
Defining a Correlative Transcriptional Signature Associated with Bulk Histone H3 Acetylation Levels in Adult Glioblastomas

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Processing of Human Samples for Western Blotting, RT-qPCR, and RNA-seq Assays
2.2. DNA Methylome Analysis
2.3. Primary Glioblastoma Cultures
2.4. Additional Statistical Analysis and Bioinformatics
3. Results
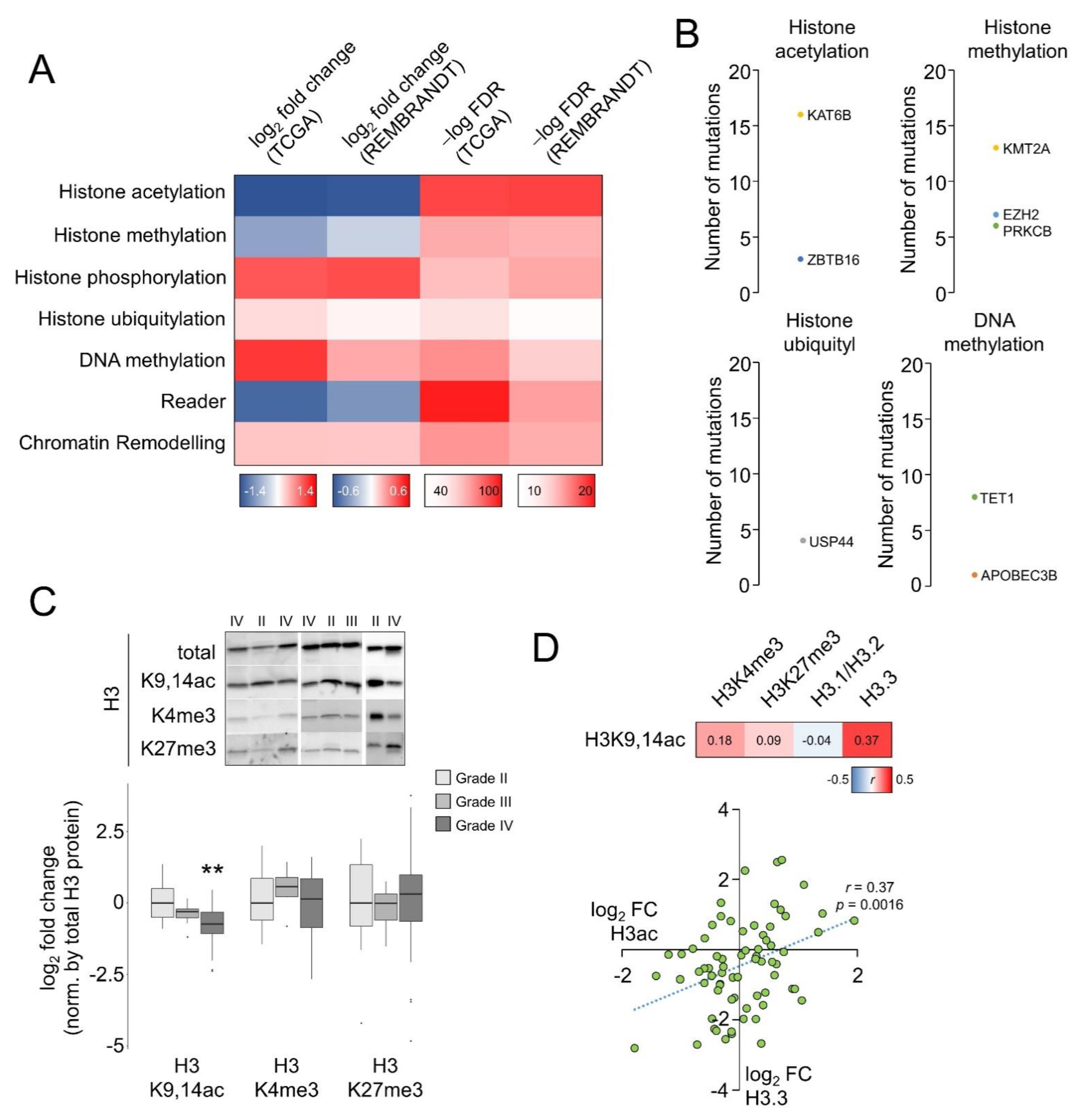
3.1. Histone Acetylation Levels Is Defective in Glioblastoma Compared to Lower-Grade Gliomas
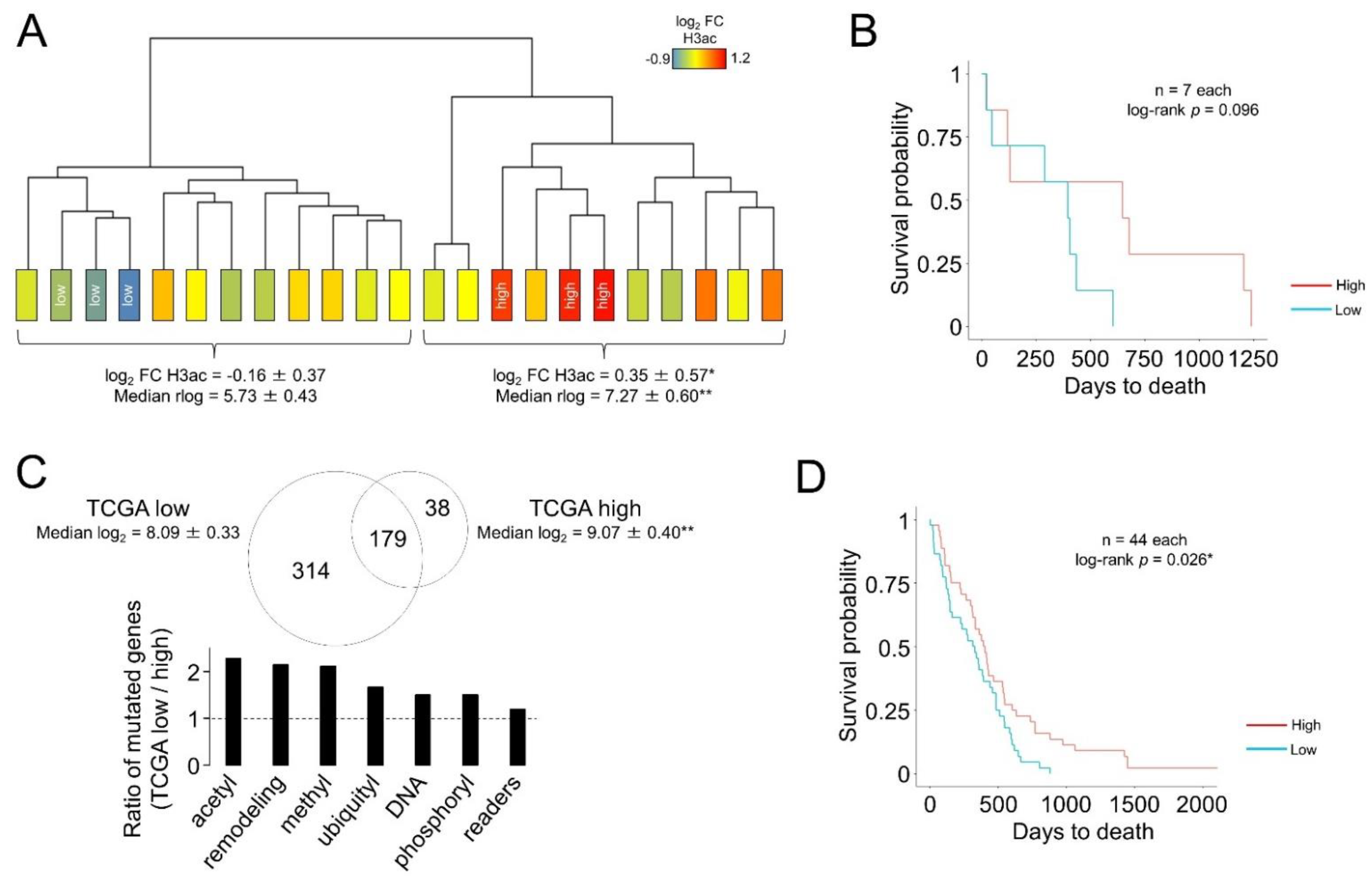
3.2. A Transcriptional Signature Can Be Correlated with Differential Bulk Levels of Histone Acetylation in Glioblastoma
3.3. The Transcriptional Signatures Associated with Histone Acetylation Can Be Linked to Differential Overall Survival Only in the Samples Showing the Most Extreme Expression Levels
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [Green Version]
- Valor, L.M.; Hervas-Corpion, I. The Epigenetics of Glioma Stem Cells: A Brief Overview. Front. Oncol. 2020, 10, 602378. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Stupp, R.; Reifenberger, G.; Brandes, A.A.; van den Bent, M.J.; Wick, W.; Hegi, M.E. MGMT promoter methylation in malignant gliomas: Ready for personalized medicine? Nat. Rev. Neurol. 2010, 6, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Turcan, S.; Rohle, D.; Goenka, A.; Walsh, L.A.; Fang, F.; Yilmaz, E.; Campos, C.; Fabius, A.W.; Lu, C.; Ward, P.S.; et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 2012, 483, 479–483. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Zhang, M.; Zhou, Y.; Guo, W.; Yi, M.; Zhang, Z.; Ding, Y.; Wang, Y. The application of histone deacetylases inhibitors in glioblastoma. J. Exp. Clin. Cancer Res. 2020, 39, 138. [Google Scholar] [CrossRef]
- Kim, M.S.; Blake, M.; Baek, J.H.; Kohlhagen, G.; Pommier, Y.; Carrier, F. Inhibition of histone deacetylase increases cytotoxicity to anticancer drugs targeting DNA. Cancer Res. 2003, 63, 7291–7300. [Google Scholar] [PubMed]
- Chinnaiyan, P.; Cerna, D.; Burgan, W.E.; Beam, K.; Williams, E.S.; Camphausen, K.; Tofilon, P.J. Postradiation sensitization of the histone deacetylase inhibitor valproic acid. Clin. Cancer Res. 2008, 14, 5410–5415. [Google Scholar] [CrossRef] [Green Version]
- Krauze, A.V.; Megan, M.; Theresa, C.Z.; Peter, M.; Shih, J.H.; Tofilon, P.J.; Rowe, L.; Gilbert, M.; Camphausen, K. The addition of Valproic acid to concurrent radiation therapy and temozolomide improves patient outcome: A Correlative analysis of RTOG 0525, SEER and a Phase II NCI trial. Cancer Stud. Ther. 2020, 5. [Google Scholar] [CrossRef] [Green Version]
- Kamitani, H.; Taniura, S.; Watanabe, K.; Sakamoto, M.; Watanabe, T.; Eling, T. Histone acetylation may suppress human glioma cell proliferation when p21 WAF/Cip1 and gelsolin are induced. Neuro-Oncology 2002, 4, 95–101. [Google Scholar] [CrossRef]
- Wang, Z.M.; Hu, J.; Zhou, D.; Xu, Z.Y.; Panasci, L.C.; Chen, Z.P. Trichostatin A inhibits proliferation and induces expression of p21WAF and p27 in human brain tumor cell lines. Ai Zheng 2002, 21, 1100–1105. [Google Scholar] [PubMed]
- Yin, D.; Ong, J.M.; Hu, J.; Desmond, J.C.; Kawamata, N.; Konda, B.M.; Black, K.L.; Koeffler, H.P. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor: Effects on gene expression and growth of glioma cells in vitro and in vivo. Clin. Cancer Res. 2007, 13, 1045–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mottet, D.; Pirotte, S.; Lamour, V.; Hagedorn, M.; Javerzat, S.; Bikfalvi, A.; Bellahcene, A.; Verdin, E.; Castronovo, V. HDAC4 represses p21(WAF1/Cip1) expression in human cancer cells through a Sp1-dependent, p53-independent mechanism. Oncogene 2009, 28, 243–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.; Berglund, A.E.; Etame, A.B. The Impact of Epigenetic Modifications on Adaptive Resistance Evolution in Glioblastoma. Int. J. Mol. Sci. 2021, 22, 8324. [Google Scholar] [CrossRef]
- Hervouet, E. The Promising Role of New Generation HDACis in Anti-Cancer Therapies. Ebiomedicine 2018, 32, 6–7. [Google Scholar] [CrossRef]
- Williams, M.J.; Singleton, W.G.; Lowis, S.P.; Malik, K.; Kurian, K.M. Therapeutic Targeting of Histone Modifications in Adult and Pediatric High-Grade Glioma. Front. Oncol. 2017, 7, 45. [Google Scholar] [CrossRef] [Green Version]
- Kitange, G.J.; Mladek, A.C.; Carlson, B.L.; Schroeder, M.A.; Pokorny, J.L.; Cen, L.; Decker, P.A.; Wu, W.; Lomberk, G.A.; Gupta, S.K.; et al. Inhibition of histone deacetylation potentiates the evolution of acquired temozolomide resistance linked to MGMT upregulation in glioblastoma xenografts. Clin. Cancer Res. 2012, 18, 4070–4079. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.Q.; Zhang, B.L.; Ni, H.B.; Liu, Z.H.; Wang, J.C.; Ren, Q.X.; Mo, J.B.; Xiong, Y.; Yao, R.Q.; Gao, D.S. Hyperacetylation of histone H3K9 involved in the promotion of abnormally high transcription of the gdnf gene in glioma cells. Mol. Neurobiol. 2014, 50, 914–922. [Google Scholar] [CrossRef]
- Ku, M.C.; Wolf, S.A.; Respondek, D.; Matyash, V.; Pohlmann, A.; Waiczies, S.; Waiczies, H.; Niendorf, T.; Synowitz, M.; Glass, R.; et al. GDNF mediates glioblastoma-induced microglia attraction but not astrogliosis. Acta Neuropathol. 2013, 125, 609–620. [Google Scholar] [CrossRef]
- Yang, W.; Xia, Y.; Hawke, D.; Li, X.; Liang, J.; Xing, D.; Aldape, K.; Hunter, T.; Alfred Yung, W.K.; Lu, Z. PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis. Cell 2012, 150, 685–696. [Google Scholar] [CrossRef]
- Yang, W.; Xia, Y.; Ji, H.; Zheng, Y.; Liang, J.; Huang, W.; Gao, X.; Aldape, K.; Lu, Z. Nuclear PKM2 regulates beta-catenin transactivation upon EGFR activation. Nature 2011, 480, 118–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.L.; Cheng, J.X.; Zhang, X.; Wang, R.; Zhang, W.; Lin, H.; Xiao, X.; Cai, S.; Chen, X.Y.; Cheng, H. Global histone modification patterns as prognostic markers to classify glioma patients. Cancer Epidemiol. Biomark. Prev. 2010, 19, 2888–2896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hervas-Corpion, I.; Gallardo-Orihuela, A.; Catalina-Fernandez, I.; Iglesias-Lozano, I.; Soto-Torres, O.; Geribaldi-Doldan, N.; Dominguez-Garcia, S.; Luna-Garcia, N.; Romero-Garcia, R.; Mora-Lopez, F.; et al. Potential Diagnostic Value of the Differential Expression of Histone H3 Variants between Low- and High-Grade Gliomas. Cancers 2021, 13, 5261. [Google Scholar] [CrossRef] [PubMed]
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 2014, 30, 1363–1369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Assenov, Y.; Muller, F.; Lutsik, P.; Walter, J.; Lengauer, T.; Bock, C. Comprehensive analysis of DNA methylation data with RnBeads. Nat. Methods 2014, 11, 1138–1140. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Rabaneda, L.G.; Carrasco, M.; Lopez-Toledano, M.A.; Murillo-Carretero, M.; Ruiz, F.A.; Estrada, C.; Castro, C. Homocysteine inhibits proliferation of neuronal precursors in the mouse adult brain by impairing the basic fibroblast growth factor signaling cascade and reducing extracellular regulated kinase 1/2-dependent cyclin E expression. FASEB J. 2008, 22, 3823–3835. [Google Scholar] [CrossRef] [PubMed]
- Pollard, S.M.; Yoshikawa, K.; Clarke, I.D.; Danovi, D.; Stricker, S.; Russell, R.; Bayani, J.; Head, R.; Lee, M.; Bernstein, M.; et al. Glioma stem cell lines expanded in adherent culture have tumor-specific phenotypes and are suitable for chemical and genetic screens. Cell Stem Cell 2009, 4, 568–580. [Google Scholar] [CrossRef] [Green Version]
- Alenda, C.; Rojas, E.; Valor, L.M. FFPE samples from cavitational ultrasonic surgical aspirates are suitable for RNA profiling of gliomas. PLoS ONE 2021, 16, e0255168. [Google Scholar] [CrossRef]
- Medvedeva, Y.A.; Lennartsson, A.; Ehsani, R.; Kulakovskiy, I.V.; Vorontsov, I.E.; Panahandeh, P.; Khimulya, G.; Kasukawa, T.; Drablos, F. EpiFactors: A comprehensive database of human epigenetic factors and complexes. Database 2015, 2015, bav067. [Google Scholar] [CrossRef] [PubMed]
- Grabowicz, I.E.; Wilczynski, B.; Kaminska, B.; Roura, A.J.; Wojtas, B.; Dabrowski, M.J. The role of epigenetic modifications, long-range contacts, enhancers and topologically associating domains in the regulation of glioma grade-specific genes. Sci. Rep. 2021, 11, 15668. [Google Scholar] [CrossRef] [PubMed]
- Deaton, A.M.; Bird, A. CpG islands and the regulation of transcription. Genes Dev. 2011, 25, 1010–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stepniak, K.; Machnicka, M.A.; Mieczkowski, J.; Macioszek, A.; Wojtas, B.; Gielniewski, B.; Poleszak, K.; Perycz, M.; Krol, S.K.; Guzik, R.; et al. Mapping chromatin accessibility and active regulatory elements reveals pathological mechanisms in human gliomas. Nat. Commun. 2021, 12, 3621. [Google Scholar] [CrossRef]
- Aras, Y.; Erguven, M.; Aktas, E.; Yazihan, N.; Bilir, A. Antagonist activity of the antipsychotic drug lithium chloride and the antileukemic drug imatinib mesylate during glioblastoma treatment in vitro. Neurol. Res. 2016, 38, 766–774. [Google Scholar] [CrossRef] [PubMed]
- Sabanciota, P.A.; Erguven, M.; Yaziotahan, N.; Aktas, E.; Aras, Y.; Civelek, E.; Aydoseli, A.; Imer, M.; Gurtekin, M.; Bilir, A. Sorafenib and lithium chloride combination treatment shows promising synergistic effects in human glioblastoma multiforme cells in vitro but midkine is not implicated. Neurol. Res. 2014, 36, 189–197. [Google Scholar] [CrossRef]
- Erguven, M.; Bilir, A.; Yazihan, N.; Ermis, E.; Sabanci, A.; Aktas, E.; Aras, Y.; Alpman, V. Decreased therapeutic effects of noscapine combined with imatinib mesylate on human glioblastoma in vitro and the effect of midkine. Cancer Cell Int. 2011, 11, 18. [Google Scholar] [CrossRef] [Green Version]
- He, M.; Chen, X.; Luo, M.; Ouyang, L.; Xie, L.; Huang, Z.; Liu, A. Suppressor of cytokine signaling 1 inhibits the maturation of dendritic cells involving the nuclear factor kappa B signaling pathway in the glioma microenvironment. Clin. Exp. Immunol. 2020, 202, 47–59. [Google Scholar] [CrossRef]
- Ogden, A.T.; Horgan, D.; Waziri, A.; Anderson, D.; Louca, J.; McKhann, G.M.; Sisti, M.B.; Parsa, A.T.; Bruce, J.N. Defective receptor expression and dendritic cell differentiation of monocytes in glioblastomas. Neurosurgery 2006, 59, 902–909; discussion 909–910. [Google Scholar] [CrossRef]
- Idbaih, A.; Carvalho Silva, R.; Criniere, E.; Marie, Y.; Carpentier, C.; Boisselier, B.; Taillibert, S.; Rousseau, A.; Mokhtari, K.; Ducray, F.; et al. Genomic changes in progression of low-grade gliomas. J. Neurooncol. 2008, 90, 133–140. [Google Scholar] [CrossRef]
- Belotti, Y.; Tolomeo, S.; Yu, R.; Lim, W.T.; Lim, C.T. Prognostic Neurotransmitter Receptors Genes Are Associated with Immune Response, Inflammation and Cancer Hallmarks in Brain Tumors. Cancers 2022, 14, 2544. [Google Scholar] [CrossRef]
- Yang, J.; Yang, Q. Identification of Core Genes and Screening of Potential Targets in Glioblastoma Multiforme by Integrated Bioinformatic Analysis. Front. Oncol. 2020, 10, 615976. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Du, M.; Lu, L. A Novel 16-Genes Signature Scoring System as Prognostic Model to Evaluate Survival Risk in Patients with Glioblastoma. Biomedicines 2022, 10, 317. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.; Chen, J.; Liu, X.; Gong, W.; Zheng, J.; Guo, X.; Liu, Y.; Liu, L.; Ma, J.; Wang, P.; et al. PVT1 regulates the malignant behaviors of human glioma cells by targeting miR-190a-5p and miR-488-3p. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1783–1794. [Google Scholar] [CrossRef] [PubMed]
- Feyissa, A.M.; Carrano, A.; Wang, X.; Allen, M.; Ertekin-Taner, N.; Dickson, D.W.; Jentoft, M.E.; Rosenfeld, S.S.; Tatum, W.O.; Ritaccio, A.L.; et al. Analysis of intraoperative human brain tissue transcriptome reveals putative risk genes and altered molecular pathways in glioma-related seizures. Epilepsy Res. 2021, 173, 106618. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.; Lee, H.; Yoon, J.G.; Madan, A.; Wayner, E.; Tonning, S.; Hothi, P.; Schroeder, B.; Ulasov, I.; Foltz, G.; et al. Global analysis of H3K4me3 and H3K27me3 profiles in glioblastoma stem cells and identification of SLC17A7 as a bivalent tumor suppressor gene. Oncotarget 2015, 6, 5369–5381. [Google Scholar] [CrossRef] [Green Version]
- Gladitz, J.; Klink, B.; Seifert, M. Network-based analysis of oligodendrogliomas predicts novel cancer gene candidates within the region of the 1p/19q co-deletion. Acta Neuropathol. Commun. 2018, 6, 49. [Google Scholar] [CrossRef] [Green Version]
- Aruga, J.; Yokota, N.; Mikoshiba, K. Human SLITRK family genes: Genomic organization and expression profiling in normal brain and brain tumor tissue. Gene 2003, 315, 87–94. [Google Scholar] [CrossRef]
- Zhou, Y.; Yang, L.; Zhang, X.; Chen, R.; Chen, X.; Tang, W.; Zhang, M. Identification of Potential Biomarkers in Glioblastoma through Bioinformatic Analysis and Evaluating Their Prognostic Value. Biomed. Res. Int. 2019, 2019, 6581576. [Google Scholar] [CrossRef] [Green Version]
- Murphy, K.A.; Lechner, M.G.; Popescu, F.E.; Bedi, J.; Decker, S.A.; Hu, P.; Erickson, J.R.; O’Sullivan, M.G.; Swier, L.; Salazar, A.M.; et al. An in vivo immunotherapy screen of costimulatory molecules identifies Fc-OX40L as a potent reagent for the treatment of established murine gliomas. Clin. Cancer Res. 2012, 18, 4657–4668. [Google Scholar] [CrossRef] [Green Version]
- Lucio-Eterovic, A.K.; Cortez, M.A.; Valera, E.T.; Motta, F.J.; Queiroz, R.G.; Machado, H.R.; Carlotti, C.G., Jr.; Neder, L.; Scrideli, C.A.; Tone, L.G. Differential expression of 12 histone deacetylase (HDAC) genes in astrocytomas and normal brain tissue: Class II and IV are hypoexpressed in glioblastomas. BMC Cancer 2008, 8, 243. [Google Scholar] [CrossRef]
- Hanisch, D.; Krumm, A.; Diehl, T.; Stork, C.M.; Dejung, M.; Butter, F.; Kim, E.; Brenner, W.; Fritz, G.; Hofmann, T.G.; et al. Class I HDAC overexpression promotes temozolomide resistance in glioma cells by regulating RAD18 expression. Cell Death Dis. 2022, 13, 293. [Google Scholar] [CrossRef] [PubMed]
- Dali-Youcef, N.; Froelich, S.; Moussallieh, F.M.; Chibbaro, S.; Noel, G.; Namer, I.J.; Heikkinen, S.; Auwerx, J. Gene expression mapping of histone deacetylases and co-factors, and correlation with survival time and 1H-HRMAS metabolomic profile in human gliomas. Sci. Rep. 2015, 5, 9087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Yan, X.; Liang, C.; Chen, H.; Liu, M.; Wu, Z.; Zheng, J.; Dang, J.; La, X.; Liu, Q. Comprehensive Analysis of the Differential Expression and Prognostic Value of Histone Deacetylases in Glioma. Front. Cell Dev. Biol. 2022, 10, 840759. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.L.; Piccolo, S.R.; Cheng, L.; Soldi, R.; Han, B.; Johnson, W.E.; Bild, A.H. Genomic pathway analysis reveals that EZH2 and HDAC4 represent mutually exclusive epigenetic pathways across human cancers. BMC Med. Genom. 2013, 6, 35. [Google Scholar] [CrossRef] [Green Version]
- Marampon, F.; Megiorni, F.; Camero, S.; Crescioli, C.; McDowell, H.P.; Sferra, R.; Vetuschi, A.; Pompili, S.; Ventura, L.; De Felice, F.; et al. HDAC4 and HDAC6 sustain DNA double strand break repair and stem-like phenotype by promoting radioresistance in glioblastoma cells. Cancer Lett. 2017, 397, 1–11. [Google Scholar] [CrossRef]
- Liu, Y.; Duan, X.; Zhang, C.; Yuan, J.; Wen, J.; Zheng, C.; Shi, J.; Yuan, M. KAT6B May Be Applied as a Potential Therapeutic Target for Glioma. J. Oncol. 2022, 2022, 2500092. [Google Scholar] [CrossRef]
- Wiesel-Motiuk, N.; Assaraf, Y.G. The key roles of the lysine acetyltransferases KAT6A and KAT6B in physiology and pathology. Drug Resist. Updat. 2020, 53, 100729. [Google Scholar] [CrossRef]
- Klein, B.J.; Jang, S.M.; Lachance, C.; Mi, W.; Lyu, J.; Sakuraba, S.; Krajewski, K.; Wang, W.W.; Sidoli, S.; Liu, J.; et al. Histone H3K23-specific acetylation by MORF is coupled to H3K14 acylation. Nat. Commun. 2019, 10, 4724. [Google Scholar] [CrossRef] [Green Version]
- Lewis, P.W.; Muller, M.M.; Koletsky, M.S.; Cordero, F.; Lin, S.; Banaszynski, L.A.; Garcia, B.A.; Muir, T.W.; Becher, O.J.; Allis, C.D. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 2013, 340, 857–861. [Google Scholar] [CrossRef] [Green Version]
- An, S.; Camarillo, J.M.; Huang, T.Y.; Li, D.; Morris, J.A.; Zoltek, M.A.; Qi, J.; Behbahani, M.; Kambhampati, M.; Kelleher, N.L.; et al. Histone tail analysis reveals H3K36me2 and H4K16ac as epigenetic signatures of diffuse intrinsic pontine glioma. J. Exp. Clin. Cancer Res. 2020, 39, 261. [Google Scholar] [CrossRef]
- Gallo, M.; Coutinho, F.J.; Vanner, R.J.; Gayden, T.; Mack, S.C.; Murison, A.; Remke, M.; Li, R.; Takayama, N.; Desai, K.; et al. MLL5 Orchestrates a Cancer Self-Renewal State by Repressing the Histone Variant H3.3 and Globally Reorganizing Chromatin. Cancer Cell 2015, 28, 715–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riva, G.; Butta, V.; Cilibrasi, C.; Baronchelli, S.; Redaelli, S.; Dalpra, L.; Lavitrano, M.; Bentivegna, A. Epigenetic targeting of glioma stem cells: Short-term and long-term treatments with valproic acid modulate DNA methylation and differentiation behavior, but not temozolomide sensitivity. Oncol. Rep. 2016, 35, 2811–2824. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Grade | Sex | Number of Samples | Median (Age) | IQR (Age) |
|---|---|---|---|---|
| II | Female | 5 | 44 | 9 |
| Male | 8 | |||
| III | Male | 7 | 47 | 11.5 |
| IV | Female | 29 | 62 | 18 |
| Male | 28 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hervás-Corpión, I.; Navarro-Calvo, J.; Martín-Climent, P.; Iriarte-Gahete, M.; Geribaldi-Doldán, N.; Castro, C.; Valor, L.M. Defining a Correlative Transcriptional Signature Associated with Bulk Histone H3 Acetylation Levels in Adult Glioblastomas. Cells 2023, 12, 374. https://doi.org/10.3390/cells12030374
Hervás-Corpión I, Navarro-Calvo J, Martín-Climent P, Iriarte-Gahete M, Geribaldi-Doldán N, Castro C, Valor LM. Defining a Correlative Transcriptional Signature Associated with Bulk Histone H3 Acetylation Levels in Adult Glioblastomas. Cells. 2023; 12(3):374. https://doi.org/10.3390/cells12030374
Chicago/Turabian StyleHervás-Corpión, Irati, Jorge Navarro-Calvo, Paula Martín-Climent, Marianela Iriarte-Gahete, Noelia Geribaldi-Doldán, Carmen Castro, and Luis M. Valor. 2023. "Defining a Correlative Transcriptional Signature Associated with Bulk Histone H3 Acetylation Levels in Adult Glioblastomas" Cells 12, no. 3: 374. https://doi.org/10.3390/cells12030374