

Miyoshi Muscular Dystrophy Type 1 with Mutated DYSF Gene Misdiagnosed as Becker Muscular Dystrophy: A Case Report and Literature Review

Abstract
:1. Introduction
2. Materials and Methods
2.1. Multiplex Ligation-Dependent Probe Amplification
2.2. Targeted Panel Sequencing
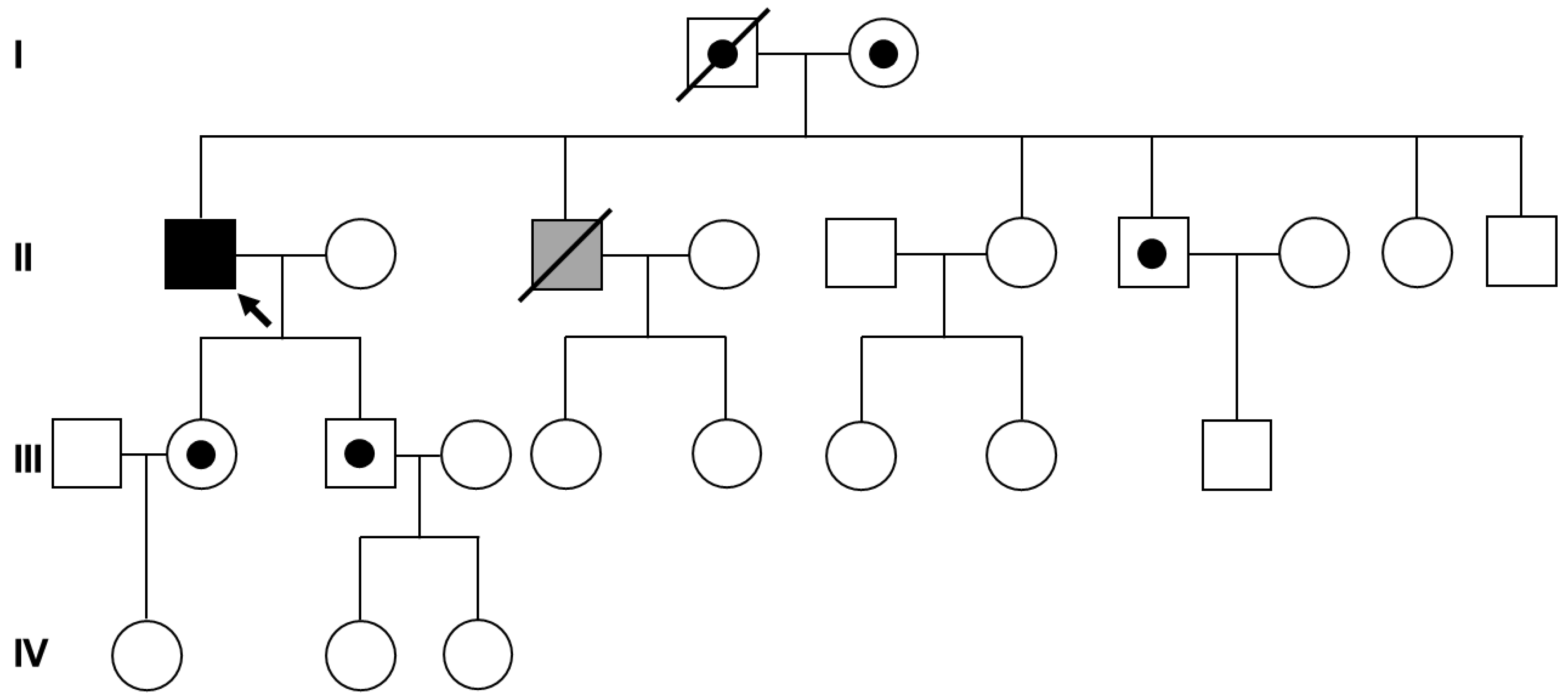
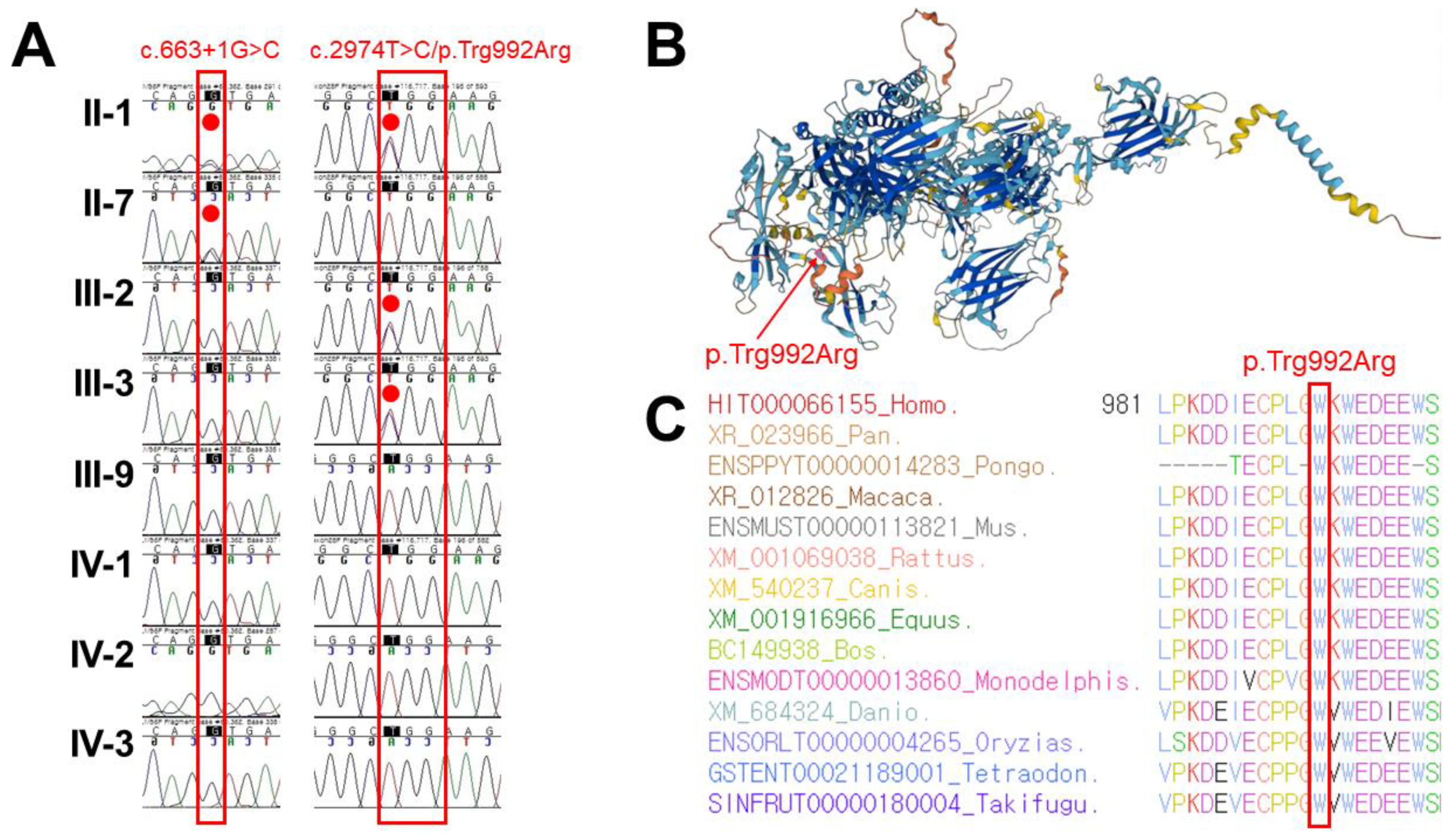
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Miyoshi, K.; Kawai, H.; Iwasa, M.; Kusaka, K.; Nishino, H. Autosomal recessive distal muscular dystrophy as a new type of progressive muscular dystrophy: Seventeen cases in eight families including an autopsied case. Brain 1986, 109 Pt 1, 31–54. [Google Scholar] [CrossRef]
- Buermans, H.P.; den Dunnen, J.T. Next generation sequencing technology: Advances and applications. Biochim. Biophys. Acta 2014, 1842, 1932–1941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, K.M.; Bhatt, A.D.; Shah, K.; Waghela, B.N.; Pandit, R.J.; Sheth, H.; Joshi, C.G.; Joshi, M.N. Molecular Diagnosis of Muscular Dystrophy Patients in Western Indian Population: A Comprehensive Mutation Analysis Using Amplicon Sequencing. Front. Genet. 2021, 12, 770350. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Quereda, L.; Rodriguez, M.J.; Diaz-Manera, J.; Alonso-Perez, J.; Gallardo, E.; Nascimento, A.; Ortez, C.; Natera-de Benito, D.; Olive, M.; Gonzalez-Mera, L.; et al. Targeted Next-Generation Sequencing in a Large Cohort of Genetically Undiagnosed Patients with Neuromuscular Disorders in Spain. Genes 2020, 11, 539. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Tan, D.; Song, D.; Zhang, X.; Chang, X.; Wang, Z.; Zhang, C.; Chan, S.H.; Wu, Q.; Wu, L.; et al. Clinical spectrum and genetic variations of LMNA-related muscular dystrophies in a large cohort of Chinese patients. J. Med. Genet. 2021, 58, 326–333. [Google Scholar] [CrossRef]
- Oh, S.H.; Kim, T.S.; Choi, Y.C. Identification of a dysferlin gene mutation in a Korean case with Miyoshi myopathy. Yonsei Med. J. 2004, 45, 927–930. [Google Scholar] [CrossRef]
- Cho, H.J.; Sung, D.H.; Kim, E.J.; Yoon, C.H.; Ki, C.S.; Kim, J.W. Clinical and genetic analysis of Korean patients with Miyoshi myopathy: Identification of three novel mutations in the DYSF gene. J. Korean Med. Sci. 2006, 21, 724–727. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.E.; Kim, H.S.; Lee, C.H.; Nam, T.S.; Choi, Y.C.; Kim, D.S. Two common mutations (p.Gln832X and c.663 + 1G > C) account for about a third of the DYSF mutations in Korean patients with dysferlinopathy. Neuromuscul. Disord. NMD 2012, 22, 505–510. [Google Scholar] [CrossRef]
- Shin, H.Y.; Jang, H.; Han, J.H.; Park, H.J.; Lee, J.H.; Kim, S.W.; Kim, S.M.; Park, Y.E.; Kim, D.S.; Bang, D.; et al. Targeted next-generation sequencing for the genetic diagnosis of dysferlinopathy. Neuromuscul. Disord. NMD 2015, 25, 502–510. [Google Scholar] [CrossRef]
- Seong, M.W.; Cho, A.; Park, H.W.; Seo, S.H.; Lim, B.C.; Seol, D.; Cho, S.I.; Park, S.S.; Chae, J.H. Clinical applications of next-generation sequencing-based gene panel in patients with muscular dystrophy: Korean experience. Clin. Genet. 2016, 89, 484–488. [Google Scholar] [CrossRef]
- Park, H.J.; Jang, H.; Kim, J.H.; Lee, J.H.; Shin, H.Y.; Kim, S.M.; Park, K.D.; Yim, S.V.; Lee, J.H.; Choi, Y.C. Discovery of pathogenic variants in a large Korean cohort of inherited muscular disorders. Clin. Genet. 2017, 91, 403–410. [Google Scholar] [CrossRef]
- Lee, S.J.; Choi, E.; Shin, S.; Park, J. Genetically confirmed limb-girdle muscular dystrophy type 2B with DYSF mutation using gene panel sequencing: A case report. Medicine 2020, 99, e20810. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Izumi, R.; Takahashi, T.; Suzuki, N.; Niihori, T.; Ono, H.; Nakamura, N.; Katada, S.; Kato, M.; Warita, H.; Tateyama, M.; et al. The genetic profile of dysferlinopathy in a cohort of 209 cases: Genotype-phenotype relationship and a hotspot on the inner DysF domain. Hum. Mutat. 2020, 41, 1540–1554. [Google Scholar] [CrossRef]
- Rosales, X.Q.; Gastier-Foster, J.M.; Lewis, S.; Vinod, M.; Thrush, D.L.; Astbury, C.; Pyatt, R.; Reshmi, S.; Sahenk, Z.; Mendell, J.R. Novel diagnostic features of dysferlinopathies. Muscle Nerve 2010, 42, 14–21. [Google Scholar] [CrossRef]
- Felice, K.J. Distal Myopathies. Neurol. Clin. 2020, 38, 637–659. [Google Scholar] [CrossRef]
- Savarese, M.; Sarparanta, J.; Vihola, A.; Jonson, P.H.; Johari, M.; Rusanen, S.; Hackman, P.; Udd, B. Panorama of the distal myopathies. Acta Myol. 2020, 39, 245–265. [Google Scholar] [CrossRef]
- Park, H.J.; Hong, J.M.; Suh, G.I.; Shin, H.Y.; Kim, S.M.; Sunwoo, I.N.; Suh, B.C.; Choi, Y.C. Heterogeneous characteristics of Korean patients with dysferlinopathy. J. Korean Med. Sci. 2012, 27, 423–429. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Guo, Y.; Fu, Y.; Jia, R.; Chen, G. Atypical Miyoshi distal myopathy: A case report. Exp. Ther. Med. 2016, 12, 3068–3072. [Google Scholar] [CrossRef]
- Xu, C.; Chen, J.; Zhang, Y.; Li, J. Limb-girdle muscular dystrophy type 2B misdiagnosed as polymyositis at the early stage: Case report and literature review. Medicine 2018, 97, e10539. [Google Scholar] [CrossRef] [PubMed]
- Contreras-Cubas, C.; Barajas-Olmos, F.; Frayre-Martínez, M.I.; Siordia-Reyes, G.; Guízar-Sánchez, C.C.; García-Ortiz, H.; Orozco, L.; Baca, V. Dysferlinopathy misdiagnosed with juvenile polymyositis in the pre-symptomatic stage of hyperCKemia: A case report and literature review. BMC Med. Genom. 2022, 15, 139. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, E.; Rojas-García, R.; de Luna, N.; Pou, A.; Brown, R.H., Jr.; Illa, I. Inflammation in dysferlin myopathy: Immunohistochemical characterization of 13 patients. Neurology 2001, 57, 2136–2138. [Google Scholar] [CrossRef] [PubMed]
- Fanin, M.; Angelini, C. Muscle pathology in dysferlin deficiency. Neuropathol. Appl. Neurobiol. 2002, 28, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Rowin, J.; Meriggioli, M.N.; Cochran, E.J.; Sanders, D.B. Prominent inflammatory changes on muscle biopsy in patients with Miyoshi myopathy. Neuromuscul. Disord. NMD 1999, 9, 417–420. [Google Scholar] [CrossRef]
- Klinge, L.; Aboumousa, A.; Eagle, M.; Hudson, J.; Sarkozy, A.; Vita, G.; Charlton, R.; Roberts, M.; Straub, V.; Barresi, R.; et al. New aspects on patients affected by dysferlin deficient muscular dystrophy. J. Neurol. Neurosurg. Psychiatry 2010, 81, 946–953. [Google Scholar] [CrossRef]
- Rawat, R.; Cohen, T.V.; Ampong, B.; Francia, D.; Henriques-Pons, A.; Hoffman, E.P.; Nagaraju, K. Inflammasome up-regulation and activation in dysferlin-deficient skeletal muscle. Am. J. Pathol. 2010, 176, 2891–2900. [Google Scholar] [CrossRef]
- Sula, A.; Cole, A.R.; Yeats, C.; Orengo, C.; Keep, N.H. Crystal structures of the human Dysferlin inner DysF domain. BMC Struct. Biol. 2014, 14, 3. [Google Scholar] [CrossRef] [Green Version]
- Aartsma-Rus, A.; Van Deutekom, J.C.; Fokkema, I.F.; Van Ommen, G.J.; Den Dunnen, J.T. Entries in the Leiden Duchenne muscular dystrophy mutation database: An overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve 2006, 34, 135–144. [Google Scholar] [CrossRef]
- Ponting, C.P.; Mott, R.; Bork, P.; Copley, R.R. Novel protein domains and repeats in Drosophila melanogaster: Insights into structure, function, and evolution. Genome Res. 2001, 11, 1996–2008. [Google Scholar] [CrossRef]
- Therrien, C.; Di Fulvio, S.; Pickles, S.; Sinnreich, M. Characterization of lipid binding specificities of dysferlin C2 domains reveals novel interactions with phosphoinositides. Biochemistry 2009, 48, 2377–2384. [Google Scholar] [CrossRef]
- Patel, P.; Harris, R.; Geddes, S.M.; Strehle, E.M.; Watson, J.D.; Bashir, R.; Bushby, K.; Driscoll, P.C.; Keep, N.H. Solution structure of the inner DysF domain of myoferlin and implications for limb girdle muscular dystrophy type 2b. J. Mol. Biol. 2008, 379, 981–990. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Parameters | Results | Normal Range | Parameters | Results | Normal Range |
|---|---|---|---|---|---|
| BMI (kg/m2) | 25.7 | 18.5–25 | Albumin (g/dL) | 4.6 | 3.5–5.2 |
| WBC (×103/μL) | 7.29 | 4.0–10.0 | Total Bilirubin (mg/dL) | 0.89 | 0–1.2 |
| RBC (×106/μL) | 5.02 | 4.2–6.3 | ALP (IU/L) | 117 | 35–130 |
| Hemoglobin (g/dL) | 15.5 | 13.0–17.0 | AST (IU/L) | 32 | 0–37 |
| Platelet (×103/μL) | 197 | 130–400 | ALT (IU/L) | 26 | 0–41 |
| BUN (mg/dL) | 9.8 | 6–20 | γ–GT (IU/L) | 43 | 6–71 |
| Creatinine (mg/dL) | 0.18 | 0.6–1.2 | LDH (IU/L) | 203 | 135–225 |
| Ca (mg/dL) | 9.4 | 8.6–10.2 | Creatine kinase (IU/L) | 352 | 50–250 |
| P (mg/dL) | 2.8 | 2.7–4.5 | Triglyceride (mg/dL) | 127 | 0–200 |
| Uric acid (mg/dL) | 5.6 | 3.4–7.0 | Cholesterol (mg/dL) | 115 | 120–220 |
| Glucose (mg/dL) | 109 | 70–110 | HDL (mg/dL) | 41 | 35–75 |
| HbA1c (%) | 5.1 | 4.2–5.9 | LDL (mg/dL) | 62 | 0–130 |
| Base Change | Codon Change | Reported VAF | rsID | gnomAD | KRGDB | References |
|---|---|---|---|---|---|---|
| c.75del | p.Ala26Argfs * 6 | 1 | N/A | 0 | 0 | [9] |
| c.313dup | p.Leu105Profs * 43 | 1 | N/A | 0 | 0 | [9] |
| c.610C > T | p.Arg204 * | 1 | rs373585652 | 0.000007953 | [9] | |
| c.663 + 1G > C | Splicing error | 19 | rs398123800 | 0.000003978 | 0.000455 | [8,9,10,11] |
| c.675G > T | p.Gln225His | 1 | N/A | 0 | 0 | [8] |
| c.757C > T | p.Arg253Trp | 1 | rs149827237 | 0.0001233 | [8] | |
| c.823del | p.Glu276Serfs * 12 | 2 | N/A | 0 | 0 | [8,11] |
| c.845T > C | p.Ile282Thr | 3 | N/A | 0 | 0 | [8,9] |
| c.895G > A | p.Gly299Arg | 1 | rs121908963 | 0.00003186 * | [9] | |
| c.937 + 1G > A | Splicing error | 5 | rs201869739 | 0.00003579 | [9,10,11] | |
| c.938-1G > A | Splicing error | 1 | N/A | 0 | 0 | [9] |
| c.1053T > G | p.(=) | 1 | rs199955501 | 0.0002864 | [8] | |
| c.1129C > T | p.Arg377 * | 2 | rs758180890 | 0.00002387 | [9,10] | |
| c.1165G > C | p.Glu389Gln | 3 | N/A | 0 | 0 | [6,8] |
| c.1284 + 2T > C | Splicing error | 20 | rs398123765 | 0.00001596 | 0 | [8,9,10,11] |
| c.1464del | p.Gly489Glufs *4 | 5 | N/A | 0 | 0 | [8,9,10] |
| c.1579G > T | p.Gly527Cys | 1 | N/A | 0 | 0 | [8] |
| c.1646del | p.Gly549Valfs * 78 | 1 | N/A | 0 | 0 | [11] |
| c.1663C > T | p.Arg555Trp | 1 | rs377735262 | 0.00002916 | 0 | [12] |
| c.1665G > C | p.(=) | 1 | N/A | 0 | 0 | [11] |
| c.2248C > T | p.Gln750 * | 1 | N/A | 0 | 0 | [8] |
| c.2494C > T | p.Gln832 * | 28 | rs199543257 | 0.00001988 | 0.000455 | [7,8,9,10,11] |
| c.2964C > A | p.Cys988 * | 1 | N/A | 0 | 0 | [8] |
| c.2974T > C | p.Trp992Arg | 3 | rs750028300 | 0.000003976 | 0.000455 | [7,9,10] |
| c.2997G > T | p.Trp999Cys | 11 | rs28937581 | 0.00001193 | 0.001818 | [7,8,9,10,11] [12] |
| c.3032-1G > A | Splicing error | 2 | N/A | 0 | 0 | [8] |
| c.3102C > G | p.Tyr1034 * | 1 | N/A | 0 | 0 | [9] |
| c.3113G > A | p.Arg1038Gln | 2 | rs150877497 | 0.00005186 | [8,11] | |
| c.3275G > A | p.Arg1092His | 2 | rs758284713 | 0.00009168 | [8] | |
| c.3276_3281dup | p.Arg1093_Trp1094insCysArg | 1 | rs758284713 | 0.000003986 | [9] | |
| c.3289C > T | p.Arg1097Cys | 1 | rs147483765 | 0.0003622 | [8] | |
| c.3307A > T | p.Lys1103 * | 1 | N/A | 0 | 0 | [7] |
| c.3407del | p.Gly1136Valfs * 2 | 2 | rs778088008 | 0.000003976 | [8,9] | |
| c.3875_3880del | p.Val1292_Gln1293del | 1 | N/A | 0 | 0 | [9] |
| c.4200dup | p.Ile1401Hisfs * 8 | 1 | N/A | 0 | 0 | [7] |
| c.4434G > A | p.Trp1478 * | 1 | rs766016391 | 0.000004045 | [8] | |
| c.4742G > A | p.Arg1581His | 1 | rs185596534 | 0.0005567 | [8] | |
| c.4795-2A > G | Splicing error | 1 | N/A | 0 | 0 | [8] |
| c.5078G > A | p.Arg1693Gln | 2 | rs779987458 | 0.000008007 | [8,10] | |
| c.5090G > C | p.Arg1697Pro | 1 | rs777701226 | 0.00002806 | [8] | |
| c.5095T > C | p.Ser1699Pro | 1 | N/A | 0 | 0 | [8] |
| c.5287G > A | p.Glu1763Lys | 1 | N/A | 0 | 0 | [10] |
| c.5607dup | p.Arg1870Glufs * 12 | 2 | N/A | 0 | 0 | [8,9] |
| c.5776C > T | p.Gln1926 * | 1 | N/A | 0 | 0 | [9] |
| c.5804C > T | p.Pro1935Leu | 1 | rs1254719758 | 0.00003185 | [10] | |
| c.5911T > C | p.Cys1971Arg | 1 | N/A | 0 | 0 | [8] |
| c.5939T > A | p.Ile1980Lys | 1 | N/A | 0 | 0 | [9] |
| c.6057-2A > C | Splicing error | 2 | N/A | 0 | 0 | [11] |
| c.6096C > G | p.Tyr2032 * | 1 | rs1012723902 | 0.000003976 | [8] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, J.; Moon, Y.J.; Kim, D.S. Miyoshi Muscular Dystrophy Type 1 with Mutated DYSF Gene Misdiagnosed as Becker Muscular Dystrophy: A Case Report and Literature Review. Genes 2023, 14, 200. https://doi.org/10.3390/genes14010200
Park J, Moon YJ, Kim DS. Miyoshi Muscular Dystrophy Type 1 with Mutated DYSF Gene Misdiagnosed as Becker Muscular Dystrophy: A Case Report and Literature Review. Genes. 2023; 14(1):200. https://doi.org/10.3390/genes14010200
Chicago/Turabian StylePark, Joonhong, Young Jae Moon, and Dal Sik Kim. 2023. "Miyoshi Muscular Dystrophy Type 1 with Mutated DYSF Gene Misdiagnosed as Becker Muscular Dystrophy: A Case Report and Literature Review" Genes 14, no. 1: 200. https://doi.org/10.3390/genes14010200