

Phenotype-Based Genetic Analysis Reveals Missing Heritability of KIF11-Related Retinopathy: Clinical and Genetic Findings

Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Targeted Exon Sequencing (TES) and Whole Genome Sequencing (WGS)
2.3. Bioinformatics Analysis
2.4. Ultra-Deep Sequencing
2.5. Minigene Assay in HEK293T Cells
3. Results
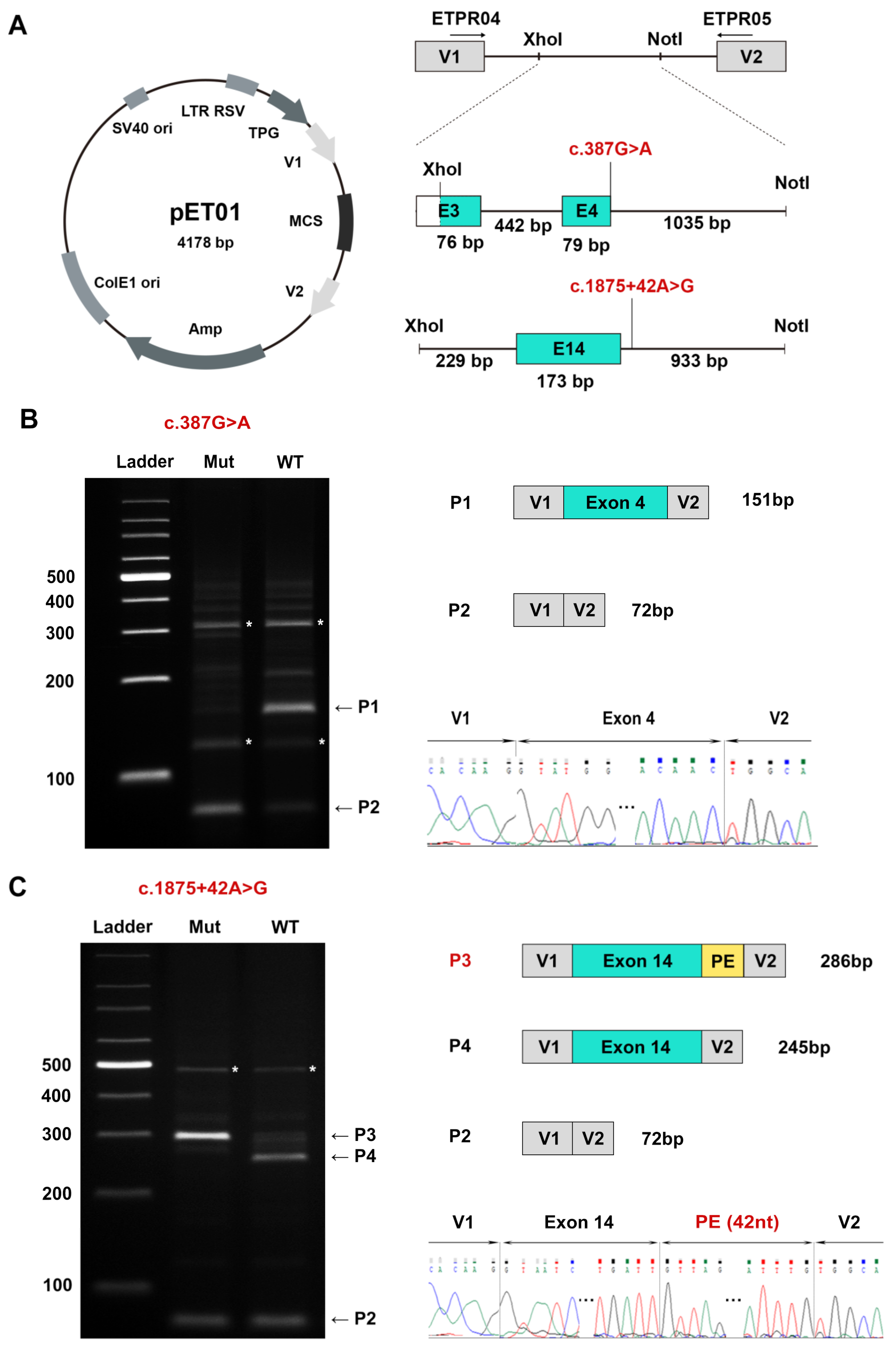
3.1. KIF11 Variants Detected
3.2. A Novel DIV and a Novel Synonymous Variant Validated by Minigene Assays
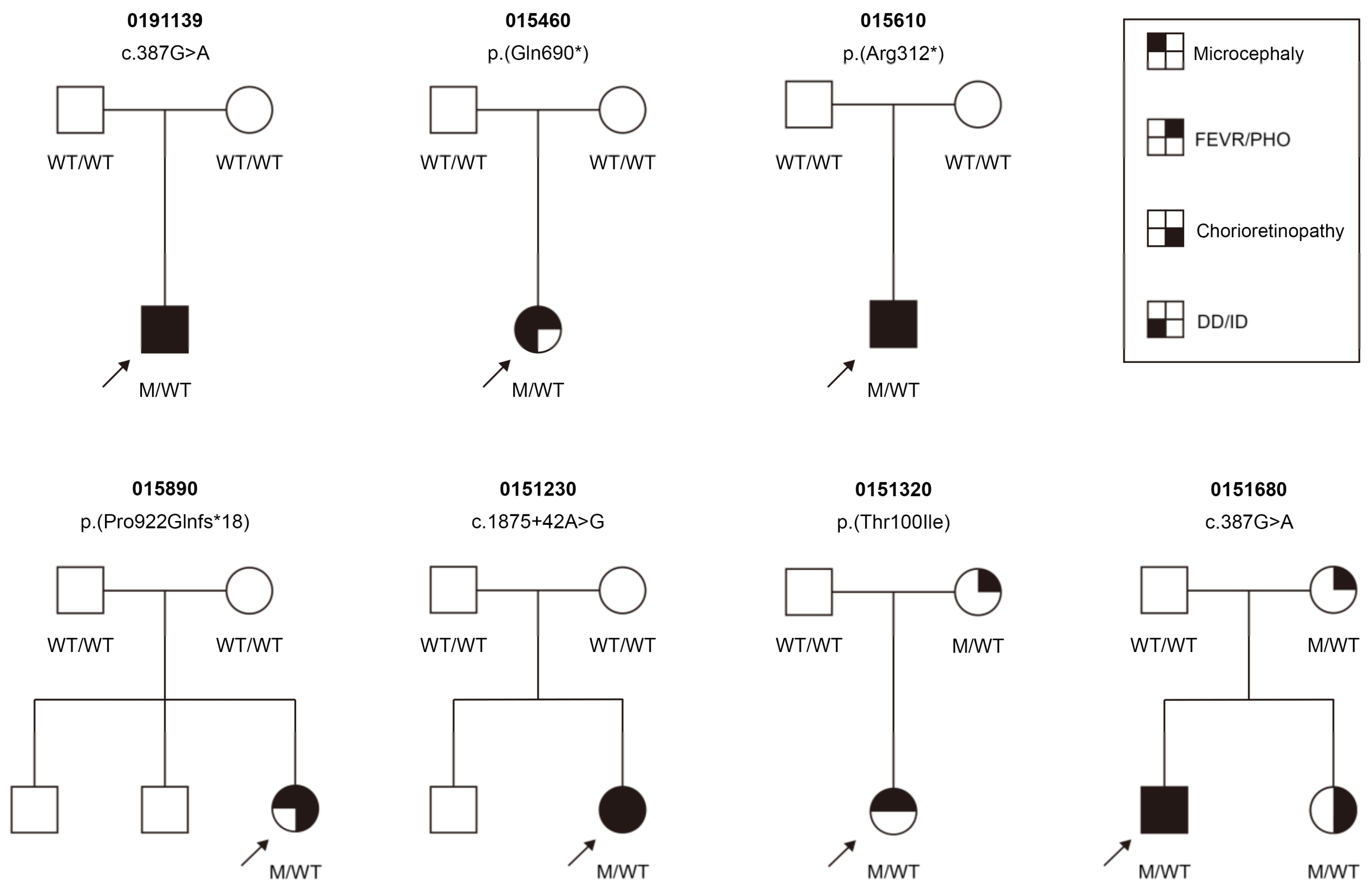
3.3. Clinical Findings
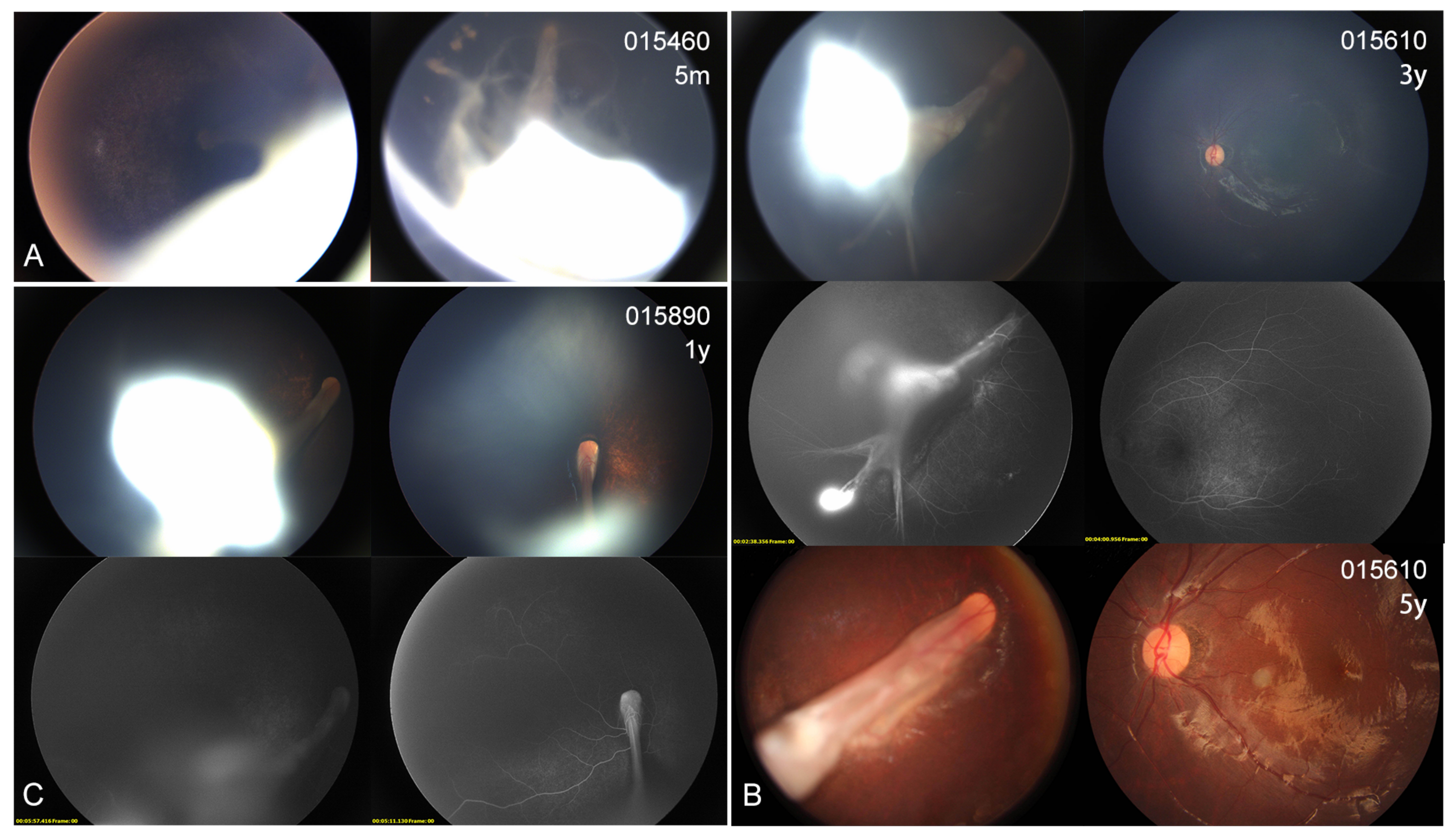
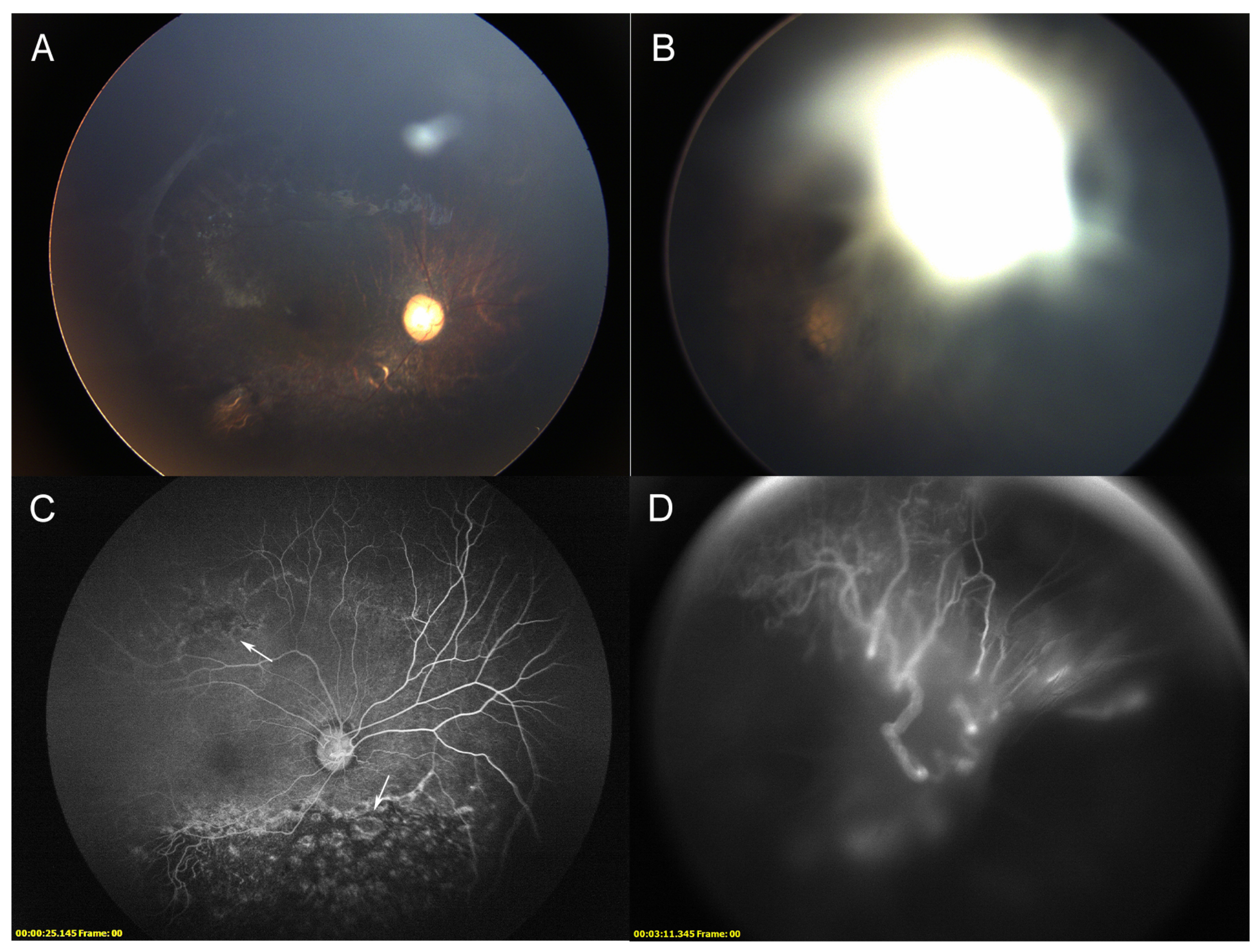
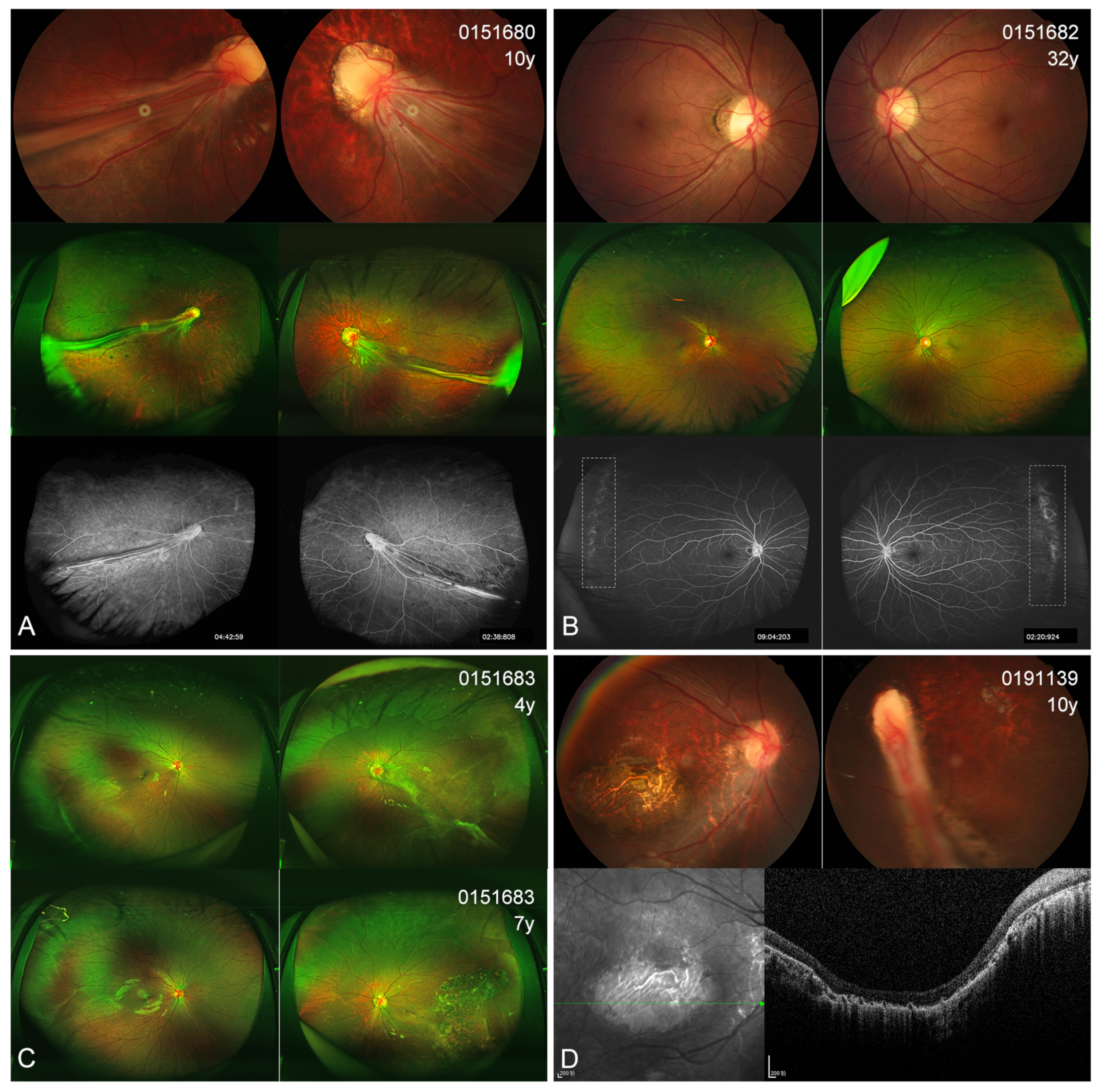
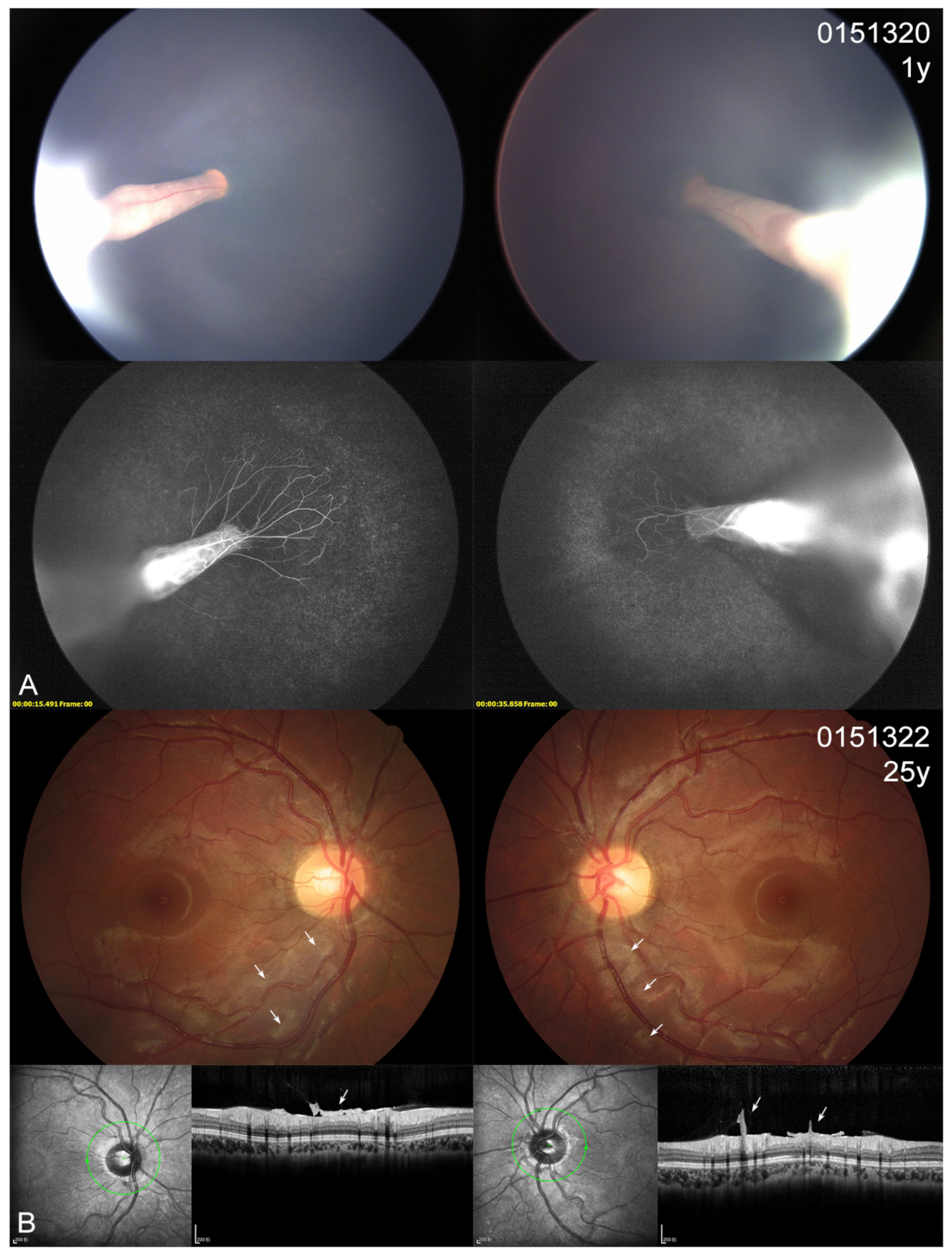
3.4. Ocular Phenotype
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Valentine, M.T.; Fordyce, P.M.; Krzysiak, T.C.; Gilbert, S.P.; Block, S.M. Individual dimers of the mitotic kinesin motor Eg5 step processively and support substantial loads in vitro. Nat. Cell Biol. 2006, 8, 470–476. [Google Scholar] [CrossRef] [Green Version]
- Exertier, P.; Javerzat, S.; Wang, B.; Franco, M.; Herbert, J.; Platonova, N.; Winandy, M.; Pujol, N.; Nivelles, O.; Ormenese, S.; et al. Impaired angiogenesis and tumor development by inhibition of the mitotic kinesin Eg5. Oncotarget 2013, 4, 2302–2316. [Google Scholar] [CrossRef] [Green Version]
- Ostergaard, P.; Simpson, M.A.; Mendola, A.; Vasudevan, P.; Connell, F.C.; van Impel, A.; Moore, A.T.; Loeys, B.L.; Ghalamkarpour, A.; Onoufriadis, A.; et al. Mutations in KIF11 cause autosomal-dominant microcephaly variably associated with congenital lymphedema and chorioretinopathy. Am. J. Hum. Genet. 2012, 90, 356–362. [Google Scholar] [CrossRef] [Green Version]
- Mirzaa, G.M.; Enyedi, L.; Parsons, G.; Collins, S.; Medne, L.; Adams, C.; Ward, T.; Davitt, B.; Bicknese, A.; Zackai, E.; et al. Congenital microcephaly and chorioretinopathy due to de novo heterozygous KIF11 mutations: Five novel mutations and review of the literature. Am. J. Med. Genet. Part A 2014, 164, 2879–2886. [Google Scholar] [CrossRef] [Green Version]
- Jones, G.E.; Ostergaard, P.; Moore, A.T.; Connell, F.C.; Williams, D.; Quarrell, O.; Brady, A.F.; Spier, I.; Hazan, F.; Moldovan, O.; et al. Microcephaly with or without chorioretinopathy, lymphoedema, or mental retardation (MCLMR): Review of phenotype associated with KIF11 mutations. Eur. J. Hum. Genet. 2014, 22, 881–887. [Google Scholar] [CrossRef]
- Robitaille, J.M.; Gillett, R.M.; LeBlanc, M.A.; Gaston, D.; Nightingale, M.; Mackley, M.P.; Parkash, S.; Hathaway, J.; Thomas, A.; Ells, A.; et al. Phenotypic overlap between familial exudative vitreoretinopathy and microcephaly, lymphedema, and chorioretinal dysplasia caused by KIF11 mutations. JAMA Ophthalmol. 2014, 132, 1393–1399. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Xiao, X.; Li, S.; Jia, X.; Guo, X.; Zhang, Q. KIF11 mutations are a common cause of autosomal dominant familial exudative vitreoretinopathy. Br. J. Ophthalmol. 2016, 100, 278–283. [Google Scholar] [CrossRef] [Green Version]
- Kashani, A.H.; Brown, K.T.; Chang, E.; Drenser, K.A.; Capone, A.; Trese, M.T. Diversity of retinal vascular anomalies in patients with familial exudative vitreoretinopathy. Ophthalmology 2014, 121, 2220–2227. [Google Scholar] [CrossRef]
- Balikova, I.; Robson, A.G.; Holder, G.E.; Ostergaard, P.; Mansour, S.; Moore, A.T. Ocular manifestations of microcephaly with or without chorioretinopathy, lymphedema or intellectual disability (MCLID) syndrome associated with mutations in KIF11. Acta Ophthalmol. 2016, 94, 92–98. [Google Scholar] [CrossRef]
- Li, J.K.; Fei, P.; Li, Y.; Huang, Q.J.; Zhang, Q.; Zhang, X.; Rao, Y.Q.; Li, J.; Zhao, P. Identification of novel KIF11 mutations in patients with familial exudative vitreoretinopathy and a phenotypic analysis. Sci. Rep. 2016, 6, 26564. [Google Scholar] [CrossRef]
- Hull, S.; Arno, G.; Ostergaard, P.; Pontikos, N.; Robson, A.G.; Webster, A.R.; Hogg, C.R.; Wright, G.A.; Henderson, R.H.H.; Martin, C.A.; et al. Clinical and Molecular Characterization of Familial Exudative Vitreoretinopathy Associated With Microcephaly. Am. J. Ophthalmol. 2019, 207, 87–98. [Google Scholar] [CrossRef]
- Schlogel, M.J.; Mendola, A.; Fastre, E.; Vasudevan, P.; Devriendt, K.; de Ravel, T.J.; Van Esch, H.; Casteels, I.; Arroyo Carrera, I.; Cristofoli, F.; et al. No evidence of locus heterogeneity in familial microcephaly with or without chorioretinopathy, lymphedema, or mental retardation syndrome. Orphanet. J. Rare Dis. 2015, 10, 52. [Google Scholar] [CrossRef] [Green Version]
- Birtel, J.; Gliem, M.; Mangold, E.; Tebbe, L.; Spier, I.; Muller, P.L.; Holz, F.G.; Neuhaus, C.; Wolfrum, U.; Bolz, H.J.; et al. Novel Insights Into the Phenotypical Spectrum of KIF11-Associated Retinopathy, Including a New Form of Retinal Ciliopathy. Invest. Ophthalmol. Vis. Sci. 2017, 58, 3950–3959. [Google Scholar] [CrossRef] [Green Version]
- Shurygina, M.F.; Simonett, J.M.; Parker, M.A.; Mitchell, A.; Grigorian, F.; Lifton, J.; Nagiel, A.; Shpak, A.A.; Dadali, E.L.; Mishina, I.A.; et al. Genotype Phenotype Correlation and Variability in Microcephaly Associated With Chorioretinopathy or Familial Exudative Vitreoretinopathy. Invest. Ophthalmol. Vis. Sci. 2020, 61, 2. [Google Scholar] [CrossRef]
- Qin, L.; Wang, J.; Tian, X.; Yu, H.; Truong, C.; Mitchell, J.J.; Wierenga, K.J.; Craigen, W.J.; Zhang, V.W.; Wong, L.C. Detection and Quantification of Mosaic Mutations in Disease Genes by Next-Generation Sequencing. J. Mol. Diagn. 2016, 18, 446–453. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Li, J.; Zhang, X.; Li, J.K.; Chen, H.J.; Zhao, P.Q.; Fei, P. Five novel copy number variations detected in patients with familial exudative vitreoretinopathy. Mol. Vis. 2021, 27, 632–642. [Google Scholar]
- Kondo, H.; Matsushita, I.; Nagata, T.; Fujihara, E.; Hosono, K.; Uchio, E.; Hotta, Y.; Kusaka, S. Retinal Features of Family Members With Familial Exudative Vitreoretinopathy Caused By Mutations in KIF11 Gene. Transl. Vis. Sci. Technol. 2021, 10, 18. [Google Scholar] [CrossRef]
- Sun, T.; Xu, K.; Ren, Y.; Xie, Y.; Zhang, X.; Tian, L.; Li, Y. Comprehensive Molecular Screening in Chinese Usher Syndrome Patients. Invest. Ophthalmol. Vis. Sci. 2018, 59, 1229–1237. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Xie, Y.; Xu, K.; Chang, H.; Zhang, X.; Li, Y. Comprehensive Genetic Analysis Unraveled the Missing Heritability in a Chinese Cohort With Wolfram Syndrome 1: Clinical and Genetic Findings. Invest. Ophthalmol. Vis. Sci. 2022, 63, 9. [Google Scholar] [CrossRef]
- Doan, R.N.; Miller, M.B.; Kim, S.N.; Rodin, R.E.; Ganz, J.; Bizzotto, S.; Morillo, K.S.; Huang, A.Y.; Digumarthy, R.; Zemmel, Z.; et al. MIPP-Seq: Ultra-sensitive rapid detection and validation of low-frequency mosaic mutations. BMC Med. Genom. 2021, 14, 47. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, Z.; Huang, L.; Sun, L.; Li, S.; Zhang, T.; Ding, X. Update on the Phenotypic and Genotypic Spectrum of KIF11-Related Retinopathy. Genes 2022, 13, 713. [Google Scholar] [CrossRef]
- Guo, Z.; Huo, X.; Wu, D.; Hao, B.; Liao, S. A Novel Variant of the KIF11 Gene, c.2922G>T, Is Associated with Microcephaly by Affecting RNA Splicing. Dev. Neurosci. 2022, 44, 113–120. [Google Scholar] [CrossRef]
- Tian, L.; Chen, C.; Song, Y.; Zhang, X.; Xu, K.; Xie, Y.; Jin, Z.B.; Li, Y. Phenotype-Based Genetic Analysis Reveals Missing Heritability of ABCA4-Related Retinopathy: Deep Intronic Variants and Copy Number Variations. Invest. Ophthalmol. Vis. Sci. 2022, 63, 5. [Google Scholar] [CrossRef]
- Yonekawa, Y.; Thomas, B.J.; Drenser, K.A.; Trese, M.T.; Capone, A., Jr. Familial Exudative Vitreoretinopathy: Spectral-Domain Optical Coherence Tomography of the Vitreoretinal Interface, Retina, and Choroid. Ophthalmology 2015, 122, 2270–2277. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Exon/ Intron | Nucleotide Change | Predicted Protein Effect # | Allele Number | Type | MT | PP2 | SIFT | HSF | MES | SpliceAI | MAF * | ACMG | Source |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 3 | c.299C>T | p.(Thr100Ile) | 1 | Missense | DC | D | D | - | - | - | - | LP | Novel |
| 4 | c.387G>A | p.(=) | 2 | Splicing | DC | - | - | SC | SC | SC | - | P | Novel |
| 8 | c.934C>T | p.(Arg312*) | 1 | Nonsense | DC | - | - | - | - | - | - | P | Novel |
| IVS14 | c.1875+42A>G | p.(Asn626Valfs*14) | 1 | Splicing | DC | - | - | SC | SC | SC | - | P | Novel |
| 16 | c.2068C>T | p.(Gln690*) | 1 | Nonsense | DC | - | - | - | - | - | - | P | Novel |
| 19 | c.2765del | p.(Pro922Glnfs*18) | 1 | Frameshift | DC | - | - | - | - | - | - | P | Novel |
| Genome Position * | Ref | Alt | Family ID | Family Member | Ref/Alt Reads | AAF | Depth # |
|---|---|---|---|---|---|---|---|
| chr10:94397210 | C | T | 015460 | proband | 98,965/96,841 | 0.495 | 226458.81 |
| 015461 | father | 184,385/0 | 0.000 | 213769.63 | |||
| 015462 | mother | 152,119/0 | 0.000 | 176279.86 | |||
| chr10:94373278 | C | T | 015610 | proband | 202,644/203,822 | 0.501 | 318693.21 |
| 015611 | father | 350,057/0 | 0.000 | 274592.15 | |||
| 015612 | mother | 354,036/0 | 0.000 | 277840.26 | |||
| chr10:94408183-94408184 | TC | T | 015890 | proband | 10,244/8895 | 0.465 | 16783.04 |
| 015891 | father | 18,053/0 | 0.000 | 15957.56 | |||
| 015892 | mother | 21,111/0 | 0.000 | 17986.41 | |||
| chr10:94393378 | A | G | 0151230 | proband | 15,908/15,686 | 0.494 | 26361.03 |
| 0151231 | father | 15,717/33 | 0.002 | 21791.67 | |||
| 0151232 | mother | 32,161/43 | 0.001 | 27411.65 | |||
| chr10:94366994 | G | A | 0191139 | proband | 17,506/18,402 | 0.512 | 23877.27 |
| 0191139-1 | father | 43,078/79 | 0.002 | 30039.17 | |||
| 0191139-2 | mother | 48,655/99 | 0.002 | 32621.80 |
| Patient ID | Gender | Exam Age (Year) | BCVA (OD/OS) | Nyst- Agmus | Ocular Manifestations (OD/OS or OU) | Micro- Cephaly | Lymph- Edema | DD/ID | CFP | Variants | Co- Segregation |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 0191139 | M | 10 | NA | + | dragged-disc, chorioretinal atrophy/esotropia, RF | + | - | + | + | c.387G>A, p.(=) | De novo |
| 015460 | F | <1 | NA | + | cataract, tractional RD | + | + | + | + | c.2068C>T, p.(Gln690*) | De novo |
| 015610 | M | 3 | HM/0.1 | + | cataract, RF, tractional RD, chorioretinal dysplasia/peripheral avascular zones | + | - | + | + | c.934C>T, p.(Arg312*) | De novo |
| 015890 | F | 3 | NA | + | RFs and tractional RD, chorioretinal dysplasia | + | - | - | + | c.2765del, p.(Pro922Glnfs*18) | De novo |
| 0151230 | F | 2 | NA | + | chorioretinal dysplasia, peripheral avascular zones/microcornea, cataract, tractional RD | + | - | + | + | c.1875+42A>G, p.(Asn626Valfs*14) | De novo |
| 0151320 | F | 1 | NA | + | cataract, RFs, tractional RD | + | - | - | + | c.299C>T, p.(Thr100Ile) | Maternal |
| 0151322 | F | 25 | 1.0/1.0 | - | tortuous vessels, posterior hyaloidal organization | - | - | - | + | c.299C>T, p.(Thr100Ile) | |
| 0151680 | M | 10 | 0.05/0.05 | + | RFs, increase or straightening of peripheral vessels, chorioretinal dysplasia | + | - | + | + | c.387G>A, p.(=) | Maternal |
| 0151682 | F | 32 | 1.0/0.8 | - | increase or straightening of peripheral vessels | - | - | - | + | c.387G>A, p.(=) | |
| 0151683 | F | 4 | 0.8/0.05 | - | increase or straightening of peripheral vessels/ERM, tractional RD, chorioretinal dysplasia | - | - | - | + | c.387G>A, p.(=) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, H.; Zhang, X.; Xu, K.; Li, N.; Xie, Y.; Yan, W.; Li, Y. Phenotype-Based Genetic Analysis Reveals Missing Heritability of KIF11-Related Retinopathy: Clinical and Genetic Findings. Genes 2023, 14, 212. https://doi.org/10.3390/genes14010212
Chang H, Zhang X, Xu K, Li N, Xie Y, Yan W, Li Y. Phenotype-Based Genetic Analysis Reveals Missing Heritability of KIF11-Related Retinopathy: Clinical and Genetic Findings. Genes. 2023; 14(1):212. https://doi.org/10.3390/genes14010212
Chicago/Turabian StyleChang, Haoyu, Xin Zhang, Ke Xu, Nien Li, Yue Xie, Weiyu Yan, and Yang Li. 2023. "Phenotype-Based Genetic Analysis Reveals Missing Heritability of KIF11-Related Retinopathy: Clinical and Genetic Findings" Genes 14, no. 1: 212. https://doi.org/10.3390/genes14010212