
Nuclear Receptor Subfamily 2 Group E Member 3 (NR2E3): Role in Retinal Development and Disease


Abstract
:1. Introduction
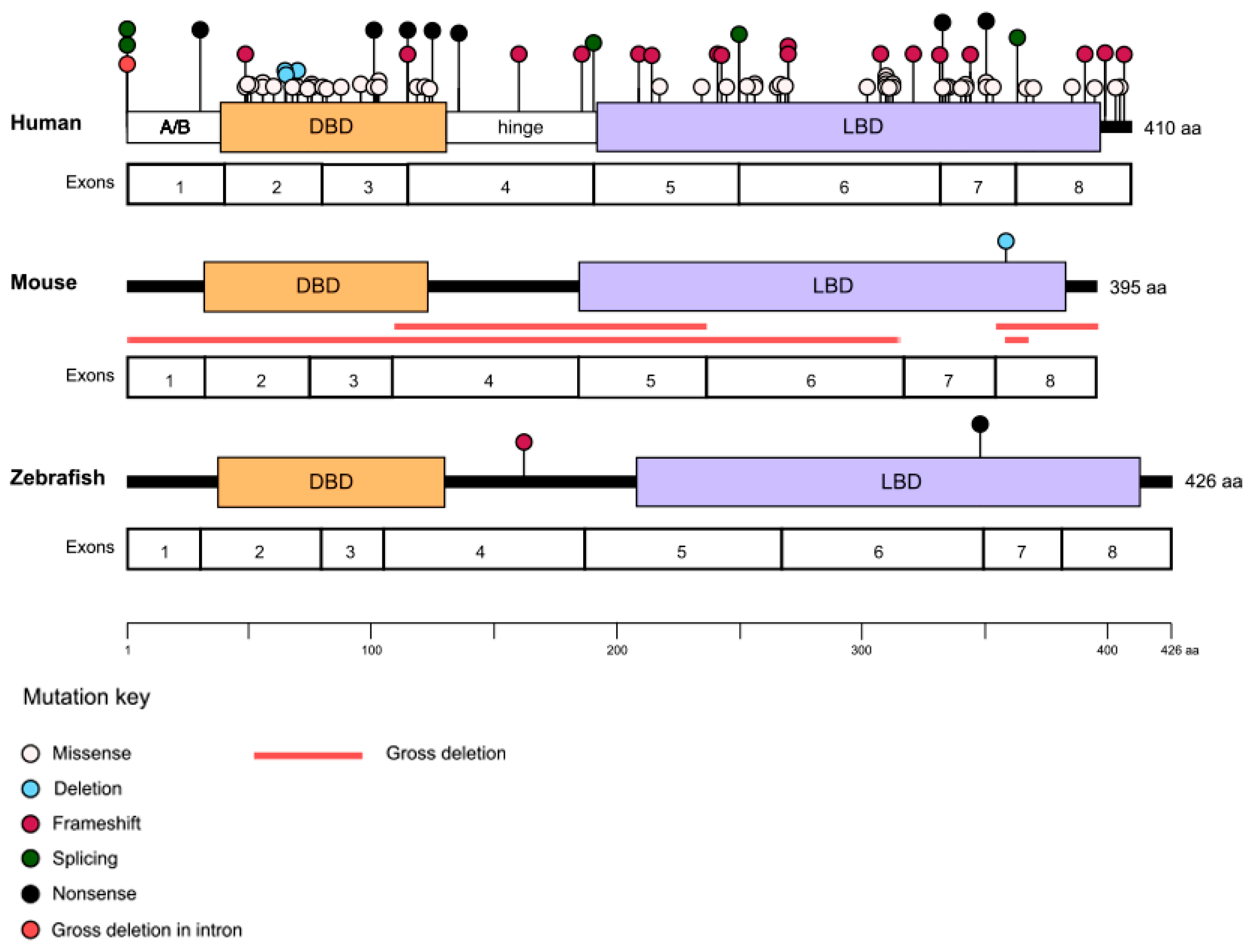
2. NR2E3 Structure
3. NR2E3 Function
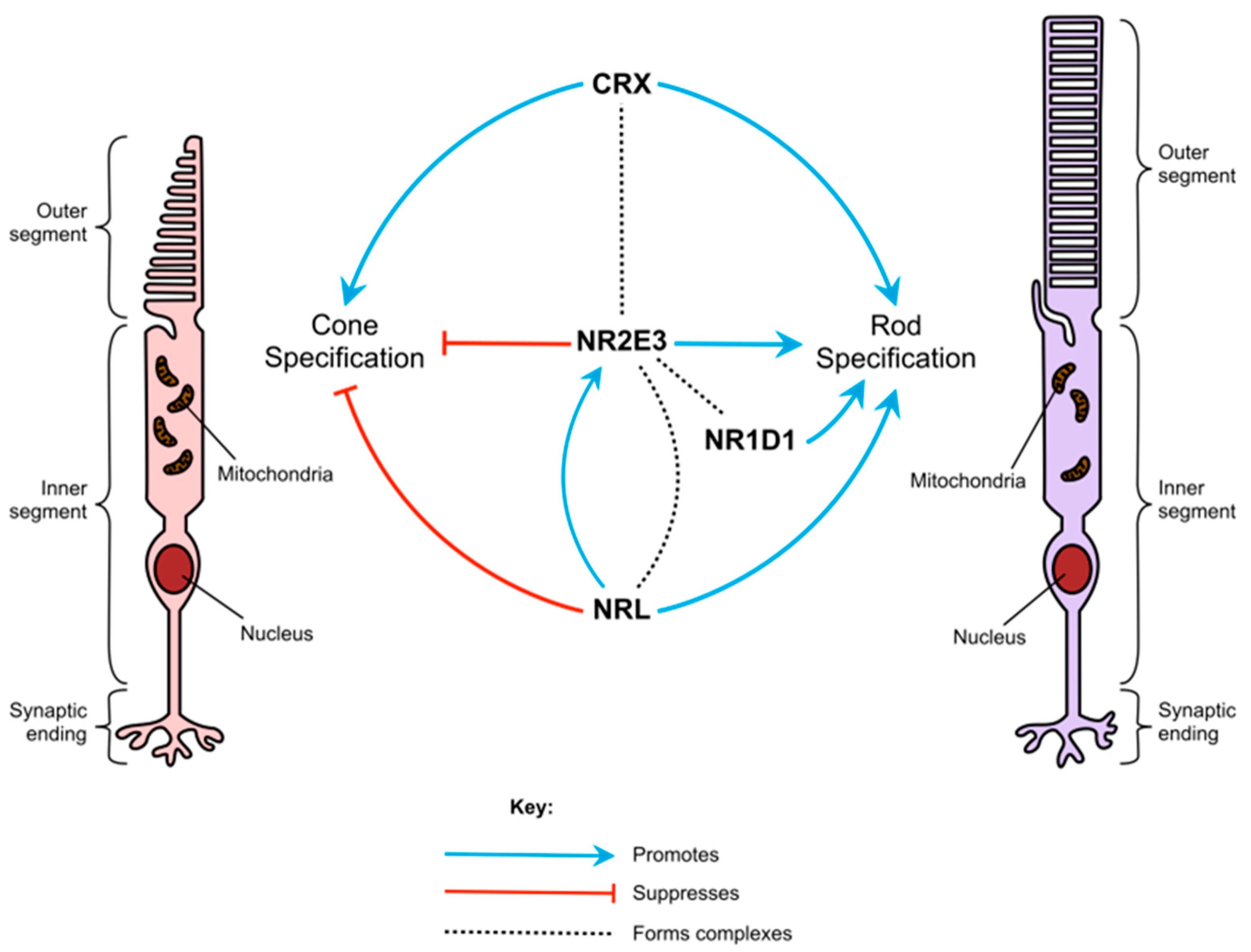
3.1. Rod and Cone Photoreceptor Differentiation
3.2. Role in the Adult Retina and Other Tissues
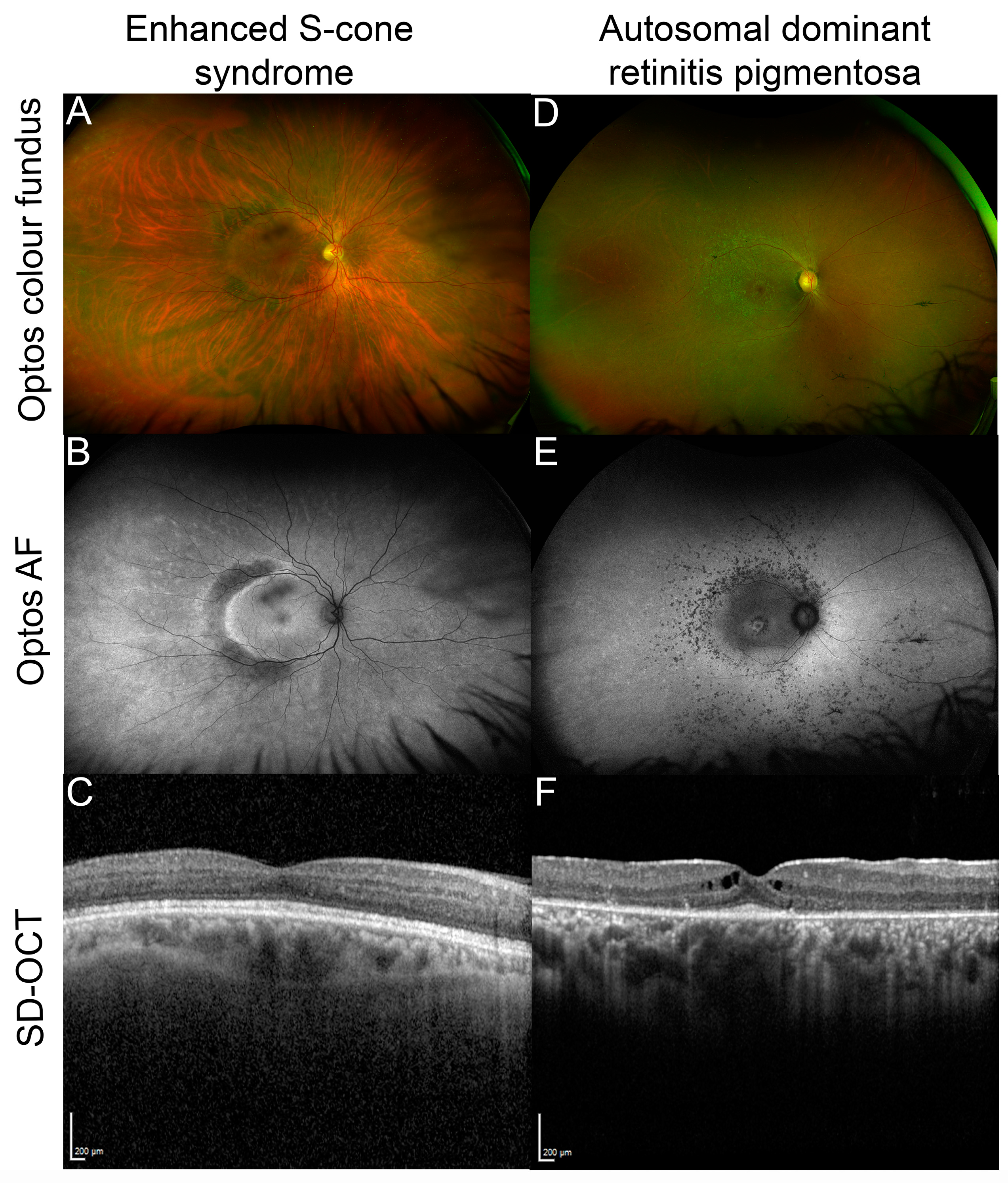
4. Clinical Phenotype

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Region | Variant | Mutation Type | Amino Acid Change | Protein Domain | Reported Phenotypes | References |
|---|---|---|---|---|---|---|
| Intron 1 | c.119-2A>C | Splicing | ESCS, GFS, CPRD, RP | [30,33,41] | ||
| c.119-3C>G | Splicing | ESCS | [46] | |||
| c.119-57_166 | Gross deletion | RP | [47] | |||
| Exon 1 | c.95G>A | Nonsense | p.Trp32 * | A/B | RP | [48] |
| Exon 2 | c.142C>T | Missense | p.Arg48Cys | DBD | ESCS | [40] |
| c.143_144delGCins25 | Indel/ Frameshift | p.Arg48Glufs*66 | DBD | RP | [49] | |
| c.145G>A | Missense | p.Val49Met | DBD | ESCS | [46] | |
| c.151G>A | Missense | p.Gly51Arg | DBD | ESCS | [40] | |
| c.166G>A | Missense | p.Gly56Arg | DBD | RP | [44] | |
| c.166G>C | Missense | p.Gly56Arg | DBD | RP | [50] | |
| c.188C>A | Missense | p.Ala63Asp | DBD | Cone–rod dystrophy | [51] | |
| c.194_202del | Deletion | p.Asn65_Cys67del | DBD | RP, ESCS | [30,52] | |
| c.196_201delGGCTGC | Deletion | p.Gly66_Cys67del | DBD | ESCS | [53] | |
| c.202A>G | Missense | p.Ser68Gly | DBD | ESCS | [54] | |
| c.211T>C | Missense | p.Phe71Leu | DBD | ESCS | [31] | |
| c.211_213delTTC | Deletion | p.Phe71del | DBD | ESCS | [42] | |
| c.223G>A | Missense | p.Val75Ile | DBD | RP | [55] | |
| c.226C>T | Missense | p.Arg76Trp | DBD | ESCS | [30] | |
| c.227G>A | Missense | p.Arg76Gln | DBD | ESCS | [30] | |
| c.242A>G | Missense | p.Tyr81Cys | DBD | ESCS | [46] | |
| Exon 3 | c.248G>A | Missense | p.Cys83Tyr | DBD | ESCS | [56] |
| c.263G>T | Missense | p.Gly88Val | DBD | ESCS | [57] | |
| c.290G>A | Missense | p.Arg97His | DBD | ESCS, RD | [30,58] | |
| c.305C>A | Missense | p.Ala102Asp | DBD | ESCS, RP, RD | [59,60,61] | |
| c.309C>A | Nonsense | p.Cys103 * | DBD | RP | [62] | |
| c.310C>T | Missense | p.Arg104Trp | DBD | ESCS | [30] | |
| c.311G>A | Missense | p.Arg104Gln | DBD | ESCS, RP, RD | [63,64,65] | |
| c.328C>T | Nonsense | p.Gln110 * | DBD | RD, RP | [16,66] | |
| c.328dupC | Insertion/ Frameshift | p.Gln110Profs*31 | DBD | GFS | [67] | |
| Exon 4 | c.352G>A | Missense | p.Val118Met | DBD | RP | [68] |
| c.364C>T | Missense | p.Arg122Cys | DBD | RP, RD | [16,69] | |
| c.371C>T | Missense | p.Pro124Leu | DBD | RP | [70] | |
| c.373C>T | Nonsense | p.Arg125 * | DBD | ESCS, RP | [71,72] | |
| c.406G>T | Nonsense | p.Glu136 * | DBD | RP | [66] | |
| c.481delA | Deletion/ Frameshift | p.Thr161Hisfs*18 | Hinge | RP, ESCS | [57,73] | |
| c.554delA | Deletion/ Frameshift | p.Lys185Serfs*66 | Hinge | RP | [74] | |
| Intron 4 | c.571+2T>C | Splicing | RP | [75] | ||
| Exon 5 | c.626dupA | Insertion/ Frameshift | p.Tyr209 * | LBD | RP | [74] |
| c.639_640insT | Insertion/ Frameshift | p.Pro214Serfs*9 | LBD | RD | [76] | |
| c.646G>A | Missense | p.Gly216Ser | LBD | GFS, RP | [72,77] | |
| c.701G>C | Missense | p.Trp234Ser | LBD | ESCS | [30] | |
| c.724_725delTC | Deletion/ Frameshift | p.Ser242Glnfs*17 | LBD | ESCS, RP, cone–rod dystrophy | [78,79,80] | |
| c.731delT | Deletion/ Frameshift | p.Leu244Argfs*7 | LBD | RP | [81] | |
| c.739C>T | Missense | p.Arg247Trp | LBD | ESCS | [31] | |
| Intron 5 | c.747+1G>C | Splicing | ESCS | [39] | ||
| Exon 6 | c.755T>C | Missense | p.Leu252Pro | LBD | ESCS | [82] |
| c.767C>A | Missense | p.Ala256Glu | LBD | ESCS, RD | [33,65] | |
| c.767C>T | Missense | p.Ala256Val | LBD | ESCS | [83] | |
| c.788T>C | Missense | p.Leu263Pro | LBD | ESCS | [57] | |
| c.790G>A | Missense | p.Gly264Arg | LBD | ESCS | [84] | |
| c.797T>A | Missense | p.Ile266Asn | LBD | RD | [61] | |
| c.808_809delCT | Deletion/ Frameshift | p.Leu270Alafs*70 | LBD | ESCS | [85] | |
| c.827_843del17 | Deletion/ Frameshift | p.Leu270Alafs*70 | LBD | ESCS, GFS, CPRD | [33] | |
| c.908T>C | Missense | p.Leu303Pro | LBD | ESCS | [31] | |
| c.919_920delAT | Deletion/ Frameshift | p.Ile307Leufs*33 | LBD | ESCS | [86] | |
| c.925C>G | Missense | p.Arg309Gly | LBD | ESCS | [30] | |
| c.925C>T | Missense | p.Arg309Trp | LBD | GFS | [87] | |
| c.926G>T | Missense | p.Arg309Leu | LBD | GFS | [72] | |
| c.926G>A | Missense | p.Arg309Gln | LBD | ESCS | [31] | |
| c.930C>G | Missense | p.Phe310Leu | LBD | ESCS | [88] | |
| c.931C>T | Missense | p.Arg311Trp | LBD | RD, RP | [66,89] | |
| c.932G>A | Missense | p.Arg311Gln | LBD | RP, ESCS, GFS, CPRD | [30,33,41,43] | |
| c.951delC | Deletion/ Frameshift | p.Thr318Argfs*6 | LBD | RP | [90] | |
| c.967dupA | Insertion/ Frameshift | p.Met323Asnfs*18 | LBD | RP | [52] | |
| Exon 7 | c.994G>A | Missense | p.Glu332Lys | LBD | ESCS | [59] |
| c.994G>T | Nonsense | p.Glu332 * | LBD | RD | [91] | |
| c.1000C>G | Missense | p.Arg334Gly | LBD | ESCS | [63] | |
| c.1007T>C | Missense | p.Leu336Pro | LBD | ESCS | [57] | |
| c.1018G>A | Missense | p.Glu340Lys | LBD | ESCS | [92] | |
| c.1025T>C | Missense | p.Val342Ala | LBD | ESCS | [59] | |
| c.1025T>G | Missense | p.Val342Gly | LBD | RP | [93] | |
| c.1034_1038delTGCAG | Deletion/ Frameshift | p.Leu345 * | LBD | RP | [41] | |
| c.1048C>G | Missense | p.Gln350Glu | LBD | RD | [76] | |
| c.1048C>T | Nonsense | p.Gln350 * | LBD | ESCS | [94] | |
| c.1049A>G | Missense | p.Gln350Arg | LBD | ESCS, RP | [42,95] | |
| c.1057C>G | Missense | p.Leu353Val | LBD | ESCS | [57] | |
| Intron 7 | c.1101-1G>A | Splicing | ESCS | [46] | ||
| Exon 8 | c.1112T>G | Missense | p.Leu371Trp | LBD | ESCS | [92] |
| c.1118T>C | Missense | p.Leu373Pro | LBD | ESCS, RD | [16,96] | |
| c.1154G>C | Missense | p.Arg385Pro | LBD | ESCS | [30] | |
| c.1171_1172delTT | Deletion/ Frameshift | p.Phe391Profs*15 | LBD | RP | [97] | |
| c.1184T>C | Missense | p.Ile395Thr | LBD | RP | [98] | |
| c.1194delT | Deletion/ Frameshift | p.Pro399Glnfs*79 | ESCS | [59] | ||
| c.1217A>G | Missense | p.Asp406Gly | GFS | [99] | ||
| c.1220T>A | Missense | p.Met407Lys | ESCS | [30] | ||
| c.1223delT | Deletion/ Frameshift | p.Phe408Serfs*7 | RD | [16] |
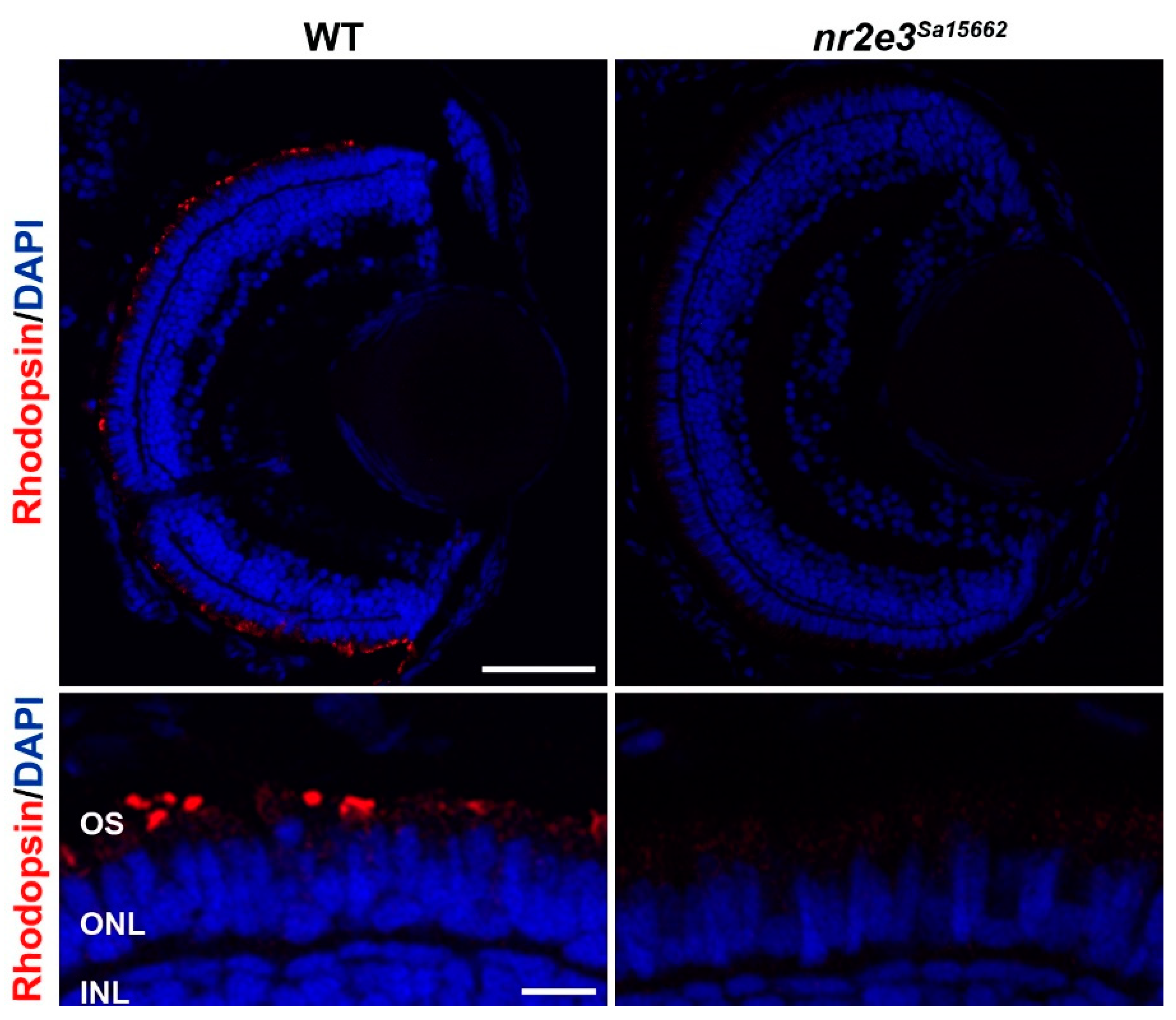
5. Animal Models
6. Treatments
7. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, J.; Rattner, A.; Nathans, J. The rod photoreceptor-specific nuclear receptor Nr2e3 represses transcription of multiple cone-specific genes. J. Neurosci. 2005, 25, 118–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aísa-Marín, I.; López-Iniesta, M.J.; Milla, S.; Lillo, J.; Navarro, G.; de la Villa, P.; Marfany, G. Nr2e3 functional domain ablation by CRISPR-Cas9D10A identifies a new isoform and generates retinitis pigmentosa and enhanced S-cone syndrome models. Neurobiol. Dis. 2020, 146, 105122. [Google Scholar] [CrossRef] [PubMed]
- Greenstein, V.C.; Zaidi, Q.; Hood, D.C.; Spehar, B.; Cideciyan, A.V.; Jacobson, S.G. The Enhanced S Cone Syndrome: An Analysis of Receptoral and Post-receptoral Changes. Vis. Res. 1996, 36, 3711–3722. [Google Scholar] [CrossRef] [Green Version]
- Hood, D.C.; Cideciyan, A.V.; Roman, A.J.; Jacobson, S.G. Enhanced S cone syndrome: Evidence for an abnormally large number of S cones. Vis. Res. 1995, 35, 1473–1481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milam, A.H.; Rose, L.; Cideciyan, A.V.; Barakat, M.R.; Tang, W.-X.; Gupta, N.; Aleman, T.S.; Wright, A.F.; Stone, E.M.; Sheffield, V.C.; et al. The nuclear receptor NR2E3 plays a role in human retinal photoreceptor differentiation and degeneration. Proc. Natl. Acad. Sci. USA 2002, 99, 473–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haider, N.B.; Naggert, J.K.; Nishina, P.M. Excess cone cell proliferation due to lack of a functional NR2E3 causes retinal dysplasia and degeneration in rd7/rd7 mice. Hum. Mol. Genet. 2001, 10, 1619–1626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, M.E.; Zhou, X.E.; Soon, F.F.; Li, X.; Li, J.; Yong, E.L.; Melcher, K.; Xu, H.E. The crystal structure of the orphan nuclear receptor NR2E3/PNR ligand binding domain reveals a dimeric auto-repressed conformation. PLoS ONE 2013, 8, e74359. [Google Scholar] [CrossRef] [Green Version]
- Schorderet, D.F.; Escher, P. NR2E3 mutations in enhanced S-cone sensitivity syndrome (ESCS), Goldmann-Favre syndrome (GFS), clumped pigmentary retinal degeneration (CPRD), and retinitis pigmentosa (RP). Hum. Mutat. 2009, 30, 1475–1485. [Google Scholar] [CrossRef]
- Mollema, N.; Haider, N.B. Focus on Molecules: Nuclear hormone receptor Nr2e3: Impact on retinal development and disease. Exp. Eye Res. 2010, 91, 116–117. [Google Scholar] [CrossRef]
- Von Alpen, D.; Tran, H.V.; Guex, N.; Venturini, G.; Munier, F.L.; Schorderet, D.F.; Haider, N.B.; Escher, P. Differential Dimerization of Variants Linked to Enhanced S-Cone Sensitivity Syndrome (ESCS) Located in the NR2E3 Ligand-Binding Domain. Hum. Mutat. 2015, 36, 599–610. [Google Scholar] [CrossRef] [Green Version]
- Bowmaker, J.K. Evolution of colour vision in vertebrates. Eye 1998, 12, 541–547. [Google Scholar] [CrossRef] [Green Version]
- Mustafi, D.; Engel, A.H.; Palczewski, K. Structure of cone photoreceptors. Prog. Retin. Eye Res. 2009, 28, 289–302. [Google Scholar] [CrossRef] [Green Version]
- Lamb, T.D. Why rods and cones? Eye 2016, 30, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Calkins, D.J. Seeing with S cones. Prog. Retin. Eye Res. 2001, 20, 255–287. [Google Scholar] [CrossRef] [PubMed]
- Curcio, C.A.; Allen, K.A.; Sloan, K.R.; Lerea, C.L.; Hurley, J.B.; Klock, I.B.; Milam, A.H. Distribution and morphology of human cone photoreceptors stained with anti-blue opsin. J. Comp. Neurol. 1991, 312, 610–624. [Google Scholar] [CrossRef]
- Murro, V.; Mucciolo, D.P.; Sodi, A.; Passerini, I.; Giorgio, D.; Virgili, G.; Rizzo, S. Novel clinical findings in autosomal recessive NR2E3-related retinal dystrophy. Graefes Arch. Clin. Exp. Ophthalmol. 2019, 257, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Haider, N.B.; Mollema, N.; Gaule, M.; Yuan, Y.; Sachs, A.J.; Nystuen, A.M.; Naggert, J.K.; Nishina, P.M. Nr2e3-directed transcriptional regulation of genes involved in photoreceptor development and cell-type specific phototransduction. Exp. Eye Res. 2009, 89, 365–372. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.; Khan, N.W.; Roger, J.E.; Swaroop, A. Excess cones in the retinal degeneration rd7 mouse, caused by the loss of function of orphan nuclear receptor Nr2e3, originate from early-born photoreceptor precursors. Hum. Mol. Genet. 2011, 20, 4102–4115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniele, L.L.; Lillo, C.; Lyubarsky, A.L.; Nikonov, S.S.; Philp, N.; Mears, A.J.; Swaroop, A.; Williams, D.S.; Pugh, E.N. Cone-like morphological, molecular, and electrophysiological features of the photoreceptors of the Nrl knockout mouse. Investig. Ophthalmol. Vis. Sci. 2005, 46, 2156–2167. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, K.M.B.; Schulte, D.; Hendrickson, A.E. Expression of photoreceptor-associated molecules during human fetal eye development. Mol. Vis. 2003, 9, 401–409. [Google Scholar]
- Cheng, H.; Khanna, H.; Oh, E.C.; Hicks, D.; Mitton, K.; Swaroop, A. Photoreceptor-specific nuclear receptor NR2E3 functions as a transcriptional activator in rod photoreceptors. Hum. Mol. Genet. 2004, 13, 1563–1575. [Google Scholar] [CrossRef]
- Freund, C.L.; Wang, Q.-L.; Chen, S.; Muskat, B.L.; Wiles, C.D.; Sheffield, V.C.; Jacobson, S.G.; Mclnnes, R.R.; Zack, D.J.; Stone, E.M. De novo mutations in the CRX homeobox gene associated with Leber congenital amaurosis. Nat. Genet. 1998, 18, 311–312. [Google Scholar] [CrossRef] [PubMed]
- Freund, C.L.; Gregory-Evans, C.Y.; Furukawa, T.; Papaioannou, M.; Looser, J.; Ploder, L.; Bellingham, J.; Ng, D.; Herbrick, J.A.S.; Duncan, A.; et al. Cone-rod dystrophy due to mutations in a novel photoreceptor-specific homeobox gene (CRX) essential for maintenance of the photoreceptor. Cell 1997, 91, 543–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, G.-H.; Ahmad, O.; Ahmad, F.; Liu, J.; Chen, S. The photoreceptor-specific nuclear receptor Nr2e3 interacts with Crx and exerts opposing effects on the transcription of rod versus cone genes. Hum. Mol. Genet. 2005, 14, 747–764. [Google Scholar] [CrossRef] [Green Version]
- Olivares, A.M.; Jelcick, A.S.; Reinecke, J.; Leehy, B.; Haider, A.; Morrison, M.A.; Cheng, L.; Chen, D.F.; DeAngelis, M.M.; Haider, N.B. Multimodal Regulation Orchestrates Normal and Complex Disease States in the Retina. Sci. Rep. 2017, 7, 690. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Datta, S.; Brabbit, E.; Love, Z.; Woytowicz, V.; Flattery, K.; Capri, J.; Yao, K.; Wu, S.; Imboden, M.; et al. Nr2e3 is a genetic modifier that rescues retinal degeneration and promotes homeostasis in multiple models of retinitis pigmentosa. Gene Ther. 2021, 28, 223–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khanal, T.; Leung, Y.-K.; Jiang, W.; Timchenko, N.; Ho, S.-M.; Kim, K. NR2E3 is a key component in p53 activation by regulating a long noncoding RNA DINO in acute liver injuries. FASEB J. 2019, 33, 8335–8348. [Google Scholar] [CrossRef] [PubMed]
- Khanal, T.; Choi, K.; Leung, Y.-K.; Wang, J.; Kim, D.; Janakiram, V.; Cho, S.-G.; Puga, A.; Ho, S.-M.; Kim, K. Loss of NR2E3 represses AHR by LSD1 reprogramming, is associated with poor prognosis in liver cancer. Sci. Rep. 2017, 7, 10662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, Y.; Kim, K.; Kim, S.; Hennessy, B.T.; Kim, S.M.; Park, E.S.; Lim, J.Y.; Li, J.; Lu, Y.; Gonzalez-Angulo, A.M.; et al. Reconstruction of nuclear receptor network reveals that NR2E3 is a novel upstream regulator of ESR1 in breast cancer. EMBO Mol. Med. 2012, 4, 52–67. [Google Scholar] [CrossRef]
- Haider, N.B.; Jacobson, S.G.; Cideciyan, A.V.; Swiderski, R.; Streb, L.M.; Searby, C.; Beck, G.; Hockey, R.; Hanna, D.B.; Gorman, S.; et al. Mutation of a nuclear receptor gene, NR2E3, causes enhanced S cone syndrome, a disorder of retinal cell fate. Nat. Genet. 2000, 24, 127–131. [Google Scholar] [CrossRef]
- de Carvalho, E.R.; Robson, A.G.; Arno, G.; Boon, C.J.; Webster, A.A.; Michaelides, M. Enhanced S-Cone Syndrome: Spectrum of Clinical, Imaging, Electrophysiologic, and Genetic Findings in a Retrospective Case Series of 56 Patients. Ophthalmol. Retina 2021, 5, 195–214. [Google Scholar] [CrossRef] [PubMed]
- Marmor, M.F.; Jacobson, S.G.; Foerster, M.H.; Kellner, U.; Weleber, R.G. Diagnostic Clinical Findings of a New Syndrome with Night Blindness, Maculopathy, and Enhanced S Cone Sensitivity. Am. J. Ophthalmol. 1990, 110, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Sharon, D.; Sandberg, M.A.; Caruso, R.C.; Berson, E.L.; Dryja, T.P. Shared mutations in NR2E3 in enhanced S-cone syndrome, Goldmann-Favre syndrome, and many cases of clumped pigmentary retinal degeneration. Arch. Ophthalmol. 2003, 121, 1316–1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobson, S.G.; Román, A.J.; Román, M.I.; Gass, J.D.M.; Parker, J.A. Relatively Enhanced S Cone Function in the Goldmann-Favre Syndrome. Am. J. Ophthalmol. 1991, 111, 446–453. [Google Scholar] [CrossRef]
- Fishman, G.A.; Jampol, L.M.; Goldberg, M.F. Diagnostic features of the Favre-Goldmann syndrome. Br. J. Ophthalmol. 1976, 60, 345–353. [Google Scholar] [CrossRef] [Green Version]
- To, K.W.; Adamian, M.; Jakobiec, F.A.; Berson, E.L. Clinical and Histopathologic Findings in Clumped Pigmentary Retinal Degeneration. Arch. Ophthalmol. 1996, 114, 950–955. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Kelly, F.; Hoyos, M.G.; Martinez, M.A.L.; Lopez-Molina, M.I.; Riveiro-Alvarez, R.; Jose, P.F.-S.; Avila-Fernandez, A.; Corton, M.; Millan, J.M.; Sandoval, B.G.; et al. Dominant Retinitis Pigmentosa, p.Gly56Arg Mutation in NR2E3: Phenotype in a Large Cohort of 24 Cases. PLoS ONE 2016, 11, e0149473. [Google Scholar] [CrossRef] [Green Version]
- Garafalo, A.V.; Calzetti, G.; Cideciyan, A.V.; Roman, A.J.; Saxena, S.; Sumaroka, A.; Choi, W.; Wright, A.F.; Jacobson, S.G. Cone Vision Changes in the Enhanced S-Cone Syndrome Caused by NR2E3Gene Mutations. Investig. Opthalmol. Vis. Sci. 2018, 59, 3209–3219. [Google Scholar] [CrossRef] [Green Version]
- Bandah, D.; Merin, S.; Ashhab, M.; Banin, E.; Sharon, D. The Spectrum of Retinal Diseases Caused by NR2E3 Mutations in Israeli and Palestinian Patients. Arch. Ophthalmol. 2009, 127, 297–302. [Google Scholar] [CrossRef] [Green Version]
- Kuniyoshi, K.; Hayashi, T.; Sakuramoto, H.; Nakao, A.; Sato, T.; Utsumi, T.; Tsuneoka, H.; Shimomura, Y. Novel Mutations in Enhanced S-cone Syndrome. Ophthalmology 2013, 120, 431.e1-6. [Google Scholar] [CrossRef]
- Bernal, S.; Solans, T.; Gamundi, M.; Hernan, I.; De Jorge, L.; Carballo, M.; Navarro, R.; Tizzano, E.; Ayuso, C.; Baiget, M. Analysis of the involvement of the NR2E3 gene in autosomal recessive retinal dystrophies. Clin. Genet. 2008, 73, 360–366. [Google Scholar] [CrossRef] [PubMed]
- Pachydaki, S.I.; Klaver, C.; Barbazetto, I.A.; Roy, M.S.; Gouras, P.; Allikmets, R.; Yannuzzi, L.A. Phenotypic features of patients with NR2E3 mutations. Arch Ophthalmol. 2009, 127, 71–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerber, S.; Rozet, J.M.; Takezawa, S.I.; Coutinho dos Santos, L.; Lopes, L.; Gribouval, O.; Penet, C.; Perrault, I.; Ducroq, D.; Souied, E.; et al. The photoreceptor cell-specific nuclear receptor gene (PNR) accounts for retinitis pigmentosa in the Crypto-Jews from Portugal (Marranos), survivors from the Spanish Inquisition. Hum. Genet. 2000, 107, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Coppieters, F.; Leroy, B.P.; Beysen, D.; Hellemans, J.; De Bosscher, K.; Haegeman, G.; Robberecht, K.; Wuyts, W.; Coucke, P.J.; De Baere, E. Recurrent Mutation in the First Zinc Finger of the Orphan Nuclear Receptor NR2E3 Causes Autosomal Dominant Retinitis Pigmentosa. Am. J. Hum. Genet. 2007, 81, 147–157. [Google Scholar] [CrossRef] [Green Version]
- Escher, P.; Gouras, P.; Tiab, L.; Bolay, S.; Delarive, T.; Chen, S.; Tsai, C.-C.; Hayashi, M.; Zernant, J.; Merriam, J.E.; et al. Mutations in NR2E3 can cause dominant or recessive retinal degenerations in the same family. Hum. Mutat. 2009, 30, 342–351. [Google Scholar] [CrossRef] [Green Version]
- Audo, I.; Michaelides, M.; Robson, A.G.; Hawlina, M.; Vaclavik, V.; Sandbach, J.M.; Neveu, M.M.; Hogg, C.R.; Hunt, D.M.; Moore, A.T.; et al. Phenotypic Variation in Enhanced S-cone Syndrome. Investig. Opthalmol. Vis. Sci. 2008, 49, 2082–2093. [Google Scholar] [CrossRef]
- van Huet, R.A.; Pierrache, L.H.; Meester-Smoor, M.A.; Klaver, C.C.; van den Born, L.I.; Hoyng, C.B.; de Wijs, I.J.; Collin, R.W.; Hoefsloot, L.H.; Klevering, B.J. The efficacy of microarray screening for autosomal recessive retinitis pigmentosa in routine clinical practice. Mol. Vis. 2015, 21, 461–476. [Google Scholar]
- Neveling, K.; Collin, R.W.; Gilissen, C.; van Huet, R.A.; Visser, L.; Kwint, M.P.; Gijsen, S.J.; Zonneveld, M.N.; Wieskamp, N.; de Ligt, J.; et al. Next-generation genetic testing for retinitis pigmentosa. Hum. Mutat. 2012, 33, 963–972. [Google Scholar] [CrossRef]
- Kannabiran, C.; Singh, H.; Sahini, N.; Jalali, S.; Mohan, G. Mutations in TULP1, NR2E3, and MFRP genes in Indian families with autosomal recessive retinitis pigmentosa. Mol. Vis. 2012, 18, 1165–1174. [Google Scholar]
- Xu, Y.; Guan, L.; Shen, T.; Zhang, J.; Xiao, X.; Jiang, H.; Li, S.; Yang, J.; Jia, X.; Yin, Y.; et al. Mutations of 60 known causative genes in 157 families with retinitis pigmentosa based on exome sequencing. Hum. Genet. 2014, 133, 1255–1271. [Google Scholar] [CrossRef]
- I Gire, A.; Sullivan, L.S.; Bowne, S.J.; Birch, D.G.; Hughbanks-Wheaton, D.; Heckenlively, J.R.; Daiger, S.P. The Gly56Arg mutation in NR2E3 accounts for 1–2% of autosomal dominant retinitis pigmentosa. Mol. Vis. 2007, 13, 1970–1975. [Google Scholar] [PubMed]
- Bravo-Gil, N.; Méndez-Vidal, C.; Romero-Pérez, L.; Pozo, M.G.-D.; la Rúa, E.R.-D.; Dopazo, J.; Borrego, S.; Antiñolo, G. Improving the management of Inherited Retinal Dystrophies by targeted sequencing of a population-specific gene panel. Sci. Rep. 2016, 6, 23910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Udar, N.; Small, K.; Chalukya, M.; Silva-Garcia, R.; Marmor, M. Developmental or degenerative—NR2E3 gene mutations in two patients with enhanced S cone syndrome. Mol. Vis. 2011, 17, 519–525. [Google Scholar]
- Park, S.P.; Hong, I.H.; Tsang, S.H.; Lee, W.; Horowitz, J.; Yzer, S.; Allikmets, R.; Chang, S. Disruption of the human cone photoreceptor mosaic from a defect in NR2E3 transcription factor function in young adults. Graefe’s Arch. Clin. Exp. Ophthalmol. 2013, 251, 2299–2309. [Google Scholar] [CrossRef]
- Maeda, A.; Yoshida, A.; Kawai, K.; Arai, Y.; Akiba, R.; Inaba, A.; Takagi, S.; Fujiki, R.; Hirami, Y.; Kurimoto, Y.; et al. Development of a molecular diagnostic test for Retinitis Pigmentosa in the Japanese population. Jpn. J. Ophthalmol. 2018, 62, 451–457. [Google Scholar] [CrossRef]
- Rocha-Sousa, A.; Hayashi, T.; Gomes, N.L.; Penas, S.; Brandão, E.; Rocha, P.; Urashima, M.; Yamada, H.; Tsuneoka, H.; Falcão-Reis, F. A novel mutation (Cys83Tyr) in the second zinc finger of NR2E3 in enhanced S-cone syndrome. Graefes Arch. Clin. Exp. Ophthalmol. 2011, 249, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.F.; Reddick, A.C.; Schwartz, S.B.; Ferguson, J.S.; Aleman, T.S.; Kellner, U.; Jurklies, B.; Schuster, A.; Zrenner, E.; Wissinger, B.; et al. Mutation analysis ofNR2E3 andNRL genes in Enhanced S Cone Syndrome. Hum. Mutat. 2004, 24, 439. [Google Scholar] [CrossRef]
- Huang, X.-F.; Huang, F.; Wu, K.-C.; Wu, J.; Chen, J.; Pang, C.-P.; Lu, F.; Qu, J.; Jin, Z.-B. Genotype–phenotype correlation and mutation spectrum in a large cohort of patients with inherited retinal dystrophy revealed by next-generation sequencing. Genet. Med. 2015, 17, 271–278. [Google Scholar] [CrossRef] [Green Version]
- Hull, S.; Arno, G.; Sergouniotis, P.I.; Tiffin, P.; Borman, A.D.; Chandra, A.; Robson, A.; Holder, G.E.; Webster, A.R.; Moore, A.T. Clinical and Molecular Characterization of Enhanced S-Cone Syndrome in Children. JAMA Ophthalmol. 2014, 132, 1341–1349. [Google Scholar] [CrossRef] [Green Version]
- Stone, E.M.; Andorf, J.L.; Whitmore, S.S.; DeLuca, A.P.; Giacalone, J.C.; Streb, L.M.; Braun, T.A.; Mullins, R.F.; Scheetz, T.E.; Sheffield, V.C.; et al. Clinically Focused Molecular Investigation of 1000 Consecutive Families with Inherited Retinal Disease. Ophthalmology 2017, 124, 1314–1331. [Google Scholar] [CrossRef]
- Lingao, M.D.; Ganesh, A.; Karthikeyan, A.S.; Al Zuhaibi, S.; Al-Hosni, A.; Al Khayat, A.; Capasso, J.; Trumler, A.A.; Stroh, E.; Al Shekaili, H.; et al. Macular cystoid spaces in patients with retinal dystrophy. Ophthalmic Genet. 2016, 37, 377–383. [Google Scholar] [CrossRef]
- Birtel, J.; Gliem, M.; Mangold, E.; Müller, P.L.; Holz, F.G.; Neuhaus, C.; Lenzner, S.; Zahnleiter, D.; Betz, C.; Eisenberger, T.; et al. Next-generation sequencing identifies unexpected genotype-phenotype correlations in patients with retinitis pigmentosa. PLoS ONE 2018, 13, e0207958. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, T.; Gekka, T.; Goto-Omoto, S.; Takeuchi, T.; Kubo, A.; Kitahara, K. Novel NR2E3 mutations (R104Q, R334G) associated with a mild form of enhanced S-cone syndrome demonstrate compound heterozygosity. Ophthalmology 2005, 112, 2115. [Google Scholar] [CrossRef]
- Kimchi, A.; Khateb, S.; Wen, R.; Guan, Z.; Obolensky, A.; Beryozkin, A.; Kurtzman, S.; Blumenfeld, A.; Pras, E.; Jacobson, S.G.; et al. Nonsyndromic Retinitis Pigmentosa in the Ashkenazi Jewish Population: Genetic and Clinical Aspects. Ophthalmology 2018, 125, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Carss, K.J.; Arno, G.; Erwood, M.; Stephens, J.; Sanchis-Juan, A.; Hull, S.; Megy, K.; Grozeva, D.; Dewhurst, E.; Malka, S.; et al. Comprehensive Rare Variant Analysis via Whole-Genome Sequencing to Determine the Molecular Pathology of Inherited Retinal Disease. Am. J. Hum. Genet. 2017, 100, 75–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weisschuh, N.; Obermaier, C.D.; Battke, F.; Bernd, A.; Kuehlewein, L.; Nasser, F.; Zobor, D.; Zrenner, E.; Weber, E.; Wissinger, B.; et al. Genetic architecture of inherited retinal degeneration in Germany: A large cohort study from a single diagnostic center over a 9-year period. Hum. Mutat. 2020, 41, 1514–1527. [Google Scholar] [CrossRef] [PubMed]
- Carrigan, M.; Duignan, E.; Malone, C.P.G.; Stephenson, K.; Saad, T.; McDermott, C.; Green, A.; Keegan, D.; Humphries, P.; Kenna, P.F.; et al. Panel-Based Population Next-Generation Sequencing for Inherited Retinal Degenerations. Sci. Rep. 2016, 6, 33248. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Zhang, X.; Chen, L.J.; Chiang, S.W.; Tam, P.O.; Lai, T.Y.; Chan, C.K.; Wang, N.; Lam, D.S.; Pang, C.P. Association of NR2E3 but not NRL mutations with retinitis pigmentosa in the Chinese population. Investigative Ophthalmol. Vis. Sci. 2010, 51, 2229–2235. [Google Scholar] [CrossRef] [Green Version]
- Bocquet, B.; Marzouka, N.A.D.; Hebrard, M.; Manes, G.; Sénéchal, A.; Meunier, I.; Hamel, C.P. Homozygosity mapping in autosomal recessive retinitis pigmentosa families detects novel mutations. Mol. Vis. 2013, 19, 2487–2500. [Google Scholar]
- Takahashi, V.K.L.; Xu, C.L.; Takiuti, J.T.; Apatoff, M.B.L.; Duong, J.K.; Mahajan, V.B.; Tsang, S.H. Comparison of structural progression between ciliopathy and non-ciliopathy associated with autosomal recessive retinitis pigmentosa. Orphanet J. Rare Dis. 2019, 14, 187. [Google Scholar] [CrossRef] [Green Version]
- Cassiman, C.; Spileers, W.; De Baere, E.; De Ravel, T.; Casteels, I. Peculiar fundus abnormalities and pathognomonic electrophysiological findings in a 14-month-old boy with NR2E3 mutations. Ophthalmic Genet. 2013, 34, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.; Aldahmesh, M.A.; Alkuraya, H.; Anazi, S.; Alsharif, H.; Khan, A.O.; Sunker, A.; Al-Mohsen, S.; Abboud, E.B.; Nowilaty, S.R.; et al. Expanding the clinical, allelic, and locus heterogeneity of retinal dystrophies. Genet. Med. 2016, 18, 554–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, Y.-C.C.; Yu, H.-C.; Martin, R.; Cirulli, E.T.; Schenker-Ahmed, N.M.; Hicks, M.; Cohen, I.V.; Jönsson, T.J.; Heister, R.; Napier, L.; et al. Precision medicine integrating whole-genome sequencing, comprehensive metabolomics, and advanced imaging. Proc. Natl. Acad. Sci. USA 2020, 117, 3053–3062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jespersgaard, C.; Fang, M.; Bertelsen, M.; Dang, X.; Jensen, H.; Chen, Y.; Bech, N.; Dai, L.; Rosenberg, T.; Zhang, J.; et al. Molecular genetic analysis using targeted NGS analysis of 677 individuals with retinal dystrophy. Sci. Rep. 2019, 9, 1219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahajan, D.; Votruba, M. A novel NR2E3 gene mutation in autosomal recessive retinitis pigmentosa with cystic maculopathy. Acta Ophthalmol. 2018, 96, e535–e536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Khuzaei, S.; Broadgate, S.; Halford, S.; Jolly, J.K.; Shanks, M.; Clouston, P.; Downes, S.M. Novel Pathogenic Sequence Variants in NR2E3 and Clinical Findings in Three Patients. Genes 2020, 11, 1288. [Google Scholar] [CrossRef]
- Seo, G.H.; Kim, T.; Choi, I.H.; Park, J.Y.; Lee, J.; Kim, S.; Won, D.G.; Oh, A.; Lee, Y.; Choi, J.; et al. Diagnostic yield and clinical utility of whole exome sequencing using an automated variant prioritization system, EVIDENCE. Clin. Genet. 2020, 98, 562–570. [Google Scholar] [CrossRef]
- Collin, R.W.J.; Born, L.I.V.D.; Klevering, B.J.; de Castro-Miró, M.; Littink, K.W.; Arimadyo, K.; Azam, M.; Yazar, V.; Zonneveld, M.N.; Paun, C.C.; et al. High-Resolution Homozygosity Mapping Is a Powerful Tool to Detect Novel Mutations Causative of Autosomal Recessive RP in the Dutch Population. Investig. Opthalmol. Vis. Sci. 2011, 52, 2227–2239. [Google Scholar] [CrossRef] [Green Version]
- Glöckle, N.; Kohl, S.; Mohr, J.; Scheurenbrand, T.; Sprecher, A.; Weisschuh, N.; Bernd, A.; Rudolph, G.; Schubach, M.; Poloschek, C.; et al. Panel-based next generation sequencing as a reliable and efficient technique to detect mutations in unselected patients with retinal dystrophies. Eur. J. Hum. Genet. 2014, 22, 99–104. [Google Scholar] [CrossRef] [Green Version]
- Haer-Wigman, L.; Van Zelst-Stams, W.A.G.; Pfundt, R.; Born, L.I.V.D.; Klaver, C.; Verheij, J.B.G.M.; Hoyng, C.B.; Breuning, M.H.; Boon, C.; Kievit, A.J.; et al. Diagnostic exome sequencing in 266 Dutch patients with visual impairment. Eur. J. Hum. Genet. 2017, 25, 591–599. [Google Scholar] [CrossRef]
- Martin-Merida, I.; Avila-Fernandez, A.; Del Pozo-Valero, M.; Blanco-Kelly, F.; Zurita, O.; Perez-Carro, R.; Aguilera-Garcia, D.; Riveiro-Alvarez, R.; Arteche, A.; Trujillo-Tiebas, M.J.; et al. Genomic Landscape of Sporadic Retinitis Pigmentosa: Findings from 877 Spanish Cases. Ophthalmology 2019, 126, 1181–1188. [Google Scholar] [CrossRef]
- Cehajic-Kapetanovic, J.; Cottriall, C.L.; Jolly, J.K.; Shanks, M.; Clouston, P.; Issa, P.C.; MacLaren, R.E. Electrophysiological verification of enhanced S-cone syndrome caused by a novel c.755T>C NR2E3 missense variant. Ophthalmic Genet. 2019, 40, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Lam, B.L.; Goldberg, J.L.; Hartley, K.L.; Stone, E.M.; Liu, M. Atypical Mild Enhanced S-Cone Syndrome with Novel Compound Heterozygosity of the NR2E3 Gene. Am. J. Ophthalmol. 2007, 144, 157–159. [Google Scholar] [CrossRef] [PubMed]
- Termühlen, J.; Alex, A.F.; Glöckle, N.; Kellner, U.; Fiedler, B.; Eter, N.; Uhlig, C.E. A new mutation in enhanced S-cone syndrome. Acta Ophthalmol. 2018, 96, e539–e540. [Google Scholar] [CrossRef] [PubMed]
- Collison, F.T.; Park, J.C.; Fishman, G.A.; Stone, E.M.; McAnany, J.J. Two-color pupillometry in enhanced S-cone syndrome caused by NR2E3 mutations. Doc. Ophthalmol. 2016, 132, 157–166. [Google Scholar] [CrossRef]
- Kuniyoshi, K.; Hayashi, T.; Sakuramoto, H.; Mishima, H.; Tsuneoka, H.; Tsunoda, K.; Iwata, T.; Shimomura, Y. New truncation mutation of the NR2E3 gene in a Japanese patient with enhanced S-cone syndrome. Jpn. J. Ophthalmol. 2016, 60, 476–485. [Google Scholar] [CrossRef]
- Bai, Z.; Xie, Y.; Liu, L.; Shao, J.; Liu, Y.; Kong, X. Genetic investigation of 211 Chinese families expands the mutational and phenotypical spectra of hereditary retinopathy genes through targeted sequencing technology. BMC Med. Genom. 2021, 14, 92. [Google Scholar] [CrossRef]
- Bechet, L.; Atia, R.; Zeitz, C.; Mohand-Saïd, S.; Sahel, J.-A.; Barale, P.-O.; Audo, I. Management of a case of Enhanced S-cone syndrome with massive foveoschisis treated with pars plana vitrectomy with silicone oil tamponade. Ophthalmic Genet. 2021, 42, 615–618. [Google Scholar] [CrossRef]
- Patel, N.; Alkuraya, H.; Alzahrani, S.; Nowailaty, S.; Seidahmed, M.; Alhemidan, A.; Ben-Omran, T.; Ghazi, N.; Al-Aqeel, A.; Al-Owain, M.; et al. Mutations in known disease genes account for the majority of autosomal recessive retinal dystrophies. Clin. Genet. 2018, 94, 554–563. [Google Scholar] [CrossRef]
- Abu-Safieh, L.; Alrashed, M.; Anazi, S.; Alkuraya, H.; Khan, A.O.; Al-Owain, M.; Al-Zahrani, J.; Al-Abdi, L.; Hashem, M.; Al-Tarimi, S.; et al. Autozygome-guided exome sequencing in retinal dystrophy patients reveals pathogenetic mutations and novel candidate disease genes. Genome Res. 2013, 23, 236–247. [Google Scholar] [CrossRef] [Green Version]
- Dockery, A.; Stephenson, K.; Keegan, D.; Wynne, N.; Silvestri, G.; Humphries, P.; Kenna, P.F.; Carrigan, M.; Farrar, G.J. Target 5000: Target Capture Sequencing for Inherited Retinal Degenerations. Genes 2017, 8, 304. [Google Scholar] [CrossRef] [Green Version]
- Ripamonti, C.; Aboshiha, J.; Henning, G.B.; Sergouniotis, P.I.; Michaelides, M.; Moore, A.T.; Webster, A.R.; Stockman, A. Vision in Observers with Enhanced S-Cone Syndrome: An Excess of S-Cones but Connected Mainly to Conventional S-Cone Pathways. Investig. Opthalmol. Vis. Sci. 2014, 55, 963–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siemiatkowska, A.M.; Arimadyo, K.; Moruz, L.M.; Astuti, G.D.; De Castro-Miro, M.; Zonneveld, M.N.; Strom, T.M.; De Wijs, I.J.; Hoefsloot, L.H.; Faradz, S.M.; et al. Molecular genetic analysis of retinitis pigmentosa in Indonesia using genome-wide homozygosity mapping. Mol. Vis. 2011, 17, 3013–3024. [Google Scholar] [PubMed]
- Nakamura, Y.; Hayashi, T.; Kozaki, K.; Kubo, A.; Omoto, S.; Watanabe, A.; Toda, K.; Takeuchi, T.; Gekka, T.; Kitahara, K. Enhanced S-cone syndrome in a Japanese family with a nonsense NR2E3 mutation (Q350X). Acta Ophthalmol. Scand. 2004, 82, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Maltese, P.E.; Colombo, L.; Martella, S.; Rossetti, L.; El Shamieh, S.; Sinibaldi, L.; Passarelli, C.; Coppè, A.M.; Buzzonetti, L.; Falsini, B.; et al. Genetics of Inherited Retinal Diseases in Understudied Ethnic Groups in Italian Hospitals. Front. Genet. 2022, 13, 914345. [Google Scholar] [CrossRef]
- Minnella, A.M.; Pagliei, V.; Savastano, M.C.; Federici, M.; Bertelli, M.; Maltese, P.E.; Placidi, G.; Corbo, G.; Falsini, B.; Caporossi, A. Swept source optical coherence tomography and optical coherence tomography angiography in pediatric enhanced S-cone syndrome: A case report. J. Med. Case Rep. 2018, 12, 287. [Google Scholar] [CrossRef]
- Song, F.; Owczarek-Lipska, M.; Ahmels, T.; Book, M.; Aisenbrey, S.; Menghini, M.; Barthelmes, D.; Schrader, S.; Spital, G.; Neidhardt, J. High-Throughput Sequencing to Identify Mutations Associated with Retinal Dystrophies. Genes 2021, 12, 1269. [Google Scholar] [CrossRef]
- Gao, F.-J.; Li, J.-K.; Chen, H.; Hu, F.-Y.; Zhang, S.-H.; Qi, Y.-H.; Xu, P.; Wang, D.-D.; Wang, L.-S.; Chang, Q.; et al. Genetic and Clinical Findings in a Large Cohort of Chinese Patients with Suspected Retinitis Pigmentosa. Ophthalmology 2019, 126, 1549–1556. [Google Scholar] [CrossRef] [Green Version]
- Manayath, G.J.; Namburi, P.; Periasamy, S.; Kale, J.A.; Narendran, V.; Ganesh, A. A novel mutation in the NR2E3 gene associated with Goldmann-Favre syndrome and vasoproliferative tumor of the retina. Mol. Vis. 2014, 20, 724–731. [Google Scholar]
- Roduit, R.; Escher, P.; Schorderet, D.F. Mutations in the DNA-binding domain of NR2E3 affect in vivo dimerization and interaction with CRX. PLoS ONE 2009, 4, e7379. [Google Scholar] [CrossRef]
- Akhmedov, N.B.; Piriev, N.I.; Chang, B.; Rapoport, A.L.; Hawes, N.L.; Nishina, P.M.; Nusinowitz, S.; Heckenlively, J.R.; Roderick, T.H.; Kozak, C.A.; et al. A deletion in a photoreceptor-specific nuclear receptor mRNA causes retinal degeneration in the rd7 mouse. Proc. Natl. Acad. Sci. USA 2000, 97, 5551–5556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Rattner, A.; Nathans, J. Effects of L1 retrotransposon insertion on transcript processing, localization and accumulation: Lessons from the retinal degeneration 7 mouse and implications for the genomic ecology of L1 elements. Hum. Mol. Genet. 2006, 15, 2146–2156. [Google Scholar] [CrossRef] [Green Version]
- Iannaccone, A.; Brabbit, E.; Lopez-Miro, C.; Love, Z.; Griffiths, V.; Kedrov, M.; Haider, N.B. Interspecies Correlations between Human and Mouse NR2E3-Associated Recessive Disease. J. Clin. Med. 2021, 10, 475. [Google Scholar] [CrossRef] [PubMed]
- Webber, A.L.; Hodor, P.; Thut, C.J.; Vogt, T.F.; Zhang, T.; Holder, D.J.; Petrukhin, K. Dual role of Nr2e3 in photoreceptor development and maintenance. Exp. Eye Res. 2008, 87, 35–48. [Google Scholar] [CrossRef]
- Xie, S.; Han, S.; Qu, Z.; Liu, F.; Li, J.; Yu, S.; Reilly, J.; Tu, J.; Liu, X.; Lu, Z.; et al. Knockout of Nr2e3 prevents rod photoreceptor differentiation and leads to selective L-/M-cone photoreceptor degeneration in zebrafish. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1273–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz, N.M.; Yuan, Y.; Leehy, B.D.; Baid, R.; Kompella, U.; DeAngelis, M.M.; Escher, P.; Haider, N.B. Modifier genes as therapeutics: The nuclear hormone receptor Rev Erb Alpha (Nr1d1) rescues Nr2e3 associated retinal disease. PLoS ONE 2014, 9, e87942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, P.A.; Tang, S.; Shimchuk, A.A.; Ding, S.; Reh, T.A. Potential of Small Molecule–Mediated Reprogramming of Rod Photoreceptors to Treat Retinitis Pigmentosa. Investig. Opthalmol. Vis. Sci. 2016, 57, 6407–6415. [Google Scholar] [CrossRef] [Green Version]
- Montana, C.L.; Kolesnikov, A.V.; Shen, S.Q.; Myers, C.A.; Kefalov, V.J.; Corbo, J.C. Reprogramming of adult rod photoreceptors prevents retinal degeneration. Proc. Natl. Acad. Sci. USA 2013, 110, 1732–1737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simunovic, M.; Shen, W.; Lin, J.; Protti, D.; Lisowski, L.; Gillies, M. Optogenetic approaches to vision restoration. Exp. Eye Res. 2019, 178, 15–26. [Google Scholar] [CrossRef]
- Tochitsky, I.; Kienzler, M.A.; Isacoff, E.; Kramer, R.H. Restoring Vision to the Blind with Chemical Photoswitches. Chem. Rev. 2018, 118, 10748–10773. [Google Scholar] [CrossRef]
- Bohrer, L.R.; Wiley, L.A.; Burnight, E.R.; Cooke, J.A.; Giacalone, J.C.; Anfinson, K.R.; Andorf, J.L.; Mullins, R.F.; Stone, E.M.; Tucker, B.A. Correction of NR2E3 Associated Enhanced S-cone Syndrome Patient-specific iPSCs using CRISPR-Cas9. Genes 2019, 10, 278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naessens, S.; Ruysschaert, L.; Lefever, S.; Coppieters, F.; De Baere, E. Antisense Oligonucleotide-Based Downregulation of the G56R Pathogenic Variant Causing NR2E3-Associated Autosomal Dominant Retinitis Pigmentosa. Genes 2019, 10, 363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terrell, D.; Comander, J. Current Stem-Cell Approaches for the Treatment of Inherited Retinal Degenerations. Semin. Ophthalmol. 2019, 34, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Gasparini, S.J.; Llonch, S.; Borsch, O.; Ader, M. Transplantation of photoreceptors into the degenerative retina: Current state and future perspectives. Prog. Retin. Eye Res. 2019, 69, 1–37. [Google Scholar] [CrossRef]
- Iannaccone, A.; Fung, K.H.; Eyestone, M.E.; Stone, E.M. Treatment of Adult-Onset Acute Macular Retinoschisis in Enhanced S-cone Syndrome with Oral Acetazolamide. Am. J. Ophthalmol. 2009, 147, 307–312.e2. [Google Scholar] [CrossRef] [Green Version]
- Chatzistergiou, V.; Papasavvas, I.; Escher, P.; Durig, J.; Vaudaux, J.; Pournaras, J.-A.; Ambresin, A. Optical Coherence Tomography Analysis of Cystoid Macular Edema in Retinal Dystrophy Treated with Oral Acetazolamide: Two Cases. Klin. Mon. Für Augenheilkd. 2020, 237, 484–486. [Google Scholar] [CrossRef]
- Wiley, L.A.; Burnight, E.R.; DeLuca, A.P.; Anfinson, K.R.; Cranston, C.M.; Kaalberg, E.E.; Penticoff, J.A.; Affatigato, L.M.; Mullins, R.F.; Stone, E.M.; et al. cGMP production of patient-specific iPSCs and photoreceptor precursor cells to treat retinal degenerative blindness. Sci. Rep. 2016, 6, 30742. [Google Scholar] [CrossRef] [Green Version]
- Terray, A.; Slembrouck, A.; Nanteau, C.; Chondroyer, C.; Zeitz, C.; Sahel, J.-A.; Audo, I.; Reichman, S.; Goureau, O. Generation of an induced pluripotent stem cell (iPSC) line from a patient with autosomal dominant retinitis pigmentosa due to a mutation in the NR2E3 gene. Stem Cell Res. 2017, 24, 1–4. [Google Scholar] [CrossRef]
- Diakatou, M.; Dubois, G.; Erkilic, N.; Sanjurjo-Soriano, C.; Meunier, I.; Kalatzis, V. Allele-Specific Knockout by CRISPR/Cas to Treat Autosomal Dominant Retinitis Pigmentosa Caused by the G56R Mutation in NR2E3. Int. J. Mol. Sci. 2021, 22, 2607. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toms, M.; Ward, N.; Moosajee, M. Nuclear Receptor Subfamily 2 Group E Member 3 (NR2E3): Role in Retinal Development and Disease. Genes 2023, 14, 1325. https://doi.org/10.3390/genes14071325
Toms M, Ward N, Moosajee M. Nuclear Receptor Subfamily 2 Group E Member 3 (NR2E3): Role in Retinal Development and Disease. Genes. 2023; 14(7):1325. https://doi.org/10.3390/genes14071325
Chicago/Turabian StyleToms, Maria, Natasha Ward, and Mariya Moosajee. 2023. "Nuclear Receptor Subfamily 2 Group E Member 3 (NR2E3): Role in Retinal Development and Disease" Genes 14, no. 7: 1325. https://doi.org/10.3390/genes14071325