, C.; Goldwurm, S.; Renieri, A.; Angelini, C.; Moggio, M.; Mora, M.; Merla, G.; Politano, L.; Garavaglia, B.; et al. Telethon Network of Genetic Biobanks: A key service for diagnosis and research on rare diseases. Orphanet J. Rare Dis. 2013, 8, 129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Amico, A.; Catteruccia, M.; Baranello, G.; Politano, L.; Govoni, A.; Previtali, S.C.; Pane, M.; D’Angelo, M.G.; Bruno, C.; Messina, S.; et al. Diagnosis of Duchenne Muscular Dystrophy in Italy in the last decade: Critical issues and areas for improvements. Neuromuscul. Disord. 2017, 27, 447–451. [Google Scholar] [CrossRef]
- Neri, M.; Rossi, R.; Trabanelli, C.; Mauro, A.; Selvatici, R.; Falzarano, M.S.; Spedicato, N.; Margutti, A.; Rimessi, P.; Fortunato, F.; et al. The Genetic Landscape of Dystrophin Mutations in Italy: A Nationwide Study. Front. Genet. 2020, 11, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flanigan, K.M.; Dunn, D.M.; von Niederhausern, A.; Soltanzadeh, P.; Gappmaier, E.; Howard, M.T.; Sampson, J.B.; Mendell, J.R.; Wall, C.; King, W.M.; et al. Mutational spectrum of DMD mutations in dystrophinopathy patients: Application of modern diagnostic techniques to a large cohort. Hum. Mutat. 2009, 30, 1657–1666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuffery-Giraud, S.; Béroud, C.; Leturcq, F.; Ben Yaou, R.; Hamroun, D.; Michel-Calemard, L.; Moizard, M.-P.; Bernard, R.; Cossée, M.; Boisseau, P.; et al. Genotype-phenotype analysis in 2,405 patients with a dystrophinopathy using the UMD-DMD database: A model of nationwide knowledgebase. Hum. Mutat. 2009, 30, 934–945. [Google Scholar] [CrossRef] [PubMed]
- The DMD Mutations Database UMD-DMD France. Available online: http://www.umd.be/DMD/ (accessed on 4 October 2021).
- Vieitez, I.; Gallano, P.; Gonzalez-Quereda, L.; Borrego, S.; Marcos, I.; Millán, J.M.; Jairo, T.; Prior, C.; Molano, J.M.; Trujillo-Tiebas, M.; et al. Mutational spectrum of Duchenne muscular dystrophy in Spain: Study of 284 cases. Neurología 2017, 32, 377–385, (In English, Spanish). [Google Scholar] [CrossRef] [PubMed]
- Garcia, S.; de Haro, T.; Zafra-Ceres, M.; Poyatos, A.; Gomez-Capilla, J.A.; Gomez-Llorente, C. Identification of de novoMutations of Duchénnè/Becker Muscular Dystrophies in Southern Spain. Int. J. Med. Sci. 2014, 11, 988–993. [Google Scholar] [CrossRef] [Green Version]
- van den Bergen, J.C.; Ginjaar, H.B.; van Essen, A.J.; Pangalila, R.; de Groot, I.J.; Wijkstra, P.J.; Zijnen, M.P.; Cobben, N.A.; Kampelmacher, M.J.; Wokke, B.H.; et al. Forty-Five Years of Duchenne Muscular Dystrophy in The Netherlands. J. Neuromuscul. Dis. 2014, 1, 99–109. [Google Scholar] [CrossRef] [Green Version]
- Selvatici, R.; Rossi, R.; Fortunato, F.; Trabanelli, C.; Sifi, Y.; Margutti, A.; Neri, M.; Gualandi, F.; Szabò, L.; Fekete, B.; et al. Ethnicity-related DMD Genotype Landscapes in European and Non-European Countries. Neurol. Genet. 2020, 7, e536. [Google Scholar] [CrossRef]
- Zimowski, J.G.; Purzycka, J.; Pawelec, M.; Ozdarska, K.; Zaremba, J. Small mutations in Duchenne/Becker muscular dystrophy in 164 unrelated Polish patients. J. Appl. Genet. 2021, 62, 289–295. [Google Scholar] [CrossRef]
- Toksoy, G.; Durmus, H.; Aghayev, A.; Bagirova, G.; Rustemoglu, B.S.; Basaran, S.; Avci, S.; Karaman, B.; Parman, Y.; Altunoglu, U.; et al. Mutation spectrum of 260 dystrophinopathy patients from Turkey and important highlights for genetic counseling. Neuromuscul. Disord. 2019, 29, 601–613. [Google Scholar] [CrossRef]
- Brabec, P.; Vondráček, P.; Klimeš, D.; Baumeister, S.; Lochmüller, H.; Pavlík, T.; Gregor, J. Characterization of the DMD/BMD patient population in Czech Republic and Slovakia using an innovative registry approach. Neuromuscul. Disord. 2009, 19, 250–254. [Google Scholar] [CrossRef]
- Elhawary, N.A.; Jiffri, E.H.; Jambi, S.; Mufti, A.H.; Dannoun, A.; Kordi, H.; Khogeer, A.; Jiffri, O.H.; Elhawary, A.N.; Tayeb, M.T. Molecular characterization of exonic rearrangements and frame shifts in the dystrophin gene in Duchenne muscular dystrophy patients in a Saudi community. Hum. Genom. 2018, 12, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tayeb, M.T. Deletion mutations in Duchenne muscular dystrophy (DMD) in Western Saudi children. Saudi J. Biol. Sci. 2010, 17, 237–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohammed, F.; Elshafey, A.; Al-Balool, H.; Alaboud, H.; Al Ben Ali, M.; Baqer, A.; Bastaki, L. Mutation spectrum analysis of Duchenne/Becker muscular dystrophy in 68 families in Kuwait: The era of personalized medicine. PLoS ONE 2018, 13, e0197205. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Zhong, X.; Liu, L.; Cui, S.; Yang, Y.; Kong, L. Genetic analysis of 1051 Chinese families with Duchenne/Becker Muscular Dystrophy. BMC Med. Genet. 2019, 20, 139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, I.F.; Lai, K.K.; Tong, T.M.; Lam, S.T. A different spectrum of DMD gene mutations in local Chinese patients with Duchenne/Becker muscular dystrophy. Chin. Med. J. 2006, 119, 1079–1087. [Google Scholar]
- Guo, R.; Zhu, G.; Zhu, H.; Ma, R.; Peng, Y.; Liang, D.; Wu, L. DMD mutation spectrum analysis in 613 Chinese patients with dystrophinopathy. J. Hum. Genet. 2015, 60, 435–442. [Google Scholar] [CrossRef]
- Wang, L.; Xu, M.; Li, H.; He, R.; Lin, J.; Zhang, C.; Zhu, Y. Genotypes and Phenotypes of DMD Small Mutations in Chinese Patients with Dystrophinopathies. Front. Genet. 2019, 10, 114. [Google Scholar] [CrossRef]
- Ma, P.; Zhang, S.; Zhang, H.; Fang, S.; Dong, Y.; Zhang, Y.; Hao, W.; Wu, S.; Zhao, Y. Comprehensive genetic characteristics of dystrophinopathies in China. Orphanet J. Rare Dis. 2018, 13, 109. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Liu, Z.; OuYang, S.; Zhu, Y.; Wang, L.; Wu, J. Distribution of dystrophin gene deletions in a Chinese population. J. Int. Med. Res. 2016, 44, 99–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Zhao, L.; Zhou, S.; Hu, C.; Shi, Y.; Shi, W.; Li, H.; Liu, F.; Wu, B.; Wang, Y. A comprehensive database of Duchenne and Becker muscular dystrophy patients (0–18 years old) in East China. Orphanet J. Rare Dis. 2015, 10, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.-M.; Yan, K.; Liu, B.; Chen, M.; Wang, L.-Y.; Huang, Y.-Z.; Qian, Y.-Q.; Sun, Y.-X.; Li, H.-G.; Dong, M.-Y. Comprehensive genetic diagnosis of patients with Duchenne/Becker muscular dystrophy (DMD/BMD) and pathogenicity analysis of splice site variants in the DMD gene. J. Zhejiang Univ. B 2019, 20, 753–765. [Google Scholar] [CrossRef] [PubMed]
- Yun, U.; Lee, S.-A.; Choi, W.A.; Kang, S.-W.; Seo, G.H.; Lee, J.H.; Park, G.; Lee, S.; Choi, Y.-C.; Park, H.J. Clinical and genetic spectra in patients with dystrophinopathy in Korea: A single-center study. PLoS ONE 2021, 16, e0255011. [Google Scholar] [CrossRef]
- Cho, A.; Seong, M.-W.; Lim, B.C.; Lee, H.J.; Byeon, J.H.; Kim, S.S.; Kim, S.Y.; Choi, S.A.; Wong, A.-L.; Lee, J.; et al. Consecutive analysis of mutation spectrum in the dystrophin gene of 507 Korean boys with Duchenne/Becker muscular dystrophy in a single center. Muscle Nerve 2017, 55, 727–734. [Google Scholar] [CrossRef] [PubMed]
- Takeshima, Y.; Yagi, M.; Okizuka, Y.; Awano, H.; Zhang, Z.; Yamauchi, Y.; Nishio, H.; Matsuo, M. Mutation spectrum of the dystrophin gene in 442 Duchenne/Becker muscular dystrophy cases from one Japanese referral center. J. Hum. Genet. 2010, 55, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Okubo, M.; Goto, K.; Komaki, H.; Nakamura, H.; Mori-Yoshimura, M.; Hayashi, Y.K.; Mitsuhashi, S.; Noguchi, S.; Kimura, E.; Nishino, I. Comprehensive analysis for genetic diagnosis of Dystrophinopathies in Japan. Orphanet J. Rare Dis. 2017, 12, 149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zamani, G.; Hosseinpour, S.; Ashrafi, M.R.; Mohammadi, M.; Badv, R.S.; Tavasoli, A.R.; Akbari, M.G.; Bereshneh, A.H.; Malamiri, R.A.; Heidari, M. Characteristics of disease progression and genetic correlation in ambulatory Iranian boys with Duchenne muscular dystrophy. BMC Neurol. 2022, 22, 162. [Google Scholar] [CrossRef] [PubMed]
- Tomar, S.; Moorthy, V.; Sethi, R.; Chai, J.; Low, P.S.; Hong, S.T.K.; Lai, P.S. Mutational spectrum of dystrophinopathies in Singapore: Insights for genetic diagnosis and precision therapy. Am. J. Med. Genet. Part C Semin. Med. Genet. 2019, 181, 230–244. [Google Scholar] [CrossRef]
- Rani, A.Q.; Sasongko, T.H.; Sulong, S.; Bunyan, D.; Salmi, A.R.; Zilfalil, B.A.; Matsuo, M.; Zabidi-Hussin, Z.A. Mutation Spectrum ofDystrophinGene in Malaysian Patients with Duchenne/Becker Muscular Dystrophy. J. Neurogenet. 2013, 27, 11–15. [Google Scholar] [CrossRef]
- Rao, M.V.; Sindhav, G.M.; Mehta, J.J. Duchenne/Becker muscular dystrophy: A report on clinical, biochemical, and genetic study in Gujarat population, India. Ann. Indian Acad. Neurol. 2014, 17, 303–307. [Google Scholar] [CrossRef] [PubMed]
- Kohli, S.; Saxena, R.; Thomas, E.; Singh, K.; BijarniaMahay, S.; Puri, R.D.; Verma, I.C. Mutation Spectrum of Dystrophinopathies in India: Implications for Therapy. Indian J. Pediatr. 2020, 87, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Polavarapu, K.; Preethish-Kumar, V.; Sekar, D.; Vengalil, S.; Nashi, S.; Mahajan, N.P.; Thomas, P.T.; Sadasivan, A.; Warrier, M.; Gupta, A.; et al. Mutation pattern in 606 Duchenne muscular dystrophy children with a comparison between familial and non-familial forms: A study in an Indian large single-center cohort. J. Neurol. 2019, 266, 2177–2185. [Google Scholar] [CrossRef] [PubMed]
- Manjunath, M.; Kiran, P.; Preethish-Kumar, V.; Nalini, A.; Singh, R.J.; Gayathri, N. A comparative study of mPCR, MLPA, and muscle biopsy results in a cohort of children with Duchenne muscular dystrophy: A first study. Neurol. India 2015, 63, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Vengalil, S.; Preethish-Kumar, V.; Polavarapu, K.; Mahadevappa, M.; Sekar, D.; Purushottam, M.; Thomas, P.T.; Nashi, S.; Nalini, A. Duchenne Muscular Dystrophy and Becker Muscular Dystrophy Confirmed by Multiplex Ligation-Dependent Probe Amplification: Genotype-Phenotype Correlation in a Large Cohort. J. Clin. Neurol. 2017, 13, 91–97. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.H.; Athimoolam, K.; Suraj, M.; Das ChristuDas, M.S.; Muralidharan, A.; Jeyam, D.; Ashokan, J.; Karthikeyan, P.; Krishna, R.; Khanna-Gupta, A.; et al. Comprehensive genetic analysis of 961 unrelated Duchenne Muscular Dystrophy patients: Focus on diagnosis, prevention and therapeutic possibilities. PLoS ONE 2020, 15, e0232654. [Google Scholar] [CrossRef]
- Tallapaka, K.; Ranganath, P.; Ramachandran, A.; Uppin, M.S.; Perala, S.; Aggarwal, S.; Lakshmi, D.; Meena, A.K.; Dalal, A.B. Molecular and Histopathological Characterization of Patients Presenting with the Duchenne Muscular Dystrophy Phenotype in a Tertiary Care Center in Southern India. Indian Pediatr. 2019, 56, 556–559. [Google Scholar] [CrossRef]
- Thakur, N.; Abeysekera, G.; Wanigasinghe, J.; Dissanayake, V.H.W. The spectrum of deletions and duplications in the dystrophin (DMD) gene in a cohort of patients with Duchenne muscular dystrophy in Sri Lanka. Neurol. India 2019, 67, 714–715. [Google Scholar]
- Elhawary, N.A.; Shawky, R.M.; Hashem, N. Frameshift deletion mechanisms in Egyptian Duchenne and Becker muscular dystrophy families. Mol. Cells 2004, 18, 141–149, Erratum in Mol. Cells 2005, 19, 155; Nasser A, Elhawary [corrected to Elhawary, Nasser A].. [Google Scholar]
- El Sherif, R.M.; Fahmy, N.A.; Nonaka, I.; A Etribi, M. Patterns of dystrophin gene deletion in Egyptian Duchenne/Becker muscular dystrophy patients. Acta Myol. 2007, 26, 145–150. [Google Scholar] [CrossRef]
- Effat, L.K.; El-Harouni, A.A.; Amr, K.; El-Minisi, T.I.; Meguid, N.; El Awady, M. Screening of Dystrophin Gene Deletions in Egyptian Patients with DMD/BMD Muscular Dystrophies. Dis. Markers 2000, 16, 125–129. [Google Scholar] [CrossRef] [Green Version]
- Sbiti, A.; El Kerch, F.; Sefiani, A. Analysis of Dystrophin Gene Deletions by Multiplex PCR in Moroccan Patients. J. Biomed. Biotechnol. 2002, 2, 158–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballo, R.; Viljoen, D.; Beighton, P. Duchenne and Becker muscular dystrophy prevalence in South Africa and molecular findings in 128 persons affected. S.Afr. Med. J. 1994, 84Pt 1, 494–497. [Google Scholar]
- Kerr, R.; Robinson, C.; Essop, F.B.; Krause, A. Genetic testing for Duchenne/Becker muscular dystrophy in Johannesburg, South Africa. S.Afr. Med. J. 2013, 103 (Suppl. S1), 999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mah, J.K.; Korngut, L.; Dykeman, J.; Day, L.; Pringsheim, T.; Jette, N. A systematic review and meta-analysis on the epidemiology of Duchenne and Becker muscular dystrophy. Neuromuscul. Disord. 2014, 24, 482–491. [Google Scholar] [CrossRef]
- Gambetta, K.E.; McCulloch, M.A.; Lal, A.K.; Knecht, K.; Butts, R.J.; Villa, C.R.; Johnson, J.N.; Conway, J.; Bock, M.J.; Schumacher, K.R.; et al. Diversity of Dystrophin Gene Mutations and Disease Progression in a Contemporary Cohort of Duchenne Muscular Dystrophy. Pediatr. Cardiol. 2022, 43, 855–867. [Google Scholar] [CrossRef] [PubMed]
- Dent, K.M.; Dunn, D.M.; von Niederhausern, A.C.; Aoyagi, A.T.; Kerr, L.; Bromberg, M.B.; Hart, K.J.; Tuohy, T.; White, S.; den Dunnen, J.T.; et al. Improved molecular diagnosis of dystrophinopathies in an unselected clinical cohort. Am. J. Med. Genet. Part A 2005, 134, 295–298. [Google Scholar] [CrossRef]
- Luce, L.N.; Dalamon, V.; Ferrer, M.; Parma, D.; Szijan, I.; Giliberto, F. MLPA analysis of an Argentine cohort of patients with dystrophinopathy: Association of intron breakpoints hot spots with STR abundance in DMD gene. J. Neurol. Sci. 2016, 365, 22–30. [Google Scholar] [CrossRef]
- Triana-Fonseca, P.; Parada-Márquez, J.F.; Silva-Aldana, C.T.; Zambrano-Arenas, D.; Arias-Gomez, L.L.; Morales-Fonseca, N.; Medina-Méndez, E.; Restrepo, C.M.; Silgado-Guzmán, D.F.; Fonseca-Mendoza, D.J. Genetic Profile of the Dystrophin Gene Reveals New Mutations in Colombian Patients Affected with Muscular Dystrophinopathy. Appl. Clin. Genet. 2021, 14, 399–408. [Google Scholar] [CrossRef]
- Ramos, E.; Conde, J.G.; Berrios, R.A.; Pardo, S.; Gómez, O.; Rodríguez, M.F.M. Prevalence and Genetic Profile of Duchene and Becker Muscular Dystrophy in Puerto Rico. J. Neuromuscul. Dis. 2016, 3, 261–266. [Google Scholar] [CrossRef] [Green Version]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Chapman, M.; Evans, K.; Azevedo, L.; Hayden, M.; Heywood, S.; Millar, D.S.; Phillips, A.D.; et al. The Human Gene Mutation Database (HGMD®): Optimizing its use in a clinical diagnostic or research setting. Hum. Genet. 2020, 139, 1197–1207. [Google Scholar] [CrossRef] [PubMed]
- Passamano, L.; Taglia, A.; Palladino, A.; Viggiano, E.; D’Ambrosio, P.; Scutifero, M.; Cecio, M.R.; Torre, V.; De Luca, F.; Picillo, E.; et al. Improvement of survival in Duchenne Muscular Dystrophy: Retrospective analysis of 835 patients. Acta Myol. 2012, 31, 121–125. [Google Scholar] [PubMed]











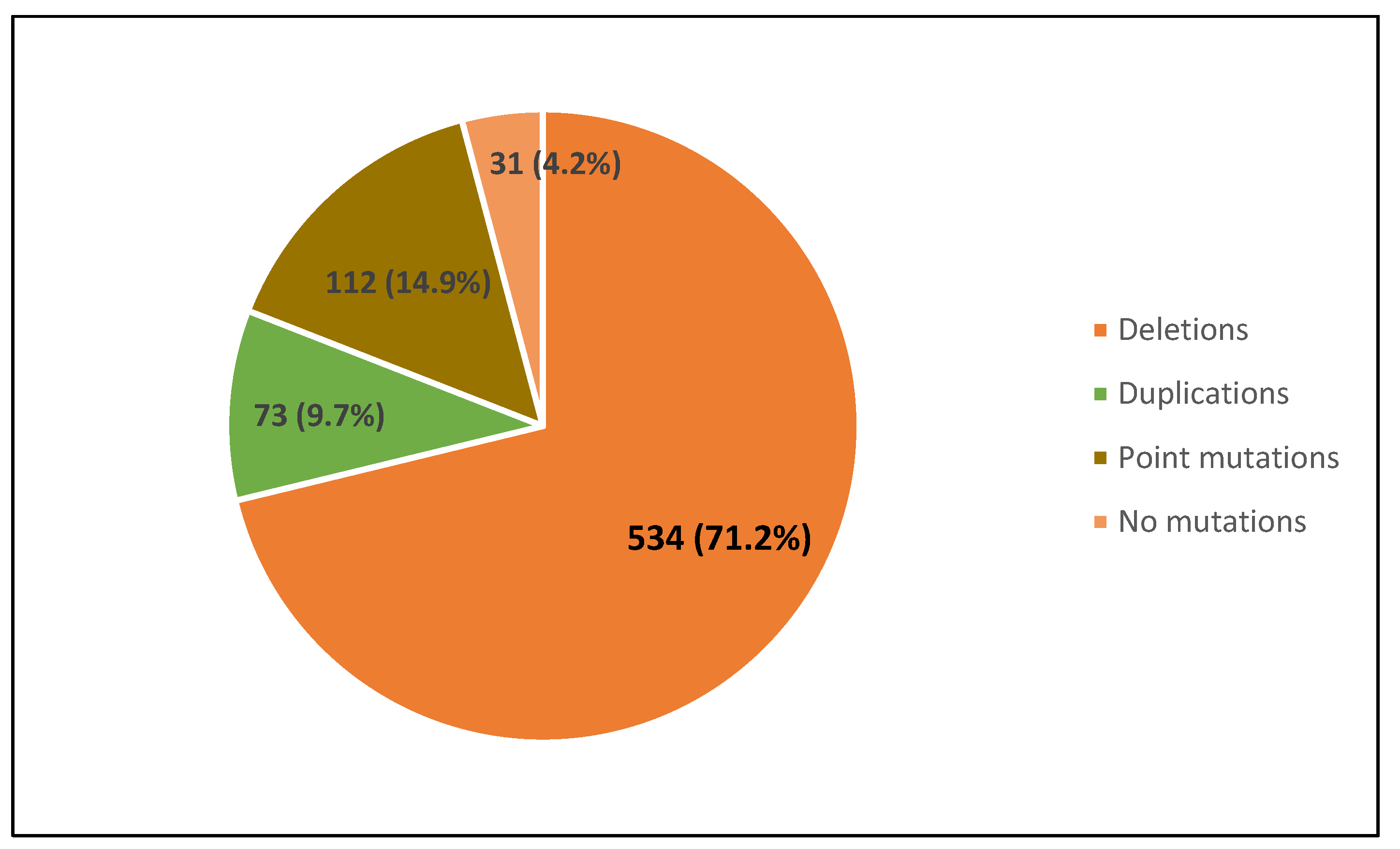{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Countries | Suspected Dystrophinopathies | Large Deletions | Large Duplications | Point Mutations/Small Rearrangments | Small Ins/Del | Nonsense | Frame-Shift | Splice Site | Missense | Reference | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| n. | n. | % | n. | % | n. | % | n. | % | n. | % | n. | % | n. | % | n. | % | ||
| EUROPE | ||||||||||||||||||
| Denmark | 182 | 87 | 47.8 | 14 | 7.7 | 4 | 2.2 | 3 | 1.6 | 1 | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | [48] |
| France | 2411 | 1404 | 58.2 | 362 | 25.6 | 465 | 19.2 | n.r. | n.r. | 187 | 7.6 | 148 | 6.1 | 125 | 8.8 | 7 | 0.4 | [66] |
| France | 2898 | 1901 | 65.5 | 323 | 11.1 | 633 | 21.8 | 201 | 6.9 | 254 | 8.7 | n.r. | n.r. | 158 | 5.4 | 20 | 0.7 | [67] |
| Spain | 284 | 131 | 46.1 | 56 | 19.7 | 97 | 34.1 | 34 | 12.0 | 49 | 17.2 | n.r. | n.r. | 10 | 3.5 | 2 | 0.7 | [68] |
| Spain | 53 | 34 | 64.2 | 4 | 7.5 | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | [69] |
| Netherlands | 462 | 212 | 45.8 | 42 | 9.0 | n.r. | n.r. | n.r. | n.r. | 49 | 10.6 | n.r. | n.r. | 18 | 3.8 | n.r. | n.r. | [70] |
| Italy | 1902 | 1242 | 65.2 | 190 | 9.9 | 469 | 24.6 | n.r. | n.r. | 200 | 10.5 | 139 | 11.1 | 67 | 5.4 | 38 | 1.9 | [64] |
| South Italyand Sicily | 750 | 534 | 71.2 | 73 | 9.7 | 112 | 14.9 | 44 * | 5.9 | 57 | 7.6 | 44 * | 5.9 | 8 | 1.1 | 0 | 0 | This study |
| EAST EUROPE | ||||||||||||||||||
| Bosnia, Bulgaria, Cyprus, Croatia, Hungary, Lithuania, Poland, Romania, Russia, Serbia, and Ukraine | 260 | 52 | 20 | 17 | 6.5 | n.r. | n.r. | n.r. | n.r. | 49 | 18.8 | 29 | 11.1 | 16 | 9 | 4 | 1.5 | [71] |
| Poland | 180 | 110 | 61.1 | 21 | 4.7 | 2 | 1.1 | n.r. | n.r. | 1 | 0.5 | 1 | 0.5 | n.r. | n.r. | n.r. | n.r. | [72] |
| Turkey | 260 | 128 | 49.2 | 25 | 9.6 | 60 | 23 | n.r. | n.r. | 32 | 12.3 | 17 | 6.5 | 1 | 0.3 | 4 | 1.5 | [73] |
| Czech Republic and Slovakia | 126 | 77 | 61.1 | 12 | 9.5 | 18 | 14.3 | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | [74] |
| ASIA | ||||||||||||||||||
| Saudi Arabia | 45 | 21 | 46.7 | 8 | 17.8 | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | [75] |
| Saudi Arabia | 15 | 6 | 40 | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | [76] |
| Kuwait | 68 | 45 | 66.1 | 3 | 4.4 | 6 | 8.8 | n.r. | n.r. | 4 | 5.8 | n.r. | n.r. | n.r. | n.r. | 2 | 2.9 | [77] |
| China | 1051 | 740 | 70.4 | 87 | 8.2 | 201 | 19.1 | 58 | 5.5 | 102 | 9.7 | n.r. | n.r. | 30 | 2.8 | 15 | 1.4 | [78] |
| China | 67 | 23 | 41.8 | 5 | 7.5 | 23 | 34.3 | 6 | 8.9 | 13 | 19.4 | n.r. | n.r. | 4 | 5.9 | n.r. | n.r. | [79] |
| China | 613 | 360 | 60.2 | 59 | 9.6 | 143 | 23.3 | 52 | 8.4 | 70 | 11.4 | n.r. | n.r. | 21 | 3.4 | n.r. | n.r. | [80] |
| China | 662 | n.r | n.r. | n.r | n.r. | 115 | 18.5 | n.r. | n.r. | 60 | 9 | 20 | 3 | 28 | 4.2 | 6 | 0.9 | [81] |
| China | 1400 | 752 | 53.7 | 92 | 6.5 | 197 | 14 | 45 | 3.2 | 124 | 8.8 | n.r. | n.r. | 22 | 1.5 | 6 | 0.4 | [82] |
| China | 401 | 238 | 59.3 | n.r. | n.r | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | [83] |
| East China | 229 | 153 | 66.8 | 22 | 9.6 | 51 | 22.2 | 23 | 10 | 26 | 11.3 | 18 | 7.8 | n.r. | n.r. | 1 | 0.4 | [84] |
| China | 100 | 69 | 69 | 9 | 9 | 22 | 22 | 6 | 6 | 13 | 13 | n.r. | 2 | 2 | 1 | 1 | [85] | |
| Korea | 218 | 147 | 67.4 | 31 | 14.2 | 40 | 18.3 | n.r. | n.r. | 18 | 8.2 | 9 | 4.1 | 11 | 5 | 1 | 0.4 | [86] |
| Korea | 507 | 319 | 65.3 | 65 | 13.3 | 104 | 21.3 | n.r. | n.r. | 60 | 12.3 | 15 | 3.1 | 22 | 4.5 | 6 | 1.2 | [87] |
| Japan | 442 | 270 | 61 | 38 | 9 | n.r. | n.r. | 34 | 7.6 | 69 | 15.6 | n.r. | n.r. | 25 | 5.4 | n.r. | n.r. | [88] |
| Japan | 1497 | 901 | 61 | 188 | 13 | 371 | 24.7 | 116 | 7.7 | 189 | 12.4 | n.r. | n.r. | 61 | 4 | 8 | 0.53 | [89] |
| Iran | 314 | 251 | 79.9 | 17 | 5.4 | 43 | 16.6 | n.r. | n.r. | 24 | 7.6 | 15 | 4.7 | 3 | 0.9 | 1 | 0.3 | [90] |
| Singapore | 145 | 95 | 65.5 | 14 | 9.6 | 31 | 21.3 | n.r. | n.r. | 18 | 12.4 | 8 | 5.5 | 4 | 2.7 | 1 | 0.6 | [91] |
| Malaysia | 35 | 27 | 77.1 | 2 | 5.7 | n.r. | n.r. | 2 | 5.7 | 3 | 8.5 | n.r. | n.r. | n.r. | n.r. | 2 | 5.7 | [92] |
| India | 88 | 65 | 73.8 | n.r | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | [93] |
| India | 1660 | 1090 | 65.6 | 98 | 5.9 | 61 | 3.6 | 25 | 1.5 | 34 | 2 | n.r. | n.r. | 5 | 0.3 | 4 | 0.2 | [94] |
| India | 606 | 492 | 81.2 | 33 | 5.4 | 70 | 11.6 | n.r. | n.r. | 40 | 6.6 | 17 | 2.8 | 12 | 1.98 | 1 | 0.16 | [95] |
| India | 83 | 60 | 72.2 | 5 | 6.5 | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | [96] |
| India | 317 | 285 | 89.9 | 26 | 8.2 | n.r. | n.r. | 317 | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | [97] |
| India | 961 | 642 | 66.8 | 55 | 5.7 | 101 | 10.5 | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | [98] |
| Southern India | 510 | 342 | 67.1 | 30 | 5.9 | 10 | 1.9 | 5 | 0.9 | 4 | 0.7 | n.r. | n.r. | n.r. | n.r. | 1 | 0.19 | [99] |
| Sri Lanka | 50 | 40 | 80 | 4 | 8 | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | [100] |
| North Africa | ||||||||||||||||||
| Algeria | 68 | 36 | 52.9 | 2 | 2.9 | n.r. | n.r. | n.r. | n.r. | 4 | 5.8 | 2 | 2.9 | 2 | 2.9 | 3 | 4.4 | [71] |
| Egypt | 152 | 78 | 51.3 | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | [101] |
| Egypt | 41 | 22 | 53.6 | 2 | 4.8 | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | [102] |
| Egypt | 100 | 55 | 55 | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | [103] |
| Morocco | 72 | 37 | 51.3 | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | [104] |
| South Africa | ||||||||||||||||||
| South Africa | 128 | 54 | 42.1 | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | [105] |
| South Africa | 261 | 90 | 34.4 | 34 | 13 | 3 | 1.1 | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | [106] |
| North America | ||||||||||||||||||
| Canada | 573 | 366 | 63.8 | 64 | 11 | 143 | 25 | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | [107] |
| USA and Canada | 436 | 256 | 79 | 23 | 7.1 | 45 | 13.9 | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | [108] |
| USA | 1014 | 460 | 45.3 | 112 | 11 | 442 | 43.5 | 113 | 10.1 | 256 | 25.2 | n.r. | 50 | 4.9 | 15 | 1.4 | [65] | |
| USA | 68 | 45 | 66.1 | 4 | 5.8 | 12 | 17.6 | n.r. | n.r. | 9 | 13.2 | 2 | 2.9 | n.r. | n.r. | 3 | 2.9 | [109] |
| Central-South America | ||||||||||||||||||
| Argentina | 81 * | 40 | 49.3 | 8 | 9.8 | 2 | 2.4 | n.r. | n.r. | 2 | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | [110] | |
| Colombia | 69 | 40 | 58.8 | 10 | 4.5 | 11 | 15.9 | n.r. | n.r. | 8 | 11.6 | n.r. | n.r. | 3 | 4.3 | n.r. | n.r. | [111] |
| Puerto Rico | 141 | 56 | 66.7 | 2 | 2.4 | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | [112] |
| World Macro-Areas | Suspected Dystrophinopathies | DMD/BMD Patients | Detection Rate | Large Deletions | Large Duplications | Point Mutations/Small Rearrangements | |||
|---|---|---|---|---|---|---|---|---|---|
| N | N | N | % | N | % | N | % | ||
| Western Europe | 8922 | 7670 | 86.0 | 5011 | 65.3 | 991 | 12.9 | 1668 | 21.7 |
| East Europe | 826 | 620 | 75.1 | 367 | 59.2 | 75 | 12.1 | 178 | 28.7 |
| Asia | 12084 | 9913 | 82.0 | 7433 | 74.9 | 891 | 9.0 | 1589 | 16.0 |
| North Africa | 433 | 236 | 54.5 | 228 | 96.6 | 4 | 0.02 | 4 | 0.02 |
| South Africa | 389 | 178 | 45.8 | 144 | 80.9 | 34 | 19.1 | n.a. | n.a. |
| North America | 2091 | 1972 | 94.3 | 1127 | 57.2 | 203 | 10.3 | 642 | 32.5 |
| Central-South America | 210 | 169 | 80.4 | 136 | 80.5 | 20 | 11.8 | 13 | 7.7 |
| Total or percentages | 25.7 | 20.7 | 80.8 | 14.4 | 69.6 | 2218 | 10.7 | 4094 | 19.7 |
| This report | 750 | 719 | 95.8 | 534 | 71.2 | 73 | 10.1 | 112 | 15.6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Viggiano, E.; Picillo, E.; Passamano, L.; Onore, M.E.; Piluso, G.; Scutifero, M.; Torella, A.; Nigro, V.; Politano, L. Spectrum of Genetic Variants in the Dystrophin Gene: A Single Centre Retrospective Analysis of 750 Duchenne and Becker Patients from Southern Italy. Genes 2023, 14, 214. https://doi.org/10.3390/genes14010214
Viggiano E, Picillo E, Passamano L, Onore ME, Piluso G, Scutifero M, Torella A, Nigro V, Politano L. Spectrum of Genetic Variants in the Dystrophin Gene: A Single Centre Retrospective Analysis of 750 Duchenne and Becker Patients from Southern Italy. Genes. 2023; 14(1):214. https://doi.org/10.3390/genes14010214
Chicago/Turabian StyleViggiano, Emanuela, Esther Picillo, Luigia Passamano, Maria Elena Onore, Giulio Piluso, Marianna Scutifero, Annalaura Torella, Vincenzo Nigro, and Luisa Politano. 2023. "Spectrum of Genetic Variants in the Dystrophin Gene: A Single Centre Retrospective Analysis of 750 Duchenne and Becker Patients from Southern Italy" Genes 14, no. 1: 214. https://doi.org/10.3390/genes14010214