
Pericyte–Glioblastoma Cell Interaction: A Key Target to Prevent Glioblastoma Progression

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Role of Vascular Co-Option in Vascular Malformation and Changes in Pericyte Contractility
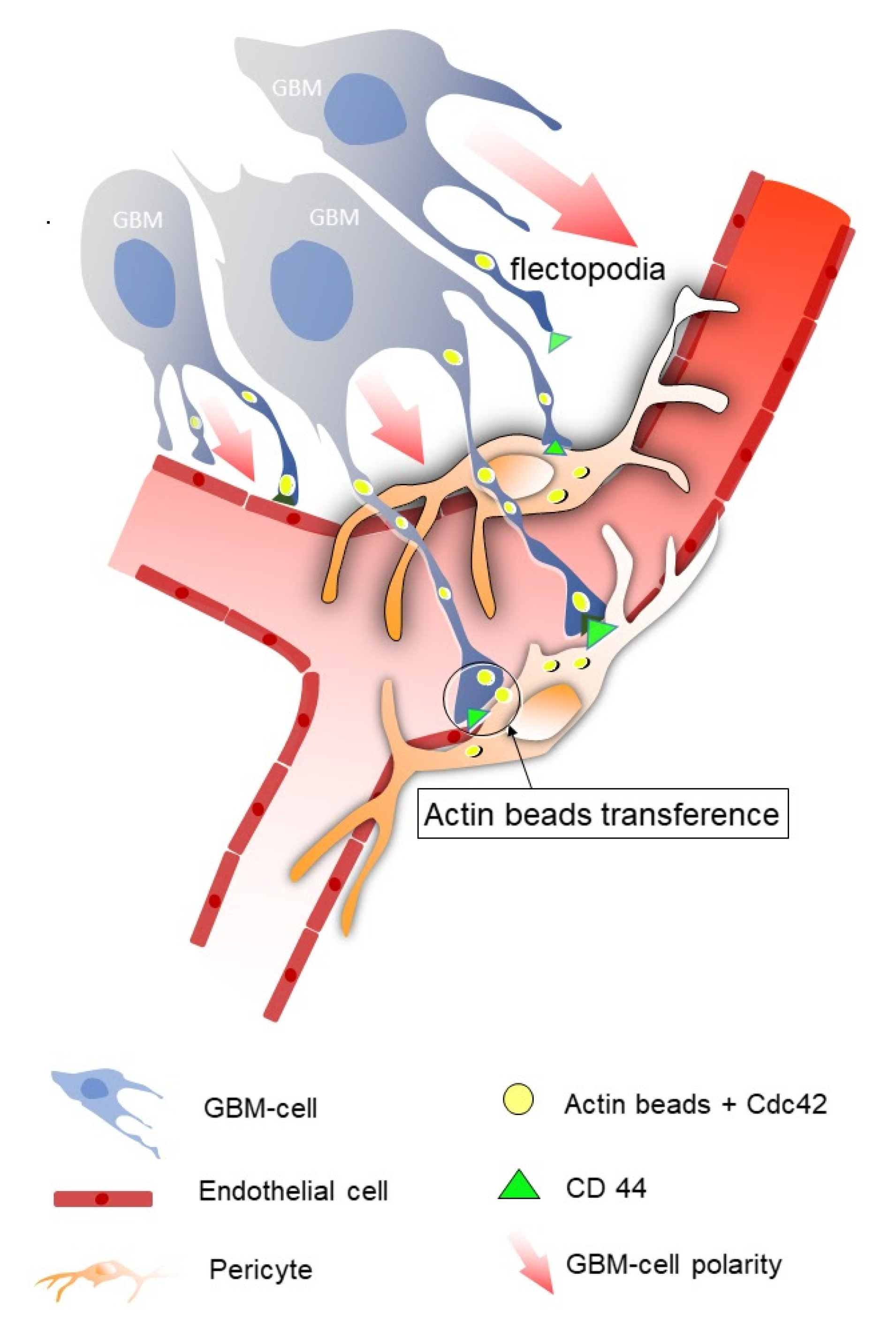
2.1. Vascular Co-Option
2.2. Flectopodia, the Execution Arm
2.3. Consequences of GBM Cell–Pericyte Interaction during Co-Option
2.4. Molecules Involved in Vessel Co-Option in GBM
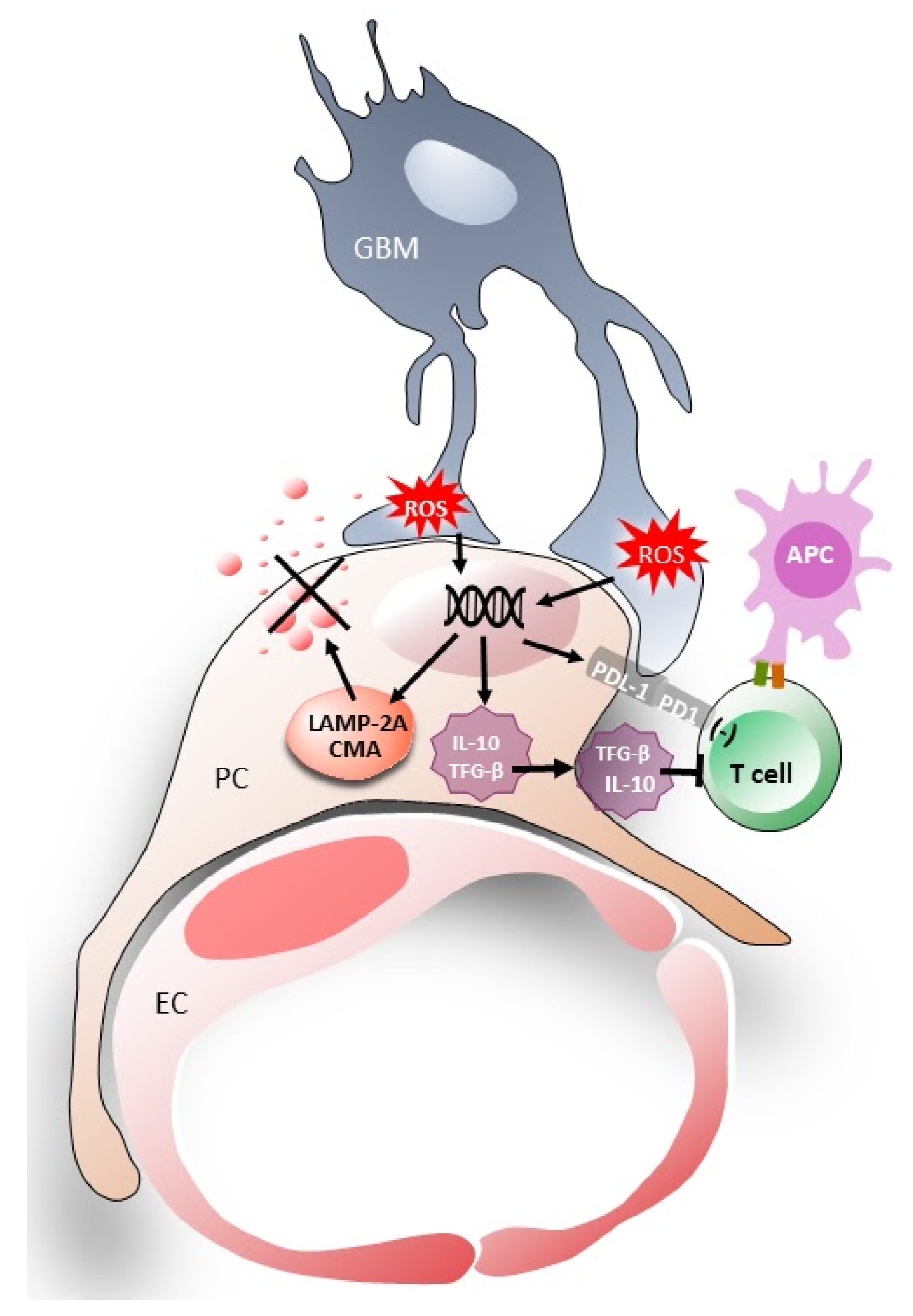
3. Immunosuppressive Properties of Glioblastoma–Pericyte Interactions
3.1. High Levels of Anti-Inflammatory Cytokines in Pericytes
3.2. The Expression of Immunosuppressive Membrane Molecules in Pericytes
3.3. Reduced Expression of Co-Stimulatory Molecules and Inhibited T Cell Activation in Conditioned Pericytes
3.4. Pericytes Interacting with GBM Cells Promote Tumor Growth
4. Induction of Chaperone-Mediated Autophagy (CMA) Activity in Pericytes
GBM-Induced CMA in Pericytes Helps Tumors Survive
5. Changes in the Microvesicular and Protein Antitumor Secretome
5.1. Pericyte Secretome
5.2. Secretome of Pericytes in Tumor Conditions
5.3. CMA-Induced Changes in Pericyte Transcriptome Profiling/Secretome
6. Future Strategies and Routes for Targeting GBM–Pericyte Interactions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Koshy, M.; Villano, J.L.; Dolecek, T.A.; Howard, A.; Mahmood, U.; Chmura, S.J.; Weichselbaum, R.R.; McCarthy, B.J. Improved Survival Time Trends for Glioblastoma Using the SEER 17 Population-Based Registries. J. Neuro-Oncol. 2012, 107, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A Randomized Trial of Bevacizumab for Newly Diagnosed Glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Philips, A.; Henshaw, D.L.; Lamburn, G.; O’Carroll, M.J. Authors’ Comment on “Brain Tumours: Rise in Glioblastoma Multiforme Incidence in England 1995–2015 Suggests an Adverse Environmental or Lifestyle Factor”. J. Environ. Public Health 2018, 2018, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Burger, P.C.; Scheithauer, B.W. Tumors of the Central Nervous System, Atlas of Tumor Pathology. Am. J. Surg. Pathol. 1995, 19, 1220. [Google Scholar] [CrossRef]
- Farin, A.; Suzuki, S.O.; Weiker, M.; Goldman, J.E.; Bruce, J.N.; Canoll, P. Transplanted Glioma Cells Migrate and Proliferate on Host Brain Vasculature: A Dynamic Analysis. Glia 2006, 53, 799–808. [Google Scholar] [CrossRef]
- Baker, G.J.; Yadav, V.N.; Motsch, S.; Koschmann, C.; Calinescu, A.-A.; Mineharu, Y.; Camelo-Piragua, S.I.; Orringer, D.; Bannykh, S.; Nichols, W.S.; et al. Mechanisms of Glioma Formation: Iterative Perivascular Glioma Growth and Invasion Leads to Tumor Progression, VEGF-Independent Vascularization, and Resistance to Antiangiogenic Therapy. Neoplasia 2014, 16, 543–561. [Google Scholar] [CrossRef]
- Holash, J.; Maisonpierre, P.C.; Compton, D.; Boland, P.; Alexander, C.R.; Zagzag, D.; Yancopoulos, G.D.; Wiegand, S.J. Vessel Cooption, Regression, and Growth in Tumors Mediated by Angiopoietins and VEGF. Science 1999, 284, 1994–1998. [Google Scholar] [CrossRef]
- Caspani, E.M.; Crossley, P.H.; Redondo-Garcia, C.; Martinez, S. Glioblastoma: A Pathogenic Crosstalk between Tumor Cells and Pericytes. PLoS ONE 2014, 9, e101402. [Google Scholar] [CrossRef]
- Zimmermann, K.W. Der Feinere Bau Der Blutcapillaren Berlin; Springer: Berlin/Heidelberg, Germany, 1923. [Google Scholar]
- Pombero, A.; Garcia-Lopez, R.; Martinez, S. Brain Mesenchymal Stem Cells: Physiology and Pathological Implications. Dev. Growth Differ. 2016, 58, 469–480. [Google Scholar] [CrossRef]
- von Tell, D.; Armulik, A.; Betsholtz, C. Pericytes and Vascular Stability. Exp. Cell Res. 2006, 312, 623–629. [Google Scholar] [CrossRef]
- Caplan, A.I. MSCs: The Sentinel and Safe-Guards of Injury. J. Cell. Physiol. 2016, 231, 1413–1416. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.D.; Winkler, E.A.; Sagare, A.P.; Singh, I.; LaRue, B.; Deane, R.; Zlokovic, B.V. Pericytes Control Key Neurovascular Functions and Neuronal Phenotype in the Adult Brain and during Brain Aging. Neuron 2010, 68, 409–427. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Joan Abbott, N.; Rönnbäck, L.; Hansson, E. Astrocyte–endothelial Interactions at the Blood–brain Barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Winkler, E.A.; Bell, R.D.; Zlokovic, B.V. Central Nervous System Pericytes in Health and Disease. Nat. Neurosci. 2011, 14, 1398–1405. [Google Scholar] [CrossRef] [PubMed]
- Tavazoie, M.; Van der Veken, L.; Silva-Vargas, V.; Louissaint, M.; Colonna, L.; Zaidi, B.; Garcia-Verdugo, J.M.; Doetsch, F. A Specialized Vascular Niche for Adult Neural Stem Cells. Cell Stem Cell 2008, 3, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Muoio, V.; Persson, P.B.; Sendeski, M.M. The Neurovascular Unit—Concept Review. Acta Physiol. 2014, 210, 790–798. [Google Scholar] [CrossRef]
- Balabanov, R.; Washington, R.; Wagnerova, J.; Dore-Duffy, P. CNS Microvascular Pericytes Express Macrophage-like Function, Cell Surface Integrin Alpha M, and Macrophage Marker ED-2. Microvasc. Res. 1996, 52, 127–142. [Google Scholar] [CrossRef]
- Thomas, W.E. Brain Macrophages: On the Role of Pericytes and Perivascular Cells. Brain Res. Brain Res. Rev. 1999, 31, 42–57. [Google Scholar] [CrossRef]
- Rustenhoven, J.; Jansson, D.; Smyth, L.C.; Dragunow, M. Brain Pericytes As Mediators of Neuroinflammation. Trends Pharmacol. Sci. 2017, 38, 291–304. [Google Scholar] [CrossRef]
- Kovac, A.; Erickson, M.A.; Banks, W.A. Brain Microvascular Pericytes Are Immunoactive in Culture: Cytokine, Chemokine, Nitric Oxide, and LRP-1 Expression in Response to Lipopolysaccharide. J. Neuroinflammation 2011, 8, 139. [Google Scholar] [CrossRef]
- Pieper, C.; Marek, J.J.; Unterberg, M.; Schwerdtle, T.; Galla, H.-J. Brain Capillary Pericytes Contribute to the Immune Defense in Response to Cytokines or LPS in Vitro. Brain Res. 2014, 1550, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Balabanov, R.; Beaumont, T.; Dore-Duffy, P. Role of Central Nervous System Microvascular Pericytes in Activation of Antigen-Primed Splenic T-Lymphocytes. J. Neurosci. Res. 1999, 55, 578–587. [Google Scholar] [CrossRef]
- Matsumoto, J.; Takata, F.; Machida, T.; Takahashi, H.; Soejima, Y.; Funakoshi, M.; Futagami, K.; Yamauchi, A.; Dohgu, S.; Kataoka, Y. Tumor Necrosis Factor-α-Stimulated Brain Pericytes Possess a Unique Cytokine and Chemokine Release Profile and Enhance Microglial Activation. Neurosci. Lett. 2014, 578, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Tigges, U.; Boroujerdi, A.; Welser-Alves, J.V.; Milner, R. TNF-α Promotes Cerebral Pericyte Remodeling in Vitro, via a Switch from α1 to α2 Integrins. J. Neuroinflammation 2013, 10, 33. [Google Scholar] [CrossRef] [PubMed]
- Rustenhoven, J.; Aalderink, M.; Scotter, E.L.; Oldfield, R.L.; Bergin, P.S.; Mee, E.W.; Graham, E.S.; Faull, R.L.M.; Curtis, M.A.; Park, T.I.-H.; et al. TGF-beta1 Regulates Human Brain Pericyte Inflammatory Processes Involved in Neurovasculature Function. J. Neuroinflammation 2016, 13, 37. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Andersson, P.; Hosaka, K.; Zhang, Y.; Cao, R.; Iwamoto, H.; Yang, X.; Nakamura, M.; Wang, J.; Zhuang, R.; et al. The PDGF-BB-SOX7 Axis-Modulated IL-33 in Pericytes and Stromal Cells Promotes Metastasis through Tumour-Associated Macrophages. Nat. Commun. 2016, 7, 11385. [Google Scholar] [CrossRef]
- Smith, A.M.; Scott Graham, E.; Feng, S.X.; Oldfield, R.L.; Bergin, P.M.; Mee, E.W.; Faull, R.L.M.; Curtis, M.A.; Dragunow, M. Adult Human Glia, Pericytes and Meningeal Fibroblasts Respond Similarly to IFNy but Not to TGFβ1 or M-CSF. PLoS ONE 2013, 8, e80463. [Google Scholar] [CrossRef]
- Nduom, E.K.; Weller, M.; Heimberger, A.B. Immunosuppressive Mechanisms in Glioblastoma. Neuro. Oncol. 2015, 17 (Suppl S7), vii9–vii14. [Google Scholar] [CrossRef]
- Zhou, W.; Ke, S.Q.; Huang, Z.; Flavahan, W.; Fang, X.; Paul, J.; Wu, L.; Sloan, A.E.; McLendon, R.E.; Li, X.; et al. Periostin Secreted by Glioblastoma Stem Cells Recruits M2 Tumour-Associated Macrophages and Promotes Malignant Growth. Nat. Cell Biol. 2015, 17, 170–182. [Google Scholar] [CrossRef]
- Preusser, M.; Lim, M.; Hafler, D.A.; Reardon, D.A.; Sampson, J.H. Prospects of Immune Checkpoint Modulators in the Treatment of Glioblastoma. Nat. Rev. Neurol. 2015, 11, 504–514. [Google Scholar] [CrossRef]
- Errico, A. CNS Cancer: Periostin-a New Potential Target for the Treatment of Glioblastoma. Nat. Rev. Clin. Oncol. 2015, 12, 128. [Google Scholar] [CrossRef] [PubMed]
- Valdor, R.; García-Bernal, D.; Bueno, C.; Ródenas, M.; Moraleda, J.M.; Macian, F.; Martínez, S. Glioblastoma Progression Is Assisted by Induction of Immunosuppressive Function of Pericytes through Interaction with Tumor Cells. Oncotarget 2017, 8, 68614–68626. [Google Scholar] [CrossRef] [PubMed]
- Crivii, C.-B.; Boșca, A.B.; Melincovici, C.S.; Constantin, A.-M.; Mărginean, M.; Dronca, E.; Suflețel, R.; Gonciar, D.; Bungărdean, M.; Șovrea, A. Glioblastoma Microenvironment and Cellular Interactions. Cancers 2022, 14, 1092. [Google Scholar] [CrossRef]
- Dapash, M.; Hou, D.; Castro, B.; Lee-Chang, C.; Lesniak, M.S. The Interplay between Glioblastoma and Its Microenvironment. Cells 2021, 10, 2257. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Liu, W.A.; Zhang, X.-J.; Shi, W.; Ren, S.-Q.; Li, Z.; Brown, K.N.; Shi, S.-H. Vascular Influence on Ventral Telencephalic Progenitors and Neocortical Interneuron Production. Dev. Cell 2016, 36, 624–638. [Google Scholar] [CrossRef] [PubMed]
- Di Marco, B.; Crouch, E.E.; Shah, B.; Duman, C.; Paredes, M.F.; Ruiz de Almodovar, C.; Huang, E.J.; Alfonso, J. Reciprocal Interaction between Vascular Filopodia and Neural Stem Cells Shapes Neurogenesis in the Ventral Telencephalon. Cell Rep. 2020, 33, 108256. [Google Scholar] [CrossRef]
- Pombero, A.; Garcia-Lopez, R.; Estirado, A.; Martinez, S. Vascular Pattern of the Dentate Gyrus Is Regulated by Neural Progenitors. Brain Struct. Funct. 2018, 223, 1971–1987. [Google Scholar] [CrossRef]
- Takashima, S.; Watanabe, C.; Ema, M.; Mizutani, K.-I. Interaction of the Nervous System and Vascular System Is Required for the Proper Assembly of the Neocortex. Neurochem. Int. 2019, 129, 104481. [Google Scholar] [CrossRef]
- Tsai, H.-H.; Niu, J.; Munji, R.; Davalos, D.; Chang, J.; Zhang, H.; Tien, A.-C.; Kuo, C.J.; Chan, J.R.; Daneman, R.; et al. Oligodendrocyte Precursors Migrate along Vasculature in the Developing Nervous System. Science 2016, 351, 379–384. [Google Scholar] [CrossRef]
- Kuczynski, E.A.; Vermeulen, P.B.; Pezzella, F.; Kerbel, R.S.; Reynolds, A.R. Vessel Co-Option in Cancer. Nat. Rev. Clin. Oncol. 2019, 16, 469–493. [Google Scholar] [CrossRef]
- Fornabaio, G.; Barnhill, R.L.; Lugassy, C.; Bentolila, L.A.; Cassoux, N.; Roman-Roman, S.; Alsafadi, S.; Del Bene, F. Angiotropism and Extravascular Migratory Metastasis in Cutaneous and Uveal Melanoma Progression in a Zebrafish Model. Sci. Rep. 2018, 8, 10448. [Google Scholar] [CrossRef]
- Frentzas, S.; Simoneau, E.; Bridgeman, V.L.; Vermeulen, P.B.; Foo, S.; Kostaras, E.; Nathan, M.; Wotherspoon, A.; Gao, Z.-H.; Shi, Y.; et al. Vessel Co-Option Mediates Resistance to Anti-Angiogenic Therapy in Liver Metastases. Nat. Med. 2016, 22, 1294–1302. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.-S.; Jones, D.; Liao, S.; Wattson, D.A.; Cui, C.H.; Duda, D.G.; Willett, C.G.; Jain, R.K.; Padera, T.P. Investigation of the Lack of Angiogenesis in the Formation of Lymph Node Metastases. JNCI J. Natl. Cancer Inst. 2015, 107, djv155. [Google Scholar] [CrossRef]
- Gaspar, L.E.; Fisher, B.J.; Macdonald, D.R.; LeBer, D.V.; Halperin, E.C.; Schold, S.C., Jr.; Cairncross, J.G. Supratentorial Malignant Glioma: Patterns of Recurrence and Implications for External Beam Local Treatment. Int. J. Radiat. Oncol. Biol. Phys. 1992, 24, 55–57. [Google Scholar] [CrossRef]
- Hou, L.C.; Veeravagu, A.; Hsu, A.R.; Tse, V.C.K. Recurrent Glioblastoma Multiforme: A Review of Natural History and Management Options. Neurosurg. Focus 2006, 20, E5. [Google Scholar] [CrossRef] [PubMed]
- Seano, G.; Jain, R.K. Vessel Co-Option in Glioblastoma: Emerging Insights and Opportunities. Angiogenesis 2020, 23, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Verhoeff, J.J.C.; van Tellingen, O.; Claes, A.; Stalpers, L.J.A.; van Linde, M.E.; Richel, D.J.; Leenders, W.P.J.; van Furth, W.R. Concerns about Anti-Angiogenic Treatment in Patients with Glioblastoma Multiforme. BMC Cancer 2009, 9, 444. [Google Scholar] [CrossRef]
- Winkler, F.; Kienast, Y.; Fuhrmann, M.; Von Baumgarten, L.; Burgold, S.; Mitteregger, G.; Kretzschmar, H.; Herms, J. Imaging Glioma Cell Invasion in Vivo Reveals Mechanisms of Dissemination and Peritumoral Angiogenesis. Glia 2009, 57, 1306–1315. [Google Scholar] [CrossRef]
- Watkins, S.; Robel, S.; Kimbrough, I.F.; Robert, S.M.; Ellis-Davies, G.; Sontheimer, H. Disruption of Astrocyte–vascular Coupling and the Blood–brain Barrier by Invading Glioma Cells. Nat. Commun. 2014, 5, 4196. [Google Scholar] [CrossRef]
- Berger, M.; Bergers, G.; Arnold, B.; Hämmerling, G.J.; Ganss, R. Regulator of G-Protein Signaling-5 Induction in Pericytes Coincides with Active Vessel Remodeling during Neovascularization. Blood 2005, 105, 1094–1101. [Google Scholar] [CrossRef]
- Mattes, B.; Scholpp, S. Emerging Role of Contact-Mediated Cell Communication in Tissue Development and Diseases. Histochem. Cell Biol. 2018, 150, 431–442. [Google Scholar]
- Junyent, S.; Garcin, C.L.; Szczerkowski, J.L.A.; Trieu, T.-J.; Reeves, J.; Habib, S.J. Specialized Cytonemes Induce Self-Organization of Stem Cells. Proc. Natl. Acad. Sci. USA 2020, 117, 7236–7244. [Google Scholar] [CrossRef] [PubMed]
- Routledge, D.; Rogers, S.; Ashktorab, H.; Phesse, T.J.; Scholpp, S. The Scaffolding Protein Flot2 Regulates Cytoneme-Based Transport of Wnt3 in Gastric Cancer. Elife 2022, 11, e77376. [Google Scholar] [CrossRef] [PubMed]
- Rotoli, D.; Morales, M.; Maeso, M.-C.; Ávila, J.; Pérez-Rodríguez, N.D.; Mobasheri, A.; van Noorden, C.J.F.; Martín-Vasallo, P. IQGAP1, AmotL2, and FKBP51 Scaffoldins in the Glioblastoma Microenvironment. J. Histochem. Cytochem. 2019, 67, 481–494. [Google Scholar] [CrossRef] [PubMed]
- Vega, S.; Kondo, A.; Suzuki, M.; Arai, H.; Jiapaer, S.; Sabit, H.; Nakada, M.; Ikeuchi, T.; Ishijima, M.; Arikawa-Hirasawa, E.; et al. Fibulin-7 Is Overexpressed in Glioblastomas and Modulates Glioblastoma Neovascularization through Interaction with angiopoietin-1. Int. J. Cancer 2019, 145, 2157–2169. [Google Scholar] [CrossRef]
- Valdor, R.; García-Bernal, D.; Riquelme, D.; Martinez, C.M.; Moraleda, J.M.; Cuervo, A.M.; Macian, F.; Martinez, S. Glioblastoma Ablates Pericytes Antitumor Immune Function through Aberrant up-Regulation of Chaperone-Mediated Autophagy. Proc. Natl. Acad. Sci. USA 2019, 116, 20655–20665. [Google Scholar] [CrossRef]
- Pichaud, F.; Walther, R.F.; Nunes de Almeida, F. Regulation of Cdc42 and Its Effectors in Epithelial Morphogenesis. J. Cell Sci. 2019, 132, jcs217869. [Google Scholar] [CrossRef]
- Glogowska, A.; Thanasupawat, T.; Beiko, J.; Pitz, M.; Hombach-Klonisch, S.; Klonisch, T. Novel CTRP8-RXFP1-JAK3-STAT3 Axis Promotes Cdc42-Dependent Actin Remodeling for Enhanced Filopodia Formation and Motility in Human Glioblastoma Cells. Mol. Oncol. 2022, 16, 368–387. [Google Scholar] [CrossRef]
- Etienne-Manneville, S. Cdc42--the Centre of Polarity. J. Cell Sci. 2004, 117, 1291–1300. [Google Scholar] [CrossRef]
- Ridley, A.J. Life at the Leading Edge. Cell 2011, 145, 1012–1022. [Google Scholar] [CrossRef]
- Kang, C.-W.; Kim, N.-H.; Jung, H.A.; Choi, H.-W.; Kang, M.-J.; Choi, J.-S.; Kim, G.-D. Desmethylanhydroicaritin Isolated from Sophora Flavescens, Shows Antitumor Activities in U87MG Cells via Inhibiting the Proliferation, Migration and Invasion. Environ. Toxicol. Pharmacol. 2016, 43, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Seifert, S.; Sontheimer, H. Bradykinin Enhances Invasion of Malignant Glioma into the Brain Parenchyma by Inducing Cells to Undergo Amoeboid Migration. J. Physiol. 2014, 592, 5109–5127. [Google Scholar] [CrossRef]
- Zagzag, D.; Esencay, M.; Mendez, O.; Yee, H.; Smirnova, I.; Huang, Y.; Chiriboga, L.; Lukyanov, E.; Liu, M.; Newcomb, E.W. Hypoxia- and Vascular Endothelial Growth Factor-Induced Stromal Cell-Derived Factor-1alpha/CXCR4 Expression in Glioblastomas: One Plausible Explanation of Scherer’s Structures. Am. J. Pathol. 2008, 173, 545–560. [Google Scholar] [CrossRef]
- Mooney, K.L.; Choy, W.; Sidhu, S.; Pelargos, P.; Bui, T.T.; Voth, B.; Barnette, N.; Yang, I. The Role of CD44 in Glioblastoma Multiforme. J. Clin. Neurosci. 2016, 34, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Dzwonek, J.; Wilczynski, G.M. CD44: Molecular Interactions, Signaling and Functions in the Nervous System. Front. Cell. Neurosci. 2015, 9, 175. [Google Scholar] [CrossRef]
- Jones, L.L.; Liu, Z.; Shen, J.; Werner, A.; Kreutzberg, G.W.; Raivich, G. Regulation of the Cell Adhesion Molecule CD44 after Nerve Transection and Direct Trauma to the Mouse Brain. J. Comp. Neurol. 2000, 426, 468–492. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, L.; Soares, R.; Laurentino, T.; Lerario, A.; Marie, S.; Oba-Shinjo, S. CD99 Expression in Glioblastoma Molecular Subtypes and Role in Migration and Invasion. Int. J. Mol. Sci. 2019, 20, 1137. [Google Scholar] [CrossRef]
- Fomchenko, E.I.; Dougherty, J.D.; Helmy, K.Y.; Katz, A.M.; Pietras, A.; Brennan, C.; Huse, J.T.; Milosevic, A.; Holland, E.C. Recruited Cells Can Become Transformed and Overtake PDGF-Induced Murine Gliomas in Vivo during Tumor Progression. PLoS ONE 2011, 6, e20605. [Google Scholar] [CrossRef]
- Garcia, C.; Dubois, L.G.; Xavier, A.L.; Geraldo, L.H.; da Fonseca, A.C.C.; Correia, A.H.; Meirelles, F.; Ventura, G.; Romão, L.; Canedo, N.H.S.; et al. The Orthotopic Xenotransplant of Human Glioblastoma Successfully Recapitulates Glioblastoma-Microenvironment Interactions in a Non-Immunosuppressed Mouse Model. BMC Cancer 2014, 14, 923. [Google Scholar] [CrossRef]
- Guillemin, G.J.; Brew, B.J. Microglia, Macrophages, Perivascular Macrophages, and Pericytes: A Review of Function and Identification. J. Leukoc. Biol. 2004, 75, 388–397. [Google Scholar] [CrossRef]
- Nduom, E.K.; Wei, J.; Yaghi, N.K.; Huang, N.; Kong, L.-Y.; Gabrusiewicz, K.; Ling, X.; Zhou, S.; Ivan, C.; Chen, J.Q.; et al. PD-L1 Expression and Prognostic Impact in Glioblastoma. Neuro. Oncol. 2016, 18, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Berghoff, A.S.; Kiesel, B.; Widhalm, G.; Rajky, O.; Ricken, G.; Wöhrer, A.; Dieckmann, K.; Filipits, M.; Brandstetter, A.; Weller, M.; et al. Programmed Death Ligand 1 Expression and Tumor-Infiltrating Lymphocytes in Glioblastoma. Neuro. Oncol. 2015, 17, 1064–1075. [Google Scholar] [CrossRef] [PubMed]
- Shimonkevitz, R.; Colon, S.; Kappler, J.W.; Marrack, P.; Grey, H.M. Antigen Recognition by H-2-Restricted T Cells. II. A Tryptic Ovalbumin Peptide That Substitutes for Processed Antigen. J. Immunol. 1984, 133, 2067–2074. [Google Scholar] [CrossRef] [PubMed]
- Domev, H.; Milkov, I.; Itskovitz-Eldor, J.; Dar, A. Immunoevasive Pericytes from Human Pluripotent Stem Cells Preferentially Modulate Induction of Allogeneic Regulatory T Cells. Stem Cells Transl. Med. 2014, 3, 1169–1181. [Google Scholar] [CrossRef]
- Bose, A.; Barik, S.; Banerjee, S.; Ghosh, T.; Mallick, A.; Bhattacharyya Majumdar, S.; Goswami, K.K.; Bhuniya, A.; Banerjee, S.; Baral, R.; et al. Tumor-Derived Vascular Pericytes Anergize Th Cells. J. Immunol. 2013, 191, 971–981. [Google Scholar] [CrossRef]
- Monks, C.R.; Freiberg, B.A.; Kupfer, H.; Sciaky, N.; Kupfer, A. Three-Dimensional Segregation of Supramolecular Activation Clusters in T Cells. Nature 1998, 395, 82–86. [Google Scholar] [CrossRef]
- Dustin, M.L.; Olszowy, M.W.; Holdorf, A.D.; Li, J.; Bromley, S.; Desai, N.; Widder, P.; Rosenberger, F.; van der Merwe, P.A.; Allen, P.M.; et al. A Novel Adaptor Protein Orchestrates Receptor Patterning and Cytoskeletal Polarity in T-Cell Contacts. Cell 1998, 94, 667–677. [Google Scholar] [CrossRef]
- Wurzer, H.; Hoffmann, C.; Al Absi, A.; Thomas, C. Actin Cytoskeleton Straddling the Immunological Synapse between Cytotoxic Lymphocytes and Cancer Cells. Cells 2019, 8, 463. [Google Scholar] [CrossRef]
- Yang, Q.; Wang, R.; Zhu, L. Chaperone-Mediated Autophagy. Autophagy Biol. Dis. 2019, 1206, 435–452. [Google Scholar]
- Kaushik, S.; Cuervo, A.M. The Coming of Age of Chaperone-Mediated Autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef]
- Salinas, M.D.; Valdor, R. Chaperone-Mediated Autophagy in Pericytes: A Key Target for the Development of New Treatments against Glioblastoma Progression. Int. J. Mol. Sci. 2022, 23, 8886. [Google Scholar] [CrossRef] [PubMed]
- Valdor, R.; Macian, F. Autophagy and the Regulation of the Immune Response. Pharmacol. Res. 2012, 66, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Valdor, R.; Mocholi, E.; Botbol, Y.; Guerrero-Ros, I.; Chandra, D.; Koga, H.; Gravekamp, C.; Cuervo, A.M.; Macian, F. Chaperone-Mediated Autophagy Regulates T Cell Responses through Targeted Degradation of Negative Regulators of T Cell Activation. Nat. Immunol. 2014, 15, 1046–1054. [Google Scholar] [CrossRef] [PubMed]
- Kiffin, R.; Christian, C.; Knecht, E.; Cuervo, A.M. Activation of Chaperone-Mediated Autophagy during Oxidative Stress. Mol. Biol. Cell 2004, 15, 4829–4840. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, B.; Wang, J.; Wu, H.; Xu, S.; Zhang, J.; Wang, L. Discovery of LAMP-2A as Potential Biomarkers for Glioblastoma Development by Modulating Apoptosis through N-CoR Degradation. Cell Commun. Signal. 2021, 19, 40. [Google Scholar] [CrossRef]
- Auzmendi-Iriarte, J.; Matheu, A. Intrinsic Role of Chaperone-Mediated Autophagy in Cancer Stem Cell Maintenance. Autophagy 2022, 18, 3035–3036. [Google Scholar] [CrossRef]
- Ding, Y.; Song, N.; Luo, Y. Role of Bone Marrow-Derived Cells in Angiogenesis: Focus on Macrophages and Pericytes. Cancer Microenviron 2012, 5, 225–236. [Google Scholar] [CrossRef]
- Gaceb, A.; Barbariga, M.; Özen, I.; Paul, G. The Pericyte Secretome: Potential Impact on Regeneration. Biochimie 2018, 155, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Gaceb, A.; Özen, I.; Padel, T.; Barbariga, M.; Paul, G. Pericytes Secrete pro-Regenerative Molecules in Response to Platelet-Derived Growth Factor-BB. J. Cereb. Blood Flow Metab. 2018, 38, 45–57. [Google Scholar] [CrossRef]
- Lv, M.; Ma, Q. Autophagy in Neurodevelopmental Disorders. Adv. Exp. Med. Biol. 2020, 1207, 171–182. [Google Scholar]
- Vogt, M.A.; Ehsaei, Z.; Knuckles, P.; Higginbottom, A.; Helmbrecht, M.S.; Kunath, T.; Eggan, K.; Williams, L.A.; Shaw, P.J.; Wurst, W.; et al. TDP-43 Induces p53-Mediated Cell Death of Cortical Progenitors and Immature Neurons. Sci. Rep. 2018, 8, 8097. [Google Scholar] [CrossRef] [PubMed]
- Bendriem, R.M.; Singh, S.; Aleem, A.A.; Antonetti, D.A.; Ross, M.E. Tight Junction Protein Occludin Regulates Progenitor Self-Renewal and Survival in Developing Cortex. Elife 2019, 8, e49376. [Google Scholar] [CrossRef] [PubMed]
- Kast, R.E.; Hill, Q.A.; Wion, D.; Mellstedt, H.; Focosi, D.; Karpel-Massler, G.; Heiland, T.; Halatsch, M.-E. Glioblastoma-Synthesized G-CSF and GM-CSF Contribute to Growth and Immunosuppression: Potential Therapeutic Benefit from Dapsone, Fenofibrate, and Ribavirin. Tumour Biol. 2017, 39, 1010428317699797. [Google Scholar] [CrossRef]
- Molina, M.L.; García-Bernal, D.; Martinez, S.; Valdor, R. Autophagy in the Immunosuppressive Perivascular Microenvironment of Glioblastoma. Cancers 2019, 12, 102. [Google Scholar] [CrossRef] [PubMed]
- Molina, M.L.; García-Bernal, D.; Salinas, M.D.; Rubio, G.; Aparicio, P.; Moraleda, J.M.; Martínez, S.; Valdor, R. Chaperone-Mediated Autophagy Ablation in Pericytes Reveals New Glioblastoma Prognostic Markers and Efficient Treatment Against Tumor Progression. Front. Cell Dev. Biol. 2022, 10, 797945. [Google Scholar] [CrossRef] [PubMed]
- Gaceb, A.; Paul, G. Pericyte Secretome. Adv. Exp. Med. Biol. 2018, 1109, 139–163. [Google Scholar]
- Verbeek, M.M.; Westphal, J.R.; Ruiter, D.J.; de Waal, R.M. T Lymphocyte Adhesion to Human Brain Pericytes Is Mediated via Very Late Antigen-4/vascular Cell Adhesion Molecule-1 Interactions. J. Immunol. 1995, 154, 5876–5884. [Google Scholar] [CrossRef]
- Beckman, J.D.; Grazul-Bilska, A.T.; Johnson, M.L.; Reynolds, L.P.; Redmer, D.A. Isolation and Characterization of Ovine Luteal Pericytes and Effects of Nitric Oxide on Pericyte Expression of Angiogenic Factors. Endocrine 2006, 29, 467–476. [Google Scholar] [CrossRef]
- Shimizu, F.; Sano, Y.; Abe, M.-A.; Maeda, T.; Ohtsuki, S.; Terasaki, T.; Kanda, T. Peripheral Nerve Pericytes Modify the Blood-Nerve Barrier Function and Tight Junctional Molecules through the Secretion of Various Soluble Factors. J. Cell. Physiol. 2011, 226, 255–266. [Google Scholar] [CrossRef]
- Tual-Chalot, S.; Leonetti, D.; Andriantsitohaina, R.; Martínez, M.C. Microvesicles: Intercellular Vectors of Biological Messages. Mol. Interv. 2011, 11, 88–94. [Google Scholar] [CrossRef]
- Ochs, K.; Sahm, F.; Opitz, C.A.; Lanz, T.V.; Oezen, I.; Couraud, P.-O.; von Deimling, A.; Wick, W.; Platten, M. Immature Mesenchymal Stem Cell-like Pericytes as Mediators of Immunosuppression in Human Malignant Glioma. J. Neuroimmunol. 2013, 265, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Ghochani, Y.; Sohrabi, A.; Muthukrishnan, S.D.; Kawaguchi, R.; Condro, M.C.; Bastola, S.; Gao, F.; Qin, Y.; Mottahedeh, J.; Luisa Iruela-Arispe, M.; et al. A Molecular Interactome of the Glioblastoma Perivascular Niche Reveals Integrin Binding Sialoprotein as a Key Mediator of Tumor Cell Migration. SSRN Electron. J. 2022, 41, 111511. [Google Scholar] [CrossRef]
- Hurtado-Alvarado, G.; Cabañas-Morales, A.M.; Gómez-Gónzalez, B. Pericytes: Brain-Immune Interface Modulators. Front. Integr. Neurosci. 2014, 7, 80. [Google Scholar] [CrossRef]
- Kaushik, S.; Bandyopadhyay, U.; Sridhar, S.; Kiffin, R.; Martinez-Vicente, M.; Kon, M.; Orenstein, S.J.; Wong, E.; Cuervo, A.M. Chaperone-Mediated Autophagy at a Glance. J. Cell Sci. 2011, 124, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Lukáš, Z.; Dvořák, K. Adhesion Molecules in Biology and Oncology. Acta Vet. Brno 2004, 73, 93–104. [Google Scholar] [CrossRef]
- Mala, U.; Baral, T.K.; Somasundaram, K. Integrative Analysis of Cell Adhesion Molecules in Glioblastoma Identified Prostaglandin F2 Receptor Inhibitor (PTGFRN) as an Essential Gene. BMC Cancer 2022, 22, 642. [Google Scholar] [CrossRef] [PubMed]
- Hall, A. The Cytoskeleton and Cancer. Cancer Metastasis Rev. 2009, 28, 5–14. [Google Scholar] [CrossRef]
- Absi, A.A.; Al Absi, A.; Wurzer, H.; Guerin, C.; Hoffmann, C.; Moreau, F.; Mao, X.; Brown-Clay, J.; Petrolli, R.; Casellas, C.P.; et al. Actin Cytoskeleton Remodeling Drives Breast Cancer Cell Escape from Natural Killer–Mediated Cytotoxicity. Cancer Res. 2018, 78, 5631–5643. [Google Scholar] [CrossRef]
- Ramsay, A.G.; Johnson, A.J.; Lee, A.M.; Gorgün, G.; Le Dieu, R.; Blum, W.; Byrd, J.C.; Gribben, J.G. Chronic Lymphocytic Leukemia T Cells Show Impaired Immunological Synapse Formation That Can Be Reversed with an Immunomodulating Drug. J. Clin. Investig. 2008, 118, 2427–2437. [Google Scholar] [CrossRef]
- Ramsay, A.G.; Evans, R.; Kiaii, S.; Svensson, L.; Hogg, N.; Gribben, J.G. Chronic Lymphocytic Leukemia Cells Induce Defective LFA-1-Directed T-Cell Motility by Altering Rho GTPase Signaling That Is Reversible with Lenalidomide. Blood 2013, 121, 2704–2714. [Google Scholar] [CrossRef]
- Jackson, S.; ElAli, A.; Virgintino, D.; Gilbert, M.R. Blood-Brain Barrier Pericyte Importance in Malignant Gliomas: What We Can Learn from Stroke and Alzheimer’s Disease. Neuro-Oncol. 2017, 19, 1173–1182. [Google Scholar] [CrossRef] [PubMed]
- Nisancioglu, M.H.; Betsholtz, C.; Genové, G. The Absence of Pericytes Does Not Increase the Sensitivity of Tumor Vasculature to Vascular Endothelial Growth Factor-A Blockade. Cancer Res. 2010, 70, 5109–5115. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Huang, Z.; Zhou, W.; Wu, Q.; Donnola, S.; Liu, J.K.; Fang, X.; Sloan, A.E.; Mao, Y.; Lathia, J.D.; et al. Glioblastoma Stem Cells Generate Vascular Pericytes to Support Vessel Function and Tumor Growth. Cell 2013, 153, 139–152. [Google Scholar] [CrossRef] [PubMed]
- King, N.E.; Courtney, J.-M.; Brown, L.S.; Foster, C.G.; Cashion, J.M.; Attrill, E.; Premilovac, D.; Howells, D.W.; Sutherland, B.A. Pharmacological PDGFRβ Inhibitors Imatinib and Sunitinib Cause Human Brain Pericyte Death in Vitro. Toxicol. Appl. Pharmacol. 2022, 444, 116025. [Google Scholar] [CrossRef]
- Zins, K.; Gunawardhana, S.; Lucas, T.; Abraham, D.; Aharinejad, S. Targeting Cdc42 with the Small Molecule Drug AZA197 Suppresses Primary Colon Cancer Growth and Prolongs Survival in a Preclinical Mouse Xenograft Model by Downregulation of PAK1 Activity. J. Transl. Med. 2013, 11, 295. [Google Scholar] [CrossRef]
- Brindani, N.; Vuong, L.M.; Acquistapace, I.M.; La Serra, M.A.; Ortega, J.A.; Veronesi, M.; Bertozzi, S.M.; Summa, M.; Girotto, S.; Bertorelli, R.; et al. Design, Synthesis, and Characterization of CDC42 GTPase Interaction Inhibitors for the Treatment of Cancer. J. Med. Chem. 2023, 66, 5981–6001. [Google Scholar] [CrossRef]
- Coelho, B.P.; Fernandes, C.F.D.L.; Boccacino, J.M.; Souza, M.C.D.S.; Melo-Escobar, M.I.; Alves, R.N.; Prado, M.B.; Iglesia, R.P.; Cangiano, G.; Mazzaro, G.L.R.; et al. Multifaceted WNT Signaling at the Crossroads Between Epithelial-Mesenchymal Transition and Autophagy in Glioblastoma. Front. Oncol. 2020, 10, 597743. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pombero, A.; Garcia-Lopez, R.; Martínez, S. Pericyte–Glioblastoma Cell Interaction: A Key Target to Prevent Glioblastoma Progression. Cells 2023, 12, 1324. https://doi.org/10.3390/cells12091324
Pombero A, Garcia-Lopez R, Martínez S. Pericyte–Glioblastoma Cell Interaction: A Key Target to Prevent Glioblastoma Progression. Cells. 2023; 12(9):1324. https://doi.org/10.3390/cells12091324
Chicago/Turabian StylePombero, Ana, Raquel Garcia-Lopez, and Salvador Martínez. 2023. "Pericyte–Glioblastoma Cell Interaction: A Key Target to Prevent Glioblastoma Progression" Cells 12, no. 9: 1324. https://doi.org/10.3390/cells12091324