
Using Cluster Analysis to Overcome the Limits of Traditional Phenotype–Genotype Correlations: The Example of RYR1-Related Myopathies
 , , , , , ,
, , , , , ,  , , , , , , , , , and add
Show full author list
, , , , , , , , , and add
Show full author list
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Standard Protocol Approvals and Patient Consents
2.2. Patients and Study Design
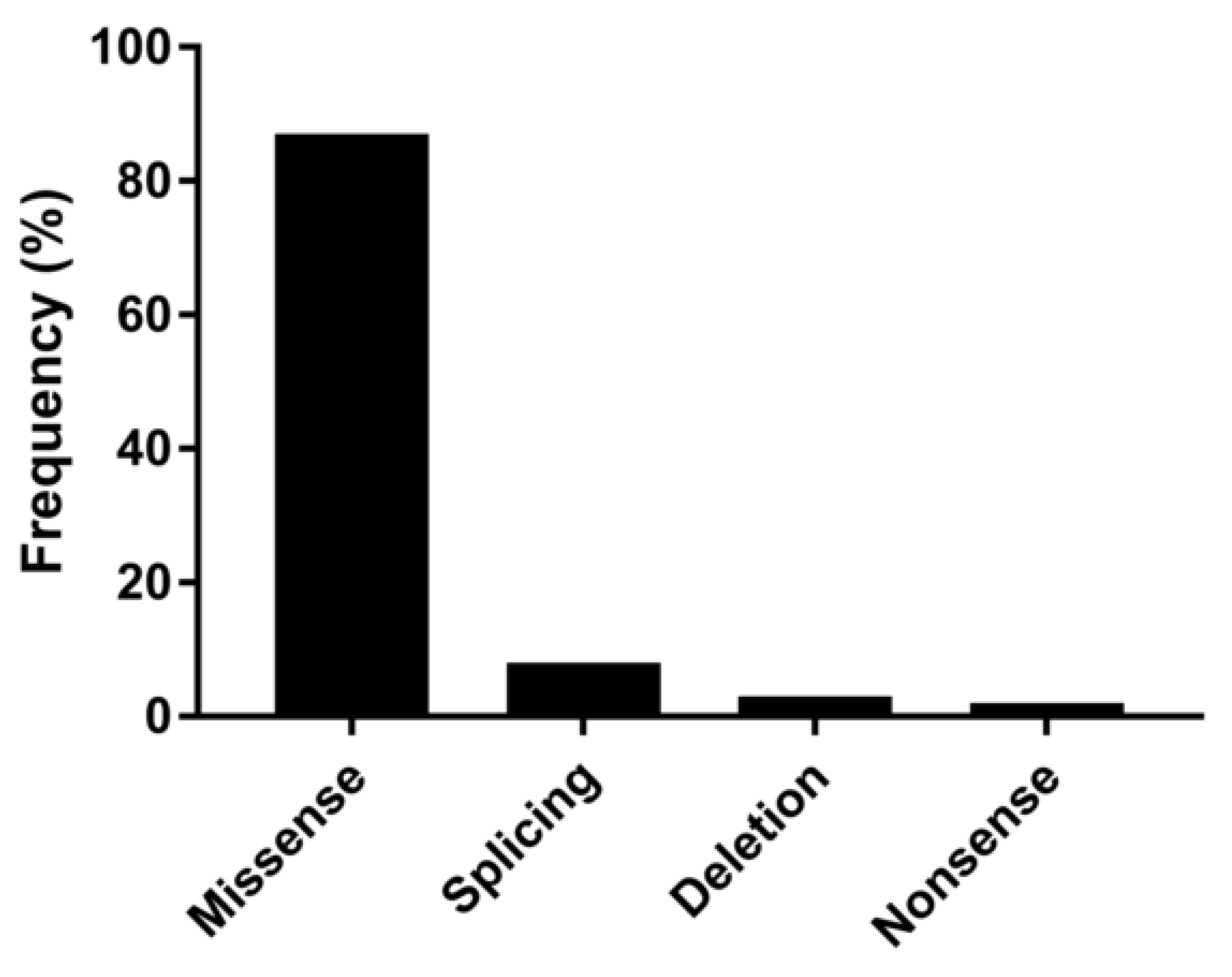
2.3. NGS Workflow and Sequencing Analyses
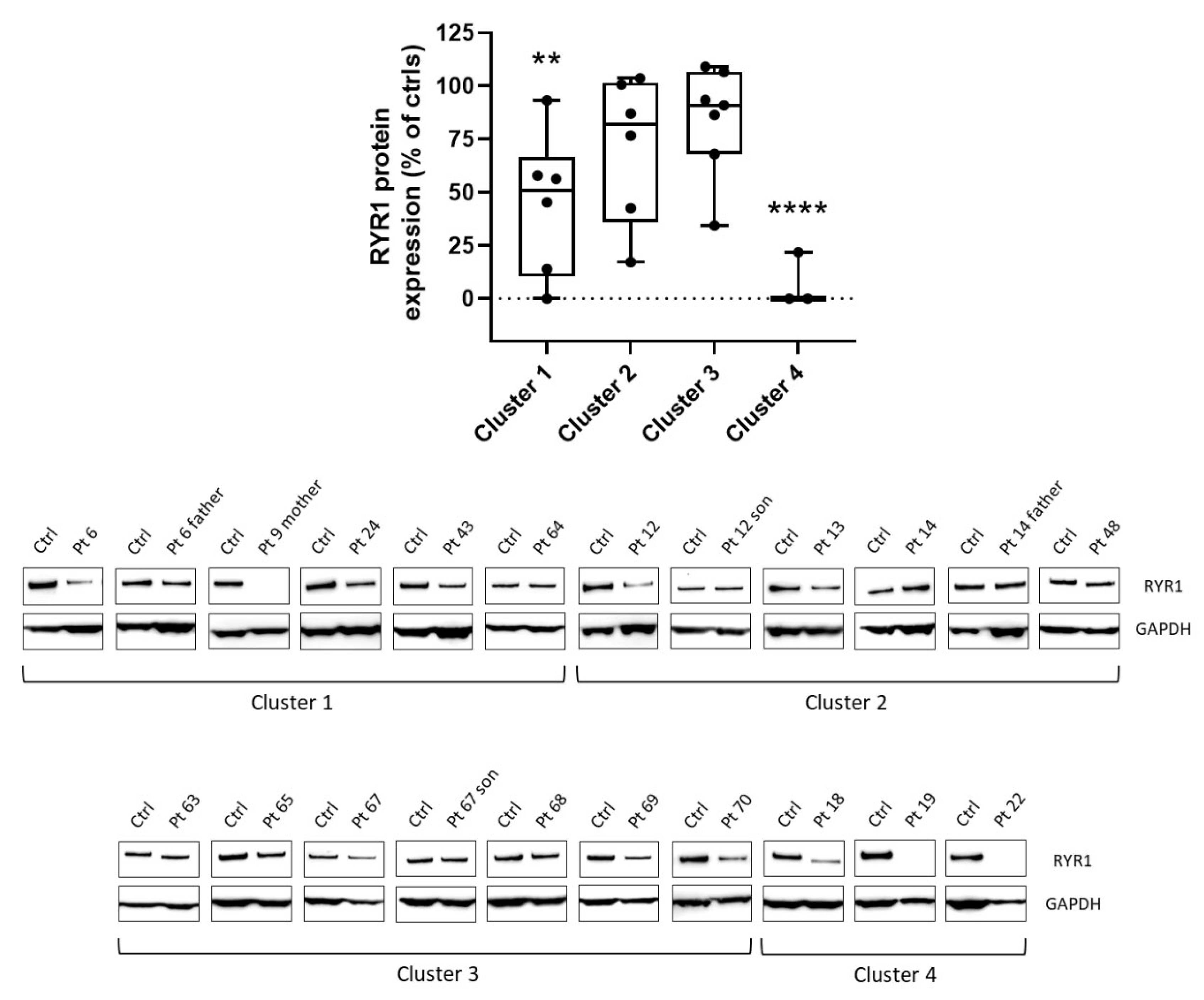
2.4. Western Blot Analysis
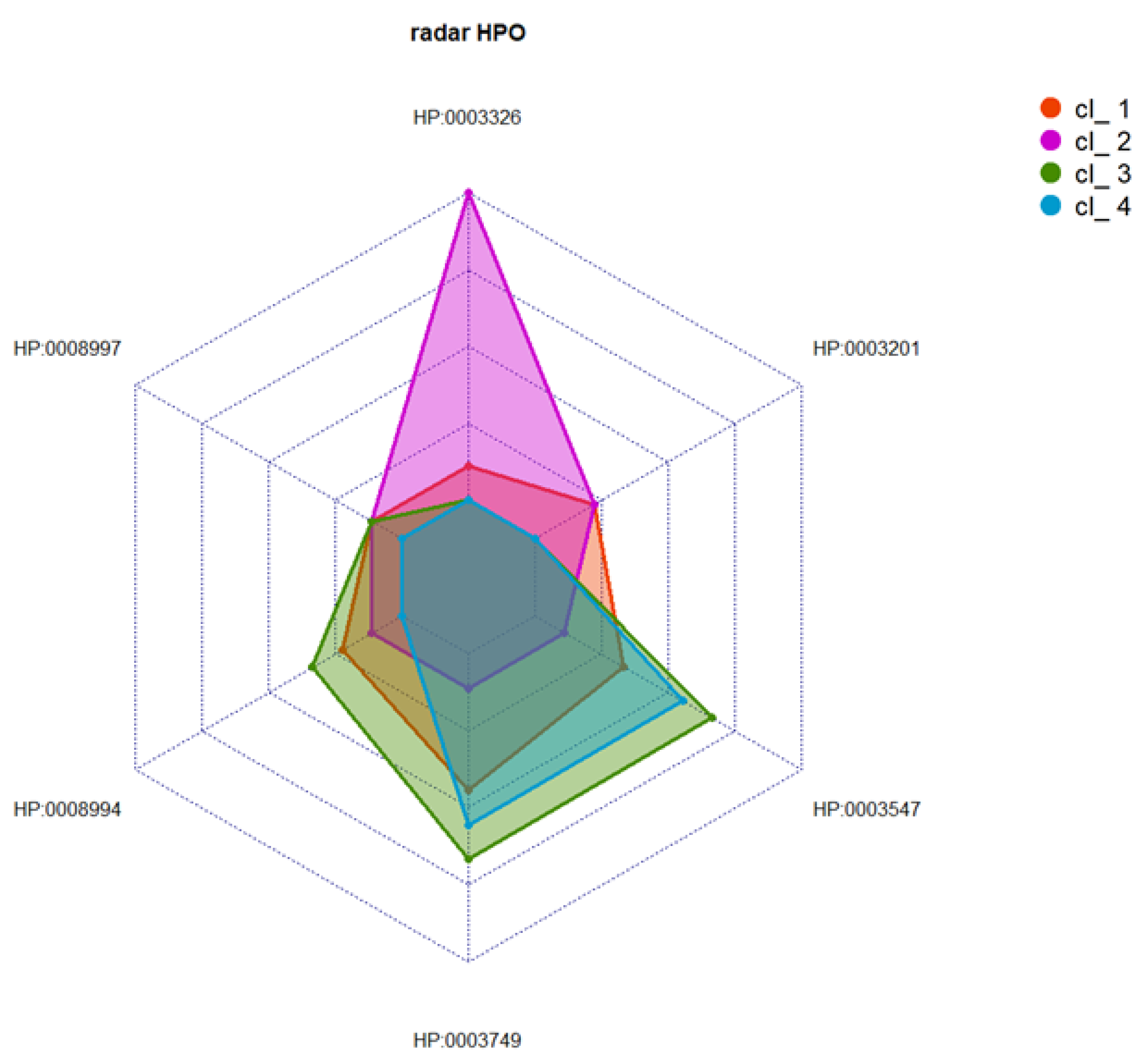
2.5. Cluster Analysis
3. Results
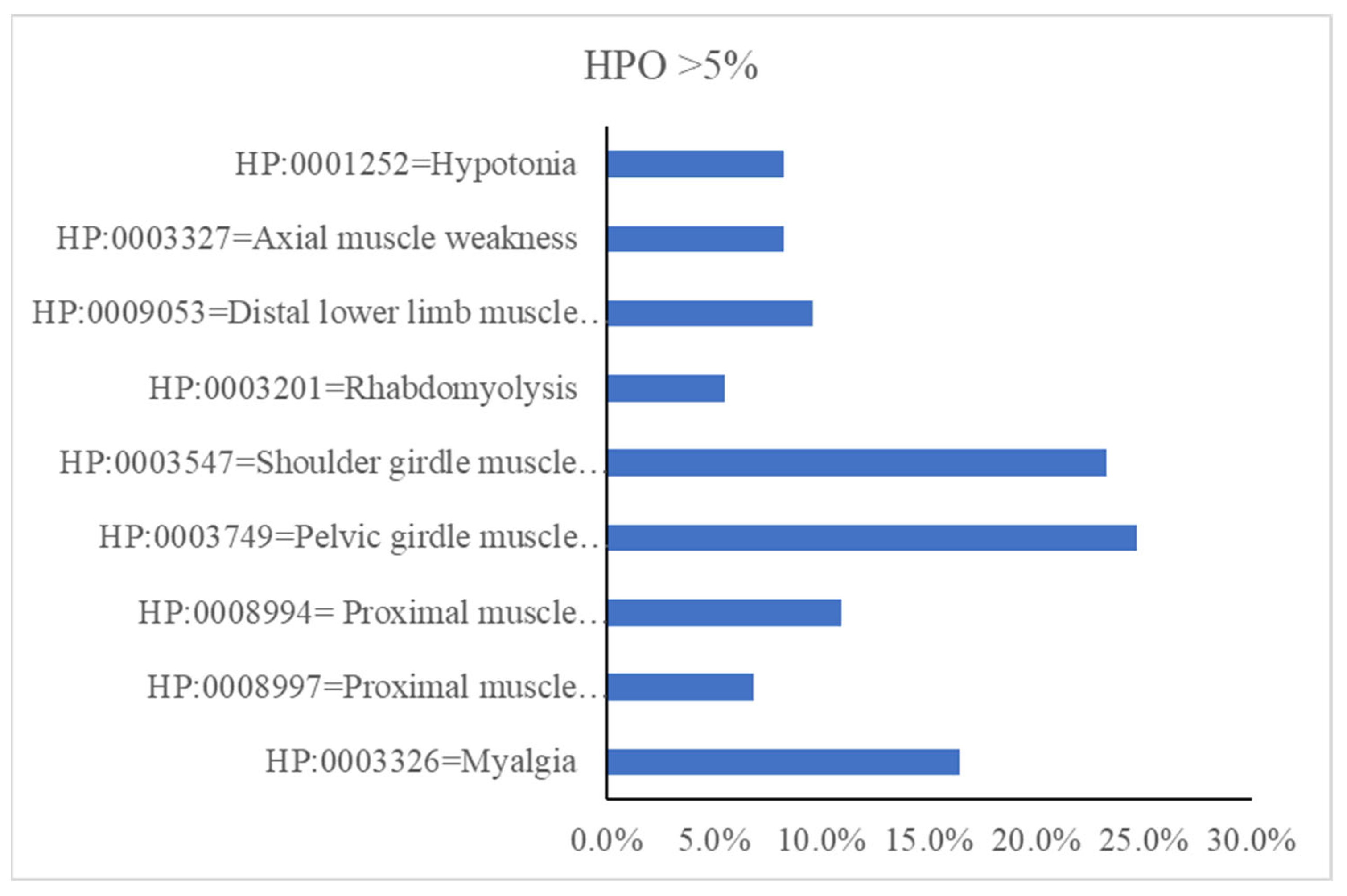
3.1. Clinical Features
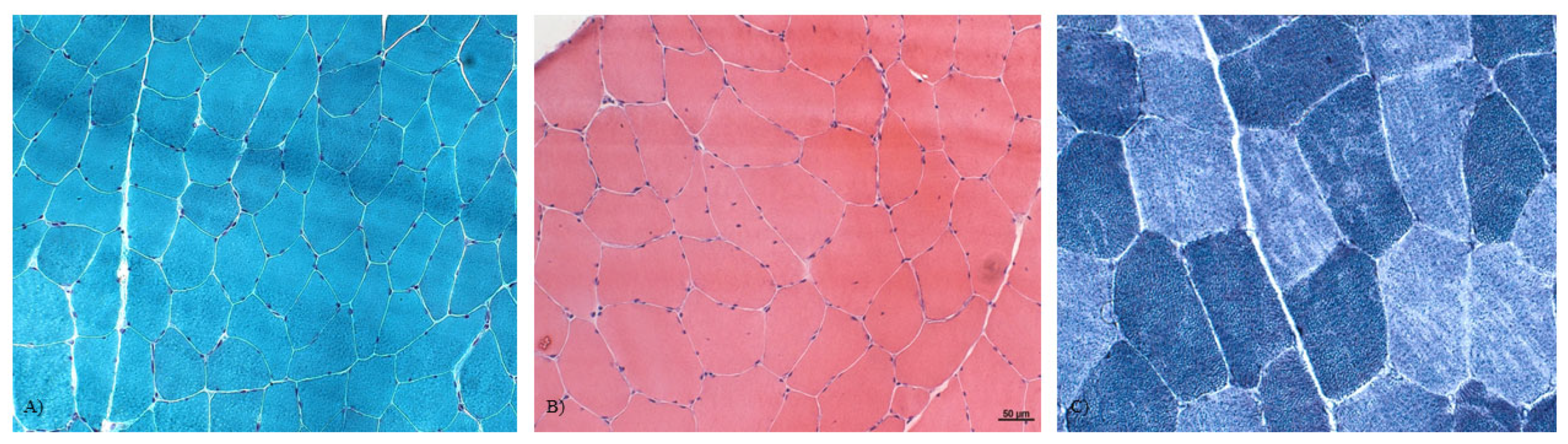
3.2. Muscle Histopathology
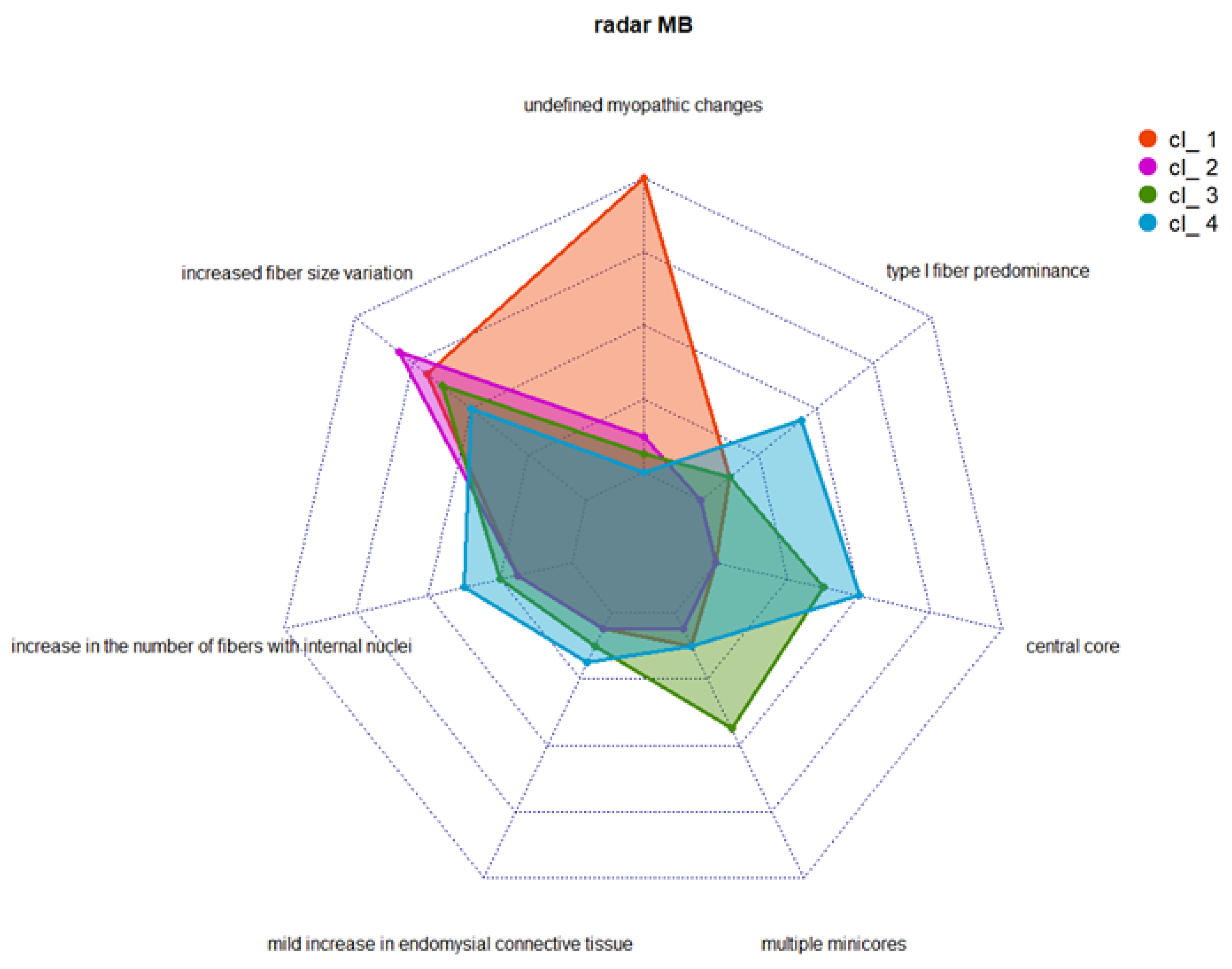
3.3. Cluster Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Gonzalez-Quereda, L.; Rodriguez, M.J.; Diaz-Manera, J.; Alonso-Perez, J.; Gallardo, E.; Nascimento, A.; Ortez, C.; Benito, D.N.-D.; Olive, M.; Gonzalez-Mera, L.; et al. Targeted next-generation sequencing in a large cohort of genetically undiagnosed patients with neuromuscular disorders in Spain. Genes 2020, 11, 539. [Google Scholar] [CrossRef] [PubMed]
- Esteva, A.; Robicquet, A.; Ramsundar, B.; Kuleshov, V.; Depristo, M.; Chou, K.; Cui, C.; Corrado, G.; Thrun, S.; Dean, J. A guide to deep learning in healthcare. Nat. Med. 2019, 25, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Todd, J.J.; Sagar, V.; Lawal, T.A.; Allen, C.; Razaqyar, M.S.; Shelton, M.S.; Chrismer, I.C.; Zhang, X.; Cosgrove, M.M.; Kuo, A.; et al. Correlation of phenotype with genotype and protein structure in RYR1-related disorders. J. Neurol. 2018, 265, 2506–2524. [Google Scholar] [CrossRef]
- Zheng, W.; Wen, H. Investigating dual Ca2+ modulation of the ryanodine receptor 1 by molecular dynamics simulation. Proteins 2020, 88, 1528–1539. [Google Scholar] [CrossRef]
- Scacheri, P.C.; Hoffman, E.P.; Fratkin, J.D.; Semino-Mora, C.; Senchak, A.; Davis, M.R.; Laing, N.G.; Vedanarayanan, V.; Subramony, S.H. A novel ryanodine receptor gene mutation causing both cores and rods in congenital myopathy. Neurology 2000, 55, 1689–1696. [Google Scholar] [CrossRef] [PubMed]
- Wilmshurst, J.; Lillis, S.; Zhou, H.; Pillay, K.; Henderson, H.; Kress, W.; Müller, C.; Ndondo, A.; Cloke, V.; Cullup, T.; et al. RYR1 mutations are a common cause of congenital myopathies with central nuclei. Ann. Neurol. 2010, 68, 717–726. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: Ajoint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Lawal, T.A.; Wires, E.S.; Terry, N.L.; Dowling, J.J.; Todd, J.J. Preclinical model systems of ryanodine receptor 1-related myopathies and malignant hyperthermia: A comprehensive scoping review of works published 1990–2019. Orphanet J. Rare Dis. 2020, 15, 113–135. [Google Scholar] [CrossRef]
- Knuiman, G.J.; Küsters, B.; Eshuis, L.; Snoeck, M.; Lammens, M.; Heytens, L.; De Ridder, W.; Baets, J.; Scalco, R.S.; Quinlivan, R.; et al. The histopathological spectrum of malignant hyperthermia and rhabdomyolysis due to RYR1 mutations. J. Neurol. 2019, 266, 876–887. [Google Scholar] [CrossRef] [Green Version]
- Shen, R.; Olshen, A.B.; Ladanyi, M. Integrative clustering of multiple genomic data types using a joint latent variable model with application to breast and lung cancer subtype analysis. Bioinformatics 2009, 25, 2906–2912. [Google Scholar] [CrossRef] [Green Version]
- Rubegni, A.; Malandrini, A.; Dosi, C.; Astrea, G.; Baldacci, J.; Battisti, C.; Bertocci, G.; Donati, M.A.; Dotti, M.T.; Federico, A.; et al. Next-generation sequencing approach to hyperCKemia: a2-year cohort study. Neurol. Genet. 2019, 5, e352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savarese, M.; Di Fruscio, G.; Mutarelli, M.; Torella, A.; Magri, F.; Santorelli, F.M.; Comi, G.P.; Bruno, C.; Nigro, V. MotorPlex provides accurate variant detection across large muscle genes both in single myopathic patients and in pools of DNA samples. Acta Neuropathol. Commun. 2014, 2, 100. [Google Scholar] [CrossRef] [PubMed]
- D’Amore, A.; Tessa, A.; Casali, C.; Dotti, M.T.; Filla, A.; Silvestri, G.; Antenora, A.; Astrea, G.; Barghigiani, M.; Battini, R.; et al. Next generation molecular diagnosis of hereditary spastic paraplegias: An Italian cross-sectional study. Front. Neurol. 2018, 9, 981. [Google Scholar] [CrossRef] [Green Version]
- Kircher, M.; Witten, D.M.; Jain, P.; O‘Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galatolo, D.; Kuo, M.E.; Mullen, P.; Meyer-Schuman, R.; Doccini, S.; Battini, R.; Lieto, M.; Tessa, A.; Filla, A.; Francklyn, C.; et al. Bi-allelic mutations in HARS1 severely impair histidyl-tRNA synthetase expression and enzymatic activity causing a novel multisystem ataxic syndrome. Hum. Mutat. 2020, 41, 1232–1237. [Google Scholar] [CrossRef]
- Cox, T.F.; Cox, M.A.A. Multidimensional Scaling; Chapman and Hall: London, UK, 2001. [Google Scholar]
- Coxon, A.P.M.; Jackson, J.E. The User’s Guide to Multidimensional Scaling: With Special Reference to the MDS(X) Library of Computer Programs. Heinemann Educational Books. 1982. Available online: https://www.tandfonline.com/doi/epdf/10.1080/00401706.1985.10488019 (accessed on 3 November 2022).
- Tan, P.N.; Steinbach, M.K. Introduction to Data Mining, 2nd ed.; Pearson: London, UK, 2018. [Google Scholar]
- Li, P.; Ding, Y.-G.; Yao, P.-P.; Xue, K.-M.; Li, C.-M. Study on high-temperature flow behavior and substructure and texture evolution of TA15 titanium alloy. J. Mater. Eng. Perform. 2016, 25, 3439–3447. [Google Scholar] [CrossRef]
- Oksanen, J.; Simpson, G.L.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’Hara, R.B.; Solymos, P.; Stevens, M.H.H.; Szoecs, E.; et al. Community Ecology Package: Ordination, diversity and dissimilarities. Community Ecol. Packag. 2019, 2, 1–297. Available online: https://cran.r-project.org/web/packages/vegan/vegan.pdf (accessed on 3 November 2022).
- Lawal, T.A.; Todd, J.J.; Meilleur, K.G. Ryanodine receptor 1-related myopathies: Diagnostic and therapeutic approaches. Neurotherapeutics 2018, 15, 885–899. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Jungbluth, H.; Sewry, C.A.; Feng, L.; Bertini, E.; Bushby, K.; Straub, V.; Roper, H.; Rose, M.R.; Brockington, M.; et al. Molecular mechanisms and phenotypic variation in RYR1-related congenital myopathies. Brain 2007, 130, 2024–2036. [Google Scholar] [CrossRef] [Green Version]
- Savarese, M.; Di Fruscio, G.; Torella, A.; Fiorillo, C.; Magri, F.; Fanin, M.; Ruggiero, L.; Ricci, G.; Astrea, G.; Passamano, L.; et al. The genetic basis of undiagnosed muscular dystrophies and myopathies. Results from 504 patients. Neurology 2016, 87, 71–76. [Google Scholar] [CrossRef] [Green Version]
- Alkhunaizi, E.; Shuster, S.; Shannon, P.; Siu, V.M.; Darilek, S.; Mohila, C.A.; Boissel, S.; Ellezam, B.; Fallet-Bianco, C.; Laberge, A.; et al. Homozygous/compound heterozygote RYR1 gene variants: Expanding the clinical spectrum. Am. J. Med. Genet. Part A 2019, 179, 386–396. [Google Scholar] [CrossRef]
- Todd, J.J.; Razaqyar, M.; Witherspoon, J.W.; Lawal, T.A.; Mankodi, A.; Chrismer, I.C.; Allen, C.; Meyer, M.D.; Kuo, A.; Shelton, M.S.; et al. Novel variants in individuals with RYR1-related congenital myopathies: Genetic, laboratory, and clinical findings. Front. Neurol. 2018, 9, 118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snoeck, M.; van Engelen, B.G.M.; Küsters, B.; Lammens, M.; Meijer, R.; Molenaar, J.P.F.; Raaphorst, J.; Verschuuren-Bemelmans, C.C.; Straathof, C.S.M.; Sie, L.T.L.; et al. RYR1-related myopathies: Awide spectrum of phenotypes throughout life. Eur. J. Neurol. 2015, 22, 1094–1112. [Google Scholar] [CrossRef]
- Dlamini, N.; Voermans, N.; Lillis, S.; Stewart, K.; Kamsteeg, E.-J.; Drost, G.; Quinlivan, R.; Snoeck, M.; Norwood, F.; Radunovic, A.; et al. Mutations in RYR1 are a common cause of exertional myalgia and rhabdomyolysis. Neuromuscul. Disord. 2013, 23, 540–548. [Google Scholar] [CrossRef]
- Haugarvoll, K.; Johansson, S.; Tzoulis, C.; Haukanes, B.I.; Bredrup, C.; Neckelmann, G.; Boman, H.; Knappskog, P.M.; Bindoff, L.A. Genotype-phenotype correlations in recessive RYR1-related myopathies. Orphanet J. Rare Dis. 2013, 8, 117. [Google Scholar] [CrossRef] [Green Version]
- Klein, A.; Lillis, S.; Munteanu, I.; Scoto, M.; Zhou, H.; Quinlivan, R.; Straub, V.; Manzur, A.Y.; Roper, H.; Jeannet, P.-Y.; et al. Clinical and genetic findings in a large cohort of patients with ryanodine receptor 1 gene-associated myopathies. Hum. Mutat. 2012, 33, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Fusto, A.; Cassandrini, D.; Fiorillo, C.; Codemo, V.; Astrea, G.; D’Amico, A.; Maggi, L.; Magri, F.; Pane, M.; Tasca, G.; et al. Expanding the clinical-pathological and genetic spectrum of RYR1-related congenital myopathies with cores and minicores: An Italian population study. Acta Neuropathol. Commun. 2022, 10, 54. [Google Scholar] [CrossRef] [PubMed]
- Ma, E.Y.; Kim, J.W.; Lee, Y.; Cho, S.W.; Kim, H.; Kim, J.K. Combined unsupervised-supervised machine learning for phenotyping complex diseases with its application to obstructive sleep apnea. Sci. Rep. 2021, 11, 4457. [Google Scholar] [CrossRef]
- Chemello, F.; Grespi, F.; Zulian, A.; Cancellara, P.; Hebert-Chatelain, E.; Martini, P.; Bean, C.; Alessio, E.; Buson, L.; Bazzega, M.; et al. Transcriptomic Analysis of Single Isolated Myofibers Identifies miR-27a-3p and miR-142-3p as Regulators of Metabolism in Skeletal Muscle. Cell Rep. 2019, 26, 3784–3797.e8. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dosi, C.; Rubegni, A.; Baldacci, J.; Galatolo, D.; Doccini, S.; Astrea, G.; Berardinelli, A.; Bruno, C.; Bruno, G.; Comi, G.P.; et al. Using Cluster Analysis to Overcome the Limits of Traditional Phenotype–Genotype Correlations: The Example of RYR1-Related Myopathies. Genes 2023, 14, 298. https://doi.org/10.3390/genes14020298
Dosi C, Rubegni A, Baldacci J, Galatolo D, Doccini S, Astrea G, Berardinelli A, Bruno C, Bruno G, Comi GP, et al. Using Cluster Analysis to Overcome the Limits of Traditional Phenotype–Genotype Correlations: The Example of RYR1-Related Myopathies. Genes. 2023; 14(2):298. https://doi.org/10.3390/genes14020298
Chicago/Turabian StyleDosi, Claudia, Anna Rubegni, Jacopo Baldacci, Daniele Galatolo, Stefano Doccini, Guja Astrea, Angela Berardinelli, Claudio Bruno, Giorgia Bruno, Giacomo Pietro Comi, and et al. 2023. "Using Cluster Analysis to Overcome the Limits of Traditional Phenotype–Genotype Correlations: The Example of RYR1-Related Myopathies" Genes 14, no. 2: 298. https://doi.org/10.3390/genes14020298