

Distinct Roles of Histone Lysine Demethylases and Methyltransferases in Developmental Eye Disease
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
3. Results
3.1. Genetic Variants Identified in Histone Lysine Methyltransferases Genes, KMT2D, KMT2C and SETD1A (also Known as KMT2F)
3.2. Genetic Variants Identified in Histone Lysine Demethylase Genes, KDM6A and KDM5C
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lavery, W.J.; Barski, A.; Wiley, S.; Schorry, E.K.; Lindsley, A.W. KMT2C/D COMPASS complex-associated diseases [K(CD)COM-ADs]: An emerging class of congenital regulopathies. Clin. Epigenetics 2020, 12, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boniel, S.; Szymanska, K.; Smigiel, R.; Szczaluba, K. Kabuki Syndrome-Clinical Review with Molecular Aspects. Genes 2021, 12, 468. [Google Scholar] [CrossRef] [PubMed]
- Barry, K.K.; Tsaparlis, M.; Hoffman, D.; Hartman, D.; Adam, M.P.; Hung, C.; Bodamer, O.A. From Genotype to Phenotype-A Review of Kabuki Syndrome. Genes 2022, 13, 1761. [Google Scholar] [CrossRef] [PubMed]
- Siano, M.A.; De Maggio, I.; Petillo, R.; Cocciadiferro, D.; Agolini, E.; Majolo, M.; Novelli, A.; Della Monica, M.; Piscopo, C. De Novo Mutation in KMT2C Manifesting as Kleefstra Syndrome 2: Case Report and Literature Review. Pediatr. Rep. 2022, 14, 131–139. [Google Scholar] [CrossRef]
- Kummeling, J.; Stremmelaar, D.E.; Raun, N.; Reijnders, M.R.F.; Willemsen, M.H.; Ruiterkamp-Versteeg, M.; Schepens, M.; Man, C.C.O.; Gilissen, C.; Cho, M.T.; et al. Characterization of SETD1A haploinsufficiency in humans and Drosophila defines a novel neurodevelopmental syndrome. Mol. Psychiatry 2021, 26, 2013–2024. [Google Scholar] [CrossRef]
- Jensen, L.R.; Amende, M.; Gurok, U.; Moser, B.; Gimmel, V.; Tzschach, A.; Janecke, A.R.; Tariverdian, G.; Chelly, J.; Fryns, J.P.; et al. Mutations in the JARID1C gene, which is involved in transcriptional regulation and chromatin remodeling, cause X-linked mental retardation. Am. J. Hum. Genet. 2005, 76, 227–236. [Google Scholar] [CrossRef] [Green Version]
- Carmignac, V.; Nambot, S.; Lehalle, D.; Callier, P.; Moortgat, S.; Benoit, V.; Ghoumid, J.; Delobel, B.; Smol, T.; Thuillier, C.; et al. Further delineation of the female phenotype with KDM5C disease causing variants: 19 new individuals and review of the literature. Clin. Genet. 2020, 98, 43–55. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Kars, M.E.; Basak, A.N.; Onat, O.E.; Bilguvar, K.; Choi, J.; Itan, Y.; Caglar, C.; Palvadeau, R.; Casanova, J.L.; Cooper, D.N.; et al. The genetic structure of the Turkish population reveals high levels of variation and admixture. Proc. Natl. Acad. Sci. USA 2021, 118, e2026076118. [Google Scholar] [CrossRef]
- Fattahi, Z.; Beheshtian, M.; Mohseni, M.; Poustchi, H.; Sellars, E.; Nezhadi, S.H.; Amini, A.; Arzhangi, S.; Jalalvand, K.; Jamali, P.; et al. Iranome: A catalog of genomic variations in the Iranian population. Hum. Mutat. 2019, 40, 1968–1984. [Google Scholar] [CrossRef]
- Deml, B.; Reis, L.M.; Lemyre, E.; Clark, R.D.; Kariminejad, A.; Semina, E.V. Novel mutations in PAX6, OTX2 and NDP in anophthalmia, microphthalmia and coloboma. Eur. J. Hum. Genet. 2016, 24, 535–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weh, E.; Reis, L.M.; Happ, H.C.; Levin, A.V.; Wheeler, P.G.; David, K.L.; Carney, E.; Angle, B.; Hauser, N.; Semina, E.V. Whole exome sequence analysis of Peters anomaly. Hum. Genet. 2014, 133, 1497–1511. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, R.; Takenouchi, T.; Uchida, K.; Sato, T.; Fukushima, H.; Yoshihashi, H.; Takahashi, T.; Tsubota, K.; Kosaki, K. Congenital corneal staphyloma as a complication of Kabuki syndrome. Am. J. Med. Genet. A 2012, 158A, 2000–2002. [Google Scholar] [CrossRef]
- Porntaveetus, T.; Abid, M.F.; Theerapanon, T.; Srichomthong, C.; Ohazama, A.; Kawasaki, K.; Kawasaki, M.; Suphapeetiporn, K.; Sharpe, P.T.; Shotelersuk, V. Expanding the Oro-Dental and Mutational Spectra of Kabuki Syndrome and Expression of KMT2D and KDM6A in Human Tooth Germs. Int. J. Biol. Sci. 2018, 14, 381–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shangguan, H.; Su, C.; Ouyang, Q.; Cao, B.; Wang, J.; Gong, C.; Chen, R. Kabuki syndrome: Novel pathogenic variants, new phenotypes and review of literature. Orphanet. J. Rare Dis. 2019, 14, 255. [Google Scholar] [CrossRef] [PubMed]
- Retterer, K.; Juusola, J.; Cho, M.T.; Vitazka, P.; Millan, F.; Gibellini, F.; Vertino-Bell, A.; Smaoui, N.; Neidich, J.; Monaghan, K.G.; et al. Clinical application of whole-exome sequencing across clinical indications. Genet. Med. Off. J. Am. Coll. Med. Genet. 2016, 18, 696–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brookes, E.; Laurent, B.; Ounap, K.; Carroll, R.; Moeschler, J.B.; Field, M.; Schwartz, C.E.; Gecz, J.; Shi, Y. Mutations in the intellectual disability gene KDM5C reduce protein stability and demethylase activity. Hum. Mol. Genet. 2015, 24, 2861–2872. [Google Scholar] [CrossRef]
- Reis, L.M.; Maheshwari, M.; Capasso, J.; Atilla, H.; Dudakova, L.; Thompson, S.; Zitano, L.; Lay-Son, G.; Lowry, R.B.; Black, J.; et al. Axenfeld-Rieger syndrome: More than meets the eye. J. Med. Genet. 2022. Online ahead of print. [Google Scholar] [CrossRef]
- Weh, E.; Reis, L.M.; Tyler, R.C.; Bick, D.; Rhead, W.J.; Wallace, S.; McGregor, T.L.; Dills, S.K.; Chao, M.C.; Murray, J.C.; et al. Novel B3GALTL mutations in classic Peters plus syndrome and lack of mutations in a large cohort of patients with similar phenotypes. Clin. Genet. 2014, 86, 142–148. [Google Scholar] [CrossRef]
- Wu, C.; Orozco, C.; Boyer, J.; Leglise, M.; Goodale, J.; Batalov, S.; Hodge, C.L.; Haase, J.; Janes, J.; Huss, J.W., 3rd; et al. BioGPS: An extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol. 2009, 10, R130. [Google Scholar] [CrossRef] [PubMed]
- Merdler-Rabinowicz, R.; Prat, D.; Pode-Shakked, B.; Abel, G.; Chorin, O.; Somech, R.; Raas-Rothschild, A. Ophthalmic manifestations in Kabuki (make-up) syndrome: A single-center pediatric cohort and systematic review of the literature. Eur. J. Med. Genet. 2021, 64, 104210. [Google Scholar] [CrossRef] [PubMed]
- Cuvertino, S.; Hartill, V.; Colyer, A.; Garner, T.; Nair, N.; Al-Gazali, L.; Canham, N.; Faundes, V.; Flinter, F.; Hertecant, J.; et al. A restricted spectrum of missense KMT2D variants cause a multiple malformations disorder distinct from Kabuki syndrome. Genet. Med. 2020, 22, 867–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwenty-Lara, J.; Nehl, D.; Borchers, A. The histone methyltransferase KMT2D, mutated in Kabuki syndrome patients, is required for neural crest cell formation and migration. Hum. Mol. Genet. 2020, 29, 305–319. [Google Scholar] [CrossRef]
- Shpargel, K.B.; Starmer, J.; Wang, C.; Ge, K.; Magnuson, T. UTX-guided neural crest function underlies craniofacial features of Kabuki syndrome. Proc. Natl. Acad. Sci. USA 2017, 114, E9046–E9055. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Jeong, Y.; Kwon, K.; Ismail, T.; Lee, H.K.; Kim, C.; Park, J.W.; Kwon, O.S.; Kang, B.S.; Lee, D.S.; et al. Physiological effects of KDM5C on neural crest migration and eye formation during vertebrate development. Epigenetics Chromatin 2018, 11, 72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindgren, A.M.; Hoyos, T.; Talkowski, M.E.; Hanscom, C.; Blumenthal, I.; Chiang, C.; Ernst, C.; Pereira, S.; Ordulu, Z.; Clericuzio, C.; et al. Haploinsufficiency of KDM6A is associated with severe psychomotor retardation, global growth restriction, seizures and cleft palate. Hum. Genet. 2013, 132, 537–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haargaard, B.; Fledelius, H.C. Down’s syndrome and early cataract. Br. J. Ophthalmol. 2006, 90, 1024–1027. [Google Scholar] [CrossRef] [Green Version]
- Anand, D.; Kakrana, A.; Siddam, A.D.; Huang, H.; Saadi, I.; Lachke, S.A. RNA sequencing-based transcriptomic profiles of embryonic lens development for cataract gene discovery. Hum. Genet. 2018, 137, 941–954. [Google Scholar] [CrossRef]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Chapman, M.; Evans, K.; Azevedo, L.; Hayden, M.; Heywood, S.; Millar, D.S.; Phillips, A.D.; et al. The Human Gene Mutation Database (HGMD((R))): Optimizing its use in a clinical diagnostic or research setting. Hum. Genet. 2020, 139, 1197–1207. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| ID | Gene | Location (hg19) | Variant Effect a | CADD Score | REVEL Score | gnomAD b | Read Depth | Inheritance | ACMG Criteria | Ocular | Cranio-Facial | Dental | Cardiac | GU | GI | Skeletal | DD/CI | Poor Growth | Additional Findings |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
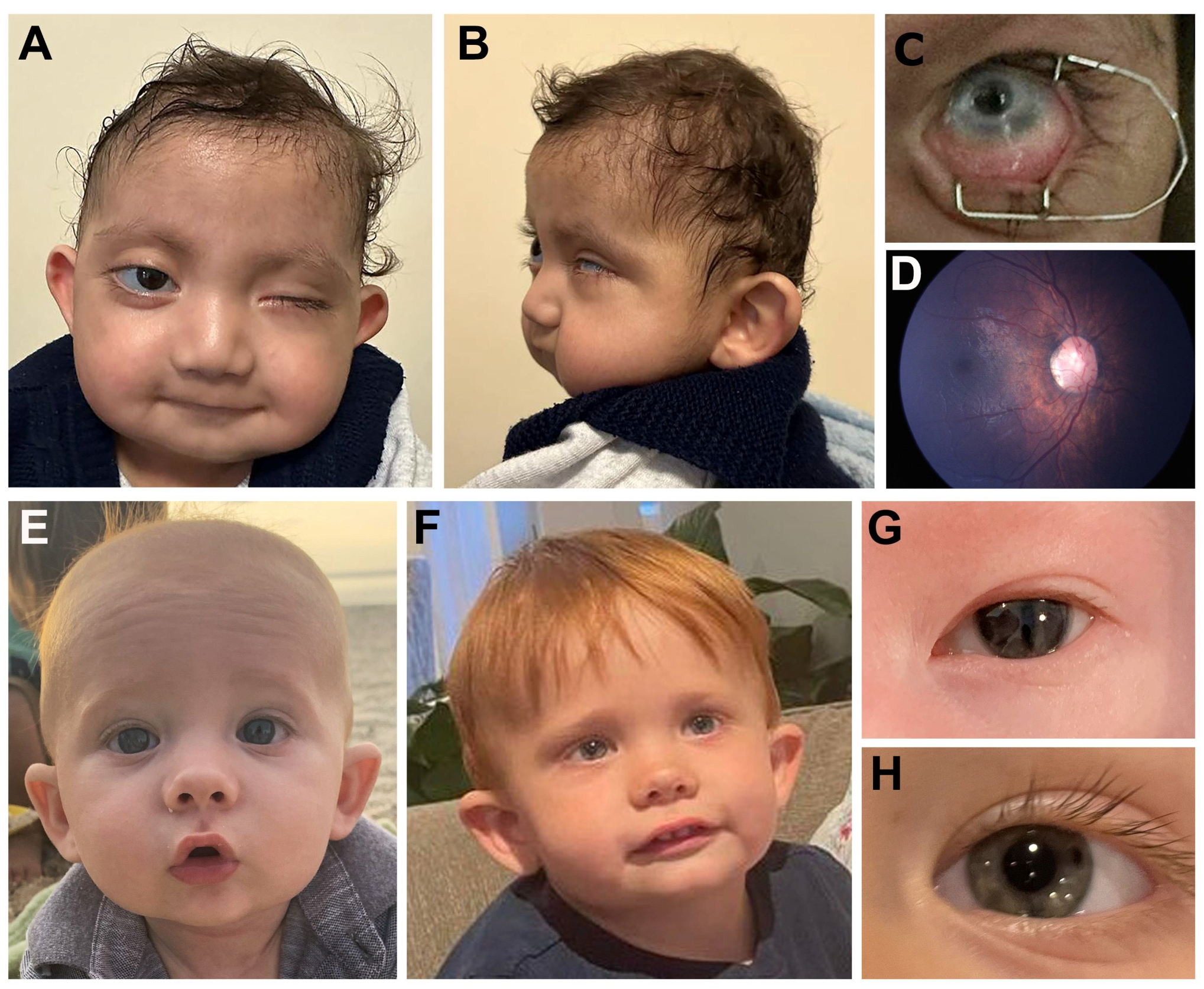
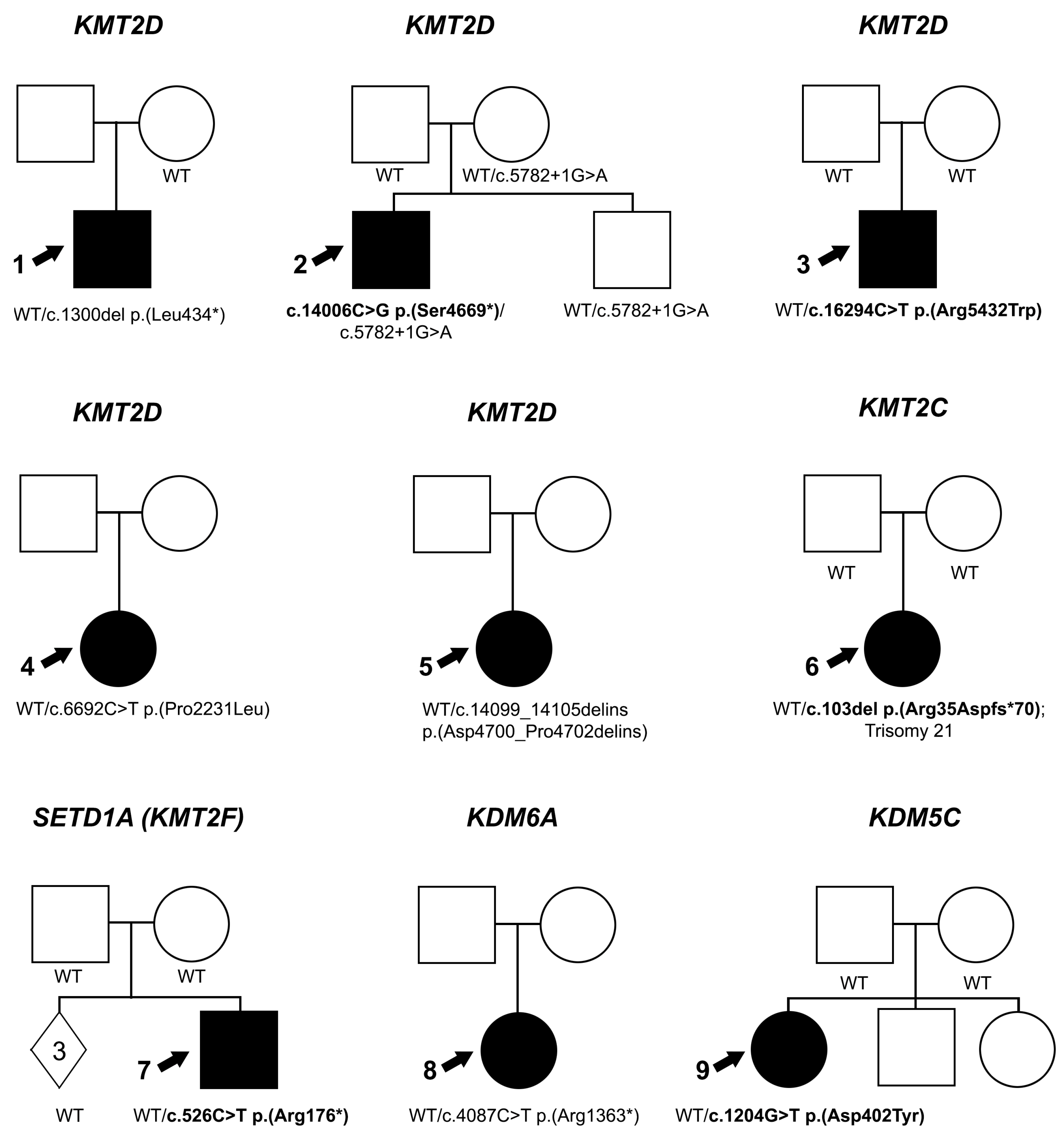
| Indiv 1 | KMT2D | 12:49446166-G- | c.1300del p.(L434*) | - | - | NP | 14/42 | NP in mother | LP (PVS1, PM2_S, PP5) | R PA, NYS and central retinal scar | + | + | + | + | + | + | + | + | Chronic lung disease |
| Indiv 2 | KMT2D | 12:49423253-G-C | c.14006C>G p.(S4669*) | 43 | - | NP | 35/72 | de novo | P (PVS1, PS2, PM2_S) | L MI, SC; R ON anomaly and retinal thinning | + | + | + | + | + | + | + | + | Hearing loss |
| KMT2D | 12:49436523-C-T | c.5782+1G>A p.(G1928_G1929insNTG) | - | - | 2/68,012 Eu | 33/59 | Inherited | LB (BS4, BP5) | |||||||||||
| Indiv 3 | KMT2D | 12:49416417-G-A | c.16294C>T p.(T5432W) | 33 | 0.907 | NP | 37/76 | de novo | P (PS1, PS2, PM2_S, PP3) | Iris hypoplasia | + | + | - | - | - | + | + | U | - |
| Indiv 4 | KMT2D | 12:49434861-G-A | c.6692C>T p.(P2231L) | 26.3 | 0.456 | NP | 21/34 | U | VUS (PM2_S) | L COL, MI | U | U | - | - | - | - | U | - | - |
| Indiv 5 | KMT2D | 12:49422990-GGAGAAT-AGAGAAC | c.14099_14105delATTCTCCinsGTTCTCT p.(N4700_P4702delinsGSL) | 29.3/32 | 0.71/0.565 | NP | 34/48 | U | VUS (PM2_S, PP3) | R mild MI, COL | U | U | + | - | - | - | U | - | - |
| Indiv 6 | KMT2C | 7:152132769-T- | c.103del p.(R35Nfs*70) | - | - | NP | 23/54 | de novo | P (PVS1, PS2, PM2_S) | CC, NYS, possible staphyloma | + | - | + | - | + | - | + | + | Trisomy 21, hearing loss, hypothyroidism |
| Indiv 7 | SETD1A (KMT2F) | 16:30974762-C-T | c.526C>T p.(R176*) | 37 | - | NP | 29/65 | de novo | P (PVS1, PS2, PM2_S) | PE, ICA, corectopia (resolved) | + | - | + | - | + | - | + | - | |
| Indiv 8 | KDM6A | X:44969405-C-T | c.4087C>T p.(R1363*) | 36 | - | NP | 75/179 | U | LP (PVS1, PM2_S) | B COL and NYS, L MI | U | - | - | - | - | - | + | + | Seizures |
| Indiv 9 | KDM5C | X:53241007-C-T | c.1204G>A p.(D402N) | 29.7 | 0.481 | NP | 35/64 | de novo | LP (PS2, PM2_S, PM5, PP2, PP5) | B PA, R GL | + | - | + | - | - | - | + | + | Pulmonary hypertension, dilated ventricles |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reis, L.M.; Atilla, H.; Kannu, P.; Schneider, A.; Thompson, S.; Bardakjian, T.; Semina, E.V. Distinct Roles of Histone Lysine Demethylases and Methyltransferases in Developmental Eye Disease. Genes 2023, 14, 216. https://doi.org/10.3390/genes14010216
Reis LM, Atilla H, Kannu P, Schneider A, Thompson S, Bardakjian T, Semina EV. Distinct Roles of Histone Lysine Demethylases and Methyltransferases in Developmental Eye Disease. Genes. 2023; 14(1):216. https://doi.org/10.3390/genes14010216
Chicago/Turabian StyleReis, Linda M., Huban Atilla, Peter Kannu, Adele Schneider, Samuel Thompson, Tanya Bardakjian, and Elena V. Semina. 2023. "Distinct Roles of Histone Lysine Demethylases and Methyltransferases in Developmental Eye Disease" Genes 14, no. 1: 216. https://doi.org/10.3390/genes14010216