
Neuroimmune Interactions in Fetal Alcohol Spectrum Disorders: Potential Therapeutic Targets and Intervention Strategies

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
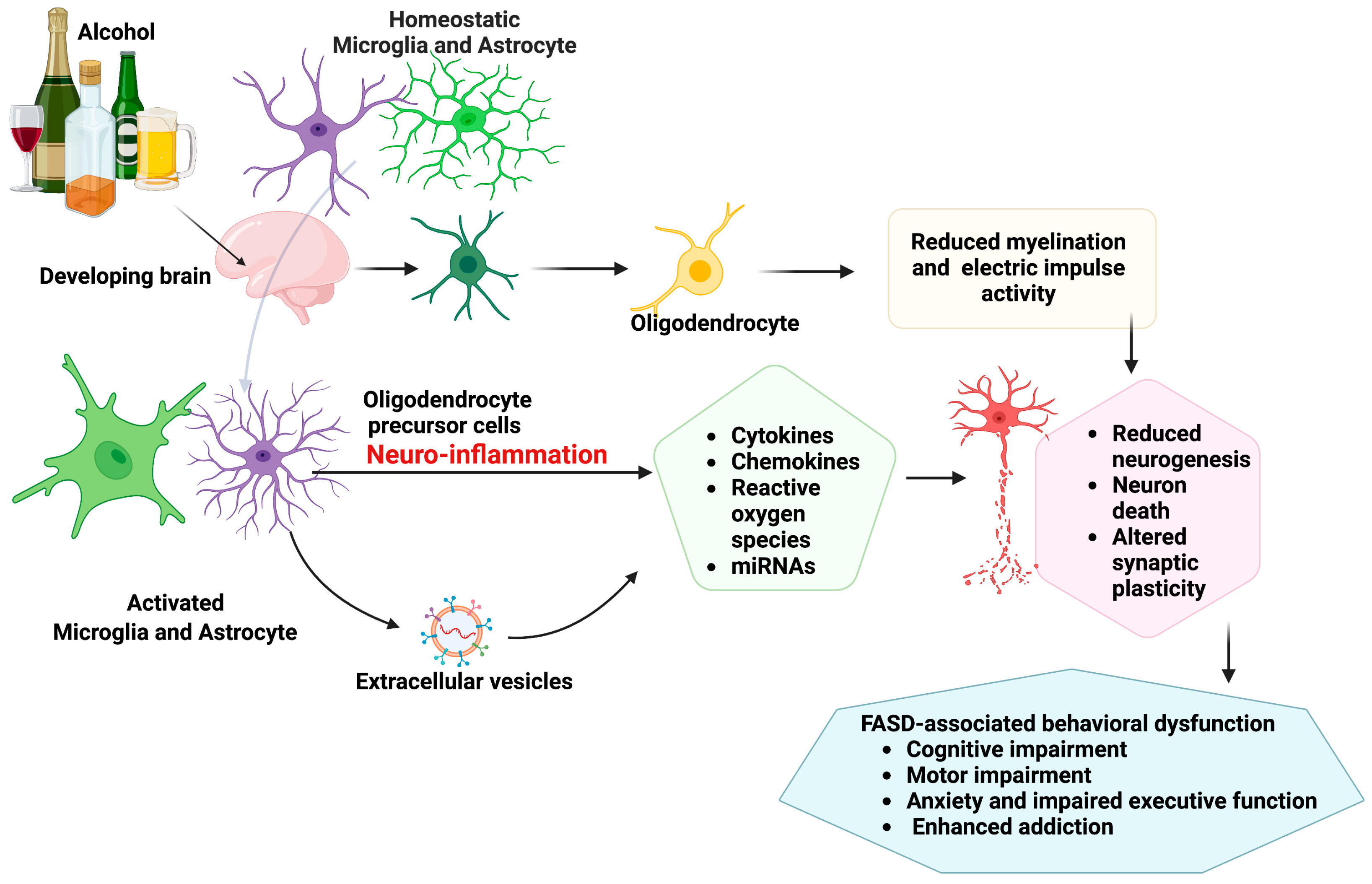
2. Glial Cell Pathologies and Neuroimmune Crosstalk in Fetal Alcohol Spectrum Disorder
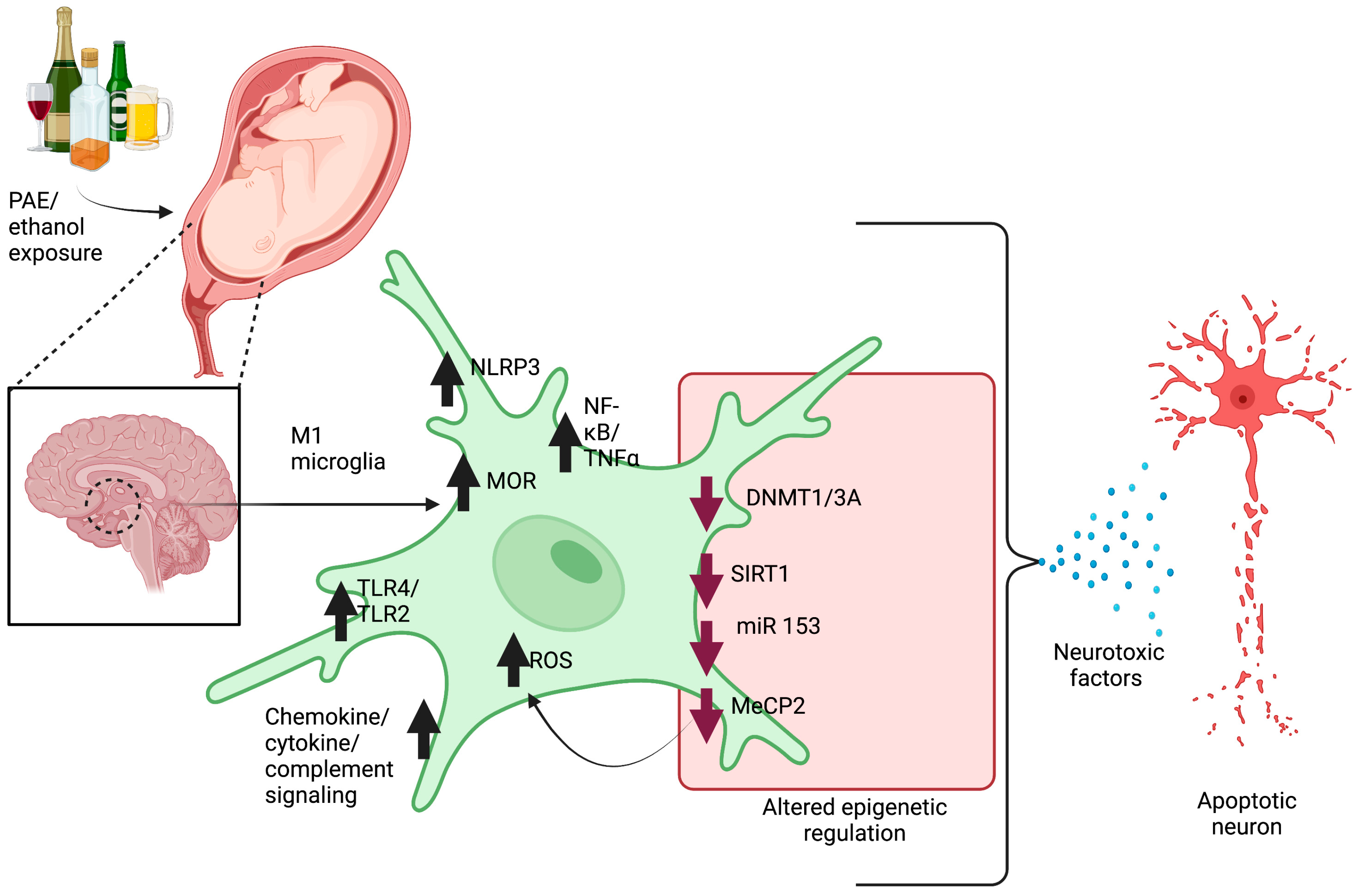
2.1. Microglia Pathology
2.2. Astrocytes Pathology
2.3. Oligodendrocyte Pathology
2.4. Neuron–Microglia Immune Interactions and Epigenetic Involvement in Pathophysiology of FASD
2.5. Extracellular Vesicles in Neuron–Glial Crosstalk
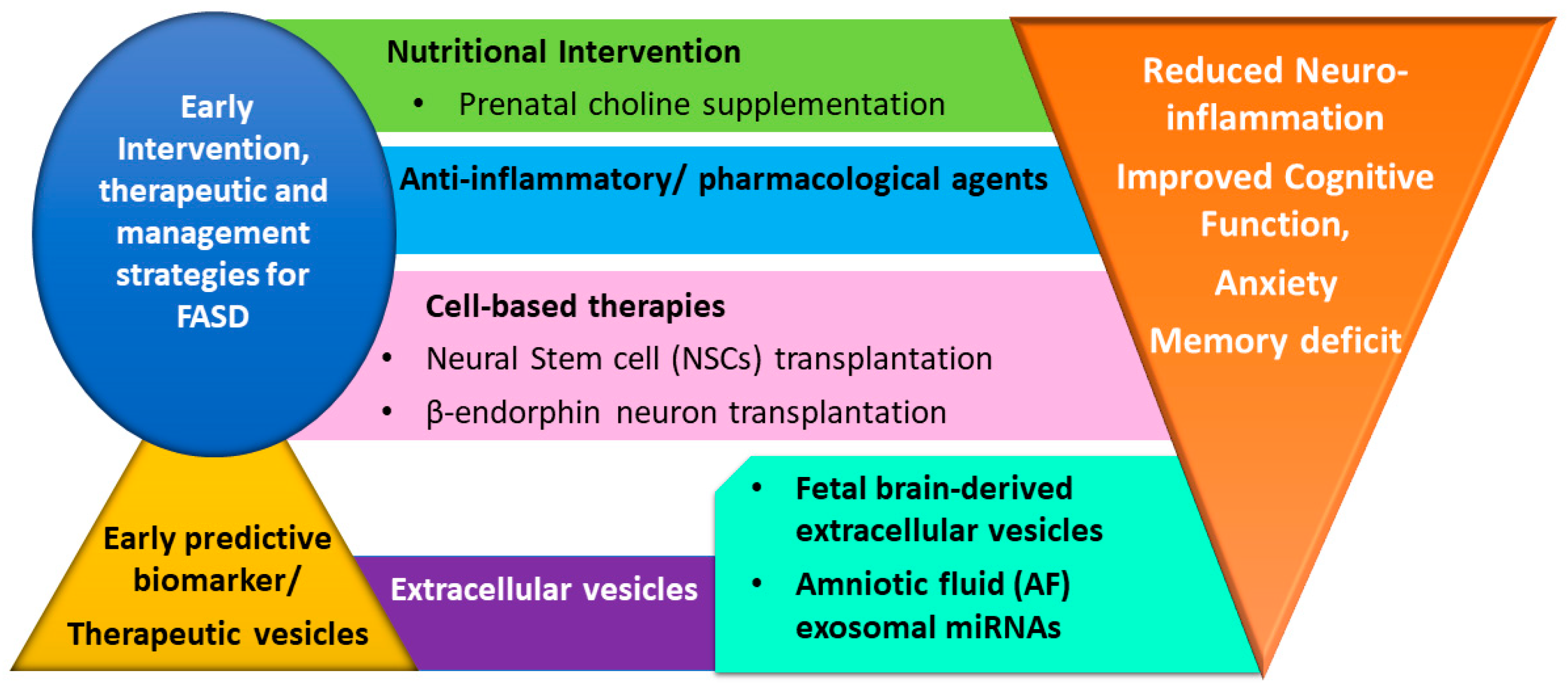
3. Therapeutic Intervention Strategies for Fetal Alcohol Spectrum Disorders (FASD)
3.1. Nutritional Supplementation
3.2. Anti-Inflammatory and Pharmacological Agents
3.3. Cell-Based Therapies
3.4. Small Extracellular Vesicles (Exosomes) as Predictive Biomarker and Therapeutic Carrier Vesicles
3.5. Outstanding Questions
- (i)
- Early EV-based intervention strategies have the potential to mitigate FASD-associated neurobehavioral abnormalities and need to be further investigated;
- (ii)
- Early diagnosis of FASD using exosome-derived proteins or miRNAs as predictive biomarkers will be helpful in devising early intervention strategies.
4. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Joseph, J.J.; Mela, M.; Pei, J. Aggressive Behaviour and Violence in Children and Adolescents with FASD: A Synthesizing Review. Clin. Psychol. Rev. 2022, 94, 102155. [Google Scholar] [CrossRef]
- Ordenewitz, L.K.; Weinmann, T.; Schlüter, J.A.; Moder, J.E.; Jung, J.; Kerber, K.; Greif-Kohistani, N.; Heinen, F.; Landgraf, M.N. Evidence-Based Interventions for Children and Adolescents with Fetal Alcohol Spectrum Disorders—A Systematic Review. Eur. J. Paediatr. Neurol. 2021, 33, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Lange, S.; Probst, C.; Gmel, G.; Rehm, J.; Burd, L.; Popova, S. Global Prevalence of Fetal Alcohol Spectrum Disorder among Children and Youth: A Systematic Review and Meta-Analysis. JAMA Pediatr. 2017, 171, 948–956. Available online: https://jamanetwork.com/journals/jamapediatrics/article-abstract/2649225 (accessed on 30 June 2023). [CrossRef]
- Stephen, J.M.; Kodituwakku, P.W.; Kodituwakku, E.L.; Romero, L.; Peters, A.M.; Sharadamma, N.M.; Caprihan, A.; Coffman, B.A. Delays in Auditory Processing Identified in Preschool Children with FASD. Alcohol. Clin. Exp. Res. 2012, 36, 1720–1727. [Google Scholar] [CrossRef] [PubMed]
- Olson, H.C.; Feldman, J.J.; Streissguth, A.P.; Sampson, P.D.; Bookstein, F.L. Neuropsychological Deficits in Adolescents with Fetal Alcohol Syndrome: Clinical Findings. Alcohol. Clin. Exp. Res. 1998, 22, 1998–2012. [Google Scholar] [CrossRef] [PubMed]
- Treit, S.; Beaulieu, C. Imaging Brain Structure in FASD. In Ethical and Legal Perspectives in Fetal Alcohol Spectrum Disorders (FASD): Foundational Issues; Jonsson, E., Clarren, S., Binnie, I., Eds.; International Library of Ethics, Law, and the New Medicine; Springer International Publishing: Cham, Switzerland, 2018; pp. 77–93. ISBN 978-3-319-71755-5. [Google Scholar]
- Wieczorek, L.; Fish, E.W.; O’Leary-Moore, S.K.; Parnell, S.E.; Sulik, K.K. Hypothalamic-Pituitary-Adrenal Axis and Behavioral Dysfunction Following Early Binge-like Prenatal Alcohol Exposure in Mice. Alcohol 2015, 49, 207–217. [Google Scholar] [CrossRef]
- Mead, E.A.; Sarkar, D.K. Fetal Alcohol Spectrum Disorders and Their Transmission through Genetic and Epigenetic Mechanisms. Front. Genet. 2014, 5, 154. [Google Scholar] [CrossRef]
- Ruffaner-Hanson, C.; Noor, S.; Sun, M.S.; Solomon, E.; Marquez, L.E.; Rodriguez, D.E.; Allan, A.M.; Caldwell, K.K.; Bakhireva, L.N.; Milligan, E.D. The Maternal-Placental-Fetal Interface: Adaptations of the HPA Axis and Immune Mediators Following Maternal Stress and Prenatal Alcohol Exposure. Exp. Neurol. 2022, 355, 114121. [Google Scholar] [CrossRef]
- Gangisetty, O.; Bekdash, R.; Maglakelidze, G.; Sarkar, D.K. Fetal Alcohol Exposure Alters Proopiomelanocortin Gene Expression and Hypothalamic-Pituitary-Adrenal Axis Function via Increasing MeCP2 Expression in the Hypothalamus. PLoS ONE 2014, 9, e113228. [Google Scholar] [CrossRef]
- Das, U.; Gangisetty, O.; Chaudhary, S.; Tarale, P.; Rousseau, B.; Price, J.; Frazier, I.; Sarkar, D.K. Epigenetic Insight into Effects of Prenatal Alcohol Exposure on Stress Axis Development: Systematic Review with Meta-Analytic Approaches. Alcohol. Clin. Exp. Res. 2023, 47, 18–35. [Google Scholar] [CrossRef]
- Jha, M.K.; Jo, M.; Kim, J.-H.; Suk, K. Microglia-Astrocyte Crosstalk: An Intimate Molecular Conversation. Neuroscientist 2019, 25, 227–240. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Choi, J.; Yoon, B.-E. Neuron-Glia Interactions in Neurodevelopmental Disorders. Cells 2020, 9, 2176. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Cottarelli, A.; Agalliu, D. Neuronal and Glial Regulation of CNS Angiogenesis and Barriergenesis. Development 2020, 147, dev182279. [Google Scholar] [CrossRef] [PubMed]
- Rigby, M.J.; Gomez, T.M.; Puglielli, L. Glial Cell-Axonal Growth Cone Interactions in Neurodevelopment and Regeneration. Front. Neurosci. 2020, 14, 203. [Google Scholar] [CrossRef] [PubMed]
- Barres, B.A.; Barde, Y.-A. Neuronal and Glial Cell Biology. Curr. Opin. Neurobiol. 2000, 10, 642–648. [Google Scholar] [CrossRef]
- Norris, G.T.; Kipnis, J. Immune Cells and CNS Physiology: Microglia and Beyond. J. Exp. Med. 2018, 216, 60–70. [Google Scholar] [CrossRef]
- Mrdjen, D.; Pavlovic, A.; Hartmann, F.J.; Schreiner, B.; Utz, S.G.; Leung, B.P.; Lelios, I.; Heppner, F.L.; Kipnis, J.; Merkler, D.; et al. High-Dimensional Single-Cell Mapping of Central Nervous System Immune Cells Reveals Distinct Myeloid Subsets in Health, Aging, and Disease. Immunity 2018, 48, 380–395. [Google Scholar] [CrossRef]
- Saito, M.; Chakraborty, G.; Hui, M.; Masiello, K.; Saito, M. Ethanol-Induced Neurodegeneration and Glial Activation in the Developing Brain. Brain Sci. 2016, 6, 31. [Google Scholar] [CrossRef]
- Guergues, J.; Wohlfahrt, J.; Zhang, P.; Liu, B.; Stevens, S.M., Jr. Deep Proteome Profiling Reveals Novel Pathways Associated with Pro-Inflammatory and Alcohol-Induced Microglial Activation Phenotypes. J. Proteom. 2020, 220, 103753. [Google Scholar] [CrossRef]
- Campbell, J.C.; Stipcevic, T.; Flores, R.E.; Perry, C.; Kippin, T.E. Alcohol Exposure Inhibits Adult Neural Stem Cell Proliferation. Exp. Brain Res. 2014, 232, 2775–2784. [Google Scholar] [CrossRef]
- Boyadjieva, N.; Advis, J.P.; Sarkar, D.K. Role of β-Endorphin, Corticotropin-Releasing Hormone, and Autonomic Nervous System in Mediation of the Effect of Chronic Ethanol on Natural Killer Cell Cytolytic Activity. Alcohol. Clin. Exp. Res. 2006, 30, 1761–1767. [Google Scholar] [CrossRef] [PubMed]
- Prinz, M.; Mildner, A. Microglia in the CNS: Immigrants from Another World. Glia 2011, 59, 177–187. [Google Scholar] [CrossRef] [PubMed]
- von Bernhardi, R.; Heredia, F.; Salgado, N.; Muñoz, P. Microglia Function in the Normal Brain. In Glial Cells in Health and Disease of the CNS; von Bernhardi, R., Ed.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2016; pp. 67–92. ISBN 978-3-319-40764-7. [Google Scholar]
- Cherry, J.D.; Tripodis, Y.; Alvarez, V.E.; Huber, B.; Kiernan, P.T.; Daneshvar, D.H.; Mez, J.; Montenigro, P.H.; Solomon, T.M.; Alosco, M.L.; et al. Microglial Neuroinflammation Contributes to Tau Accumulation in Chronic Traumatic Encephalopathy. Acta Neuropathol. Commun. 2016, 4, 112. [Google Scholar] [CrossRef] [PubMed]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic Pruning by Microglia Is Necessary for Normal Brain Development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef] [PubMed]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia Sculpt Postnatal Neural Circuits in an Activity and Complement-Dependent Manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef]
- Cornell, J.; Salinas, S.; Huang, H.-Y.; Zhou, M. Microglia Regulation of Synaptic Plasticity and Learning and Memory. Neural Regen. Res. 2022, 17, 705–716. [Google Scholar] [CrossRef]
- Peng, H.; Geil Nickell, C.R.; Chen, K.Y.; McClain, J.A.; Nixon, K. Increased Expression of M1 and M2 Phenotypic Markers in Isolated Microglia after Four-Day Binge Alcohol Exposure in Male Rats. Alcohol 2017, 62, 29–40. [Google Scholar] [CrossRef]
- Henriques, J.F.; Portugal, C.C.; Canedo, T.; Relvas, J.B.; Summavielle, T.; Socodato, R. Microglia and Alcohol Meet at the Crossroads: Microglia as Critical Modulators of Alcohol Neurotoxicity. Toxicol. Lett. 2018, 283, 21–31. [Google Scholar] [CrossRef]
- Drew, P.D.; Johnson, J.W.; Douglas, J.C.; Phelan, K.D.; Kane, C.J.M. Pioglitazone Blocks Ethanol Induction of Microglial Activation and Immune Responses in the Hippocampus, Cerebellum, and Cerebral Cortex in a Mouse Model of Fetal Alcohol Spectrum Disorders. Alcohol. Clin. Exp. Res. 2015, 39, 445–454. [Google Scholar] [CrossRef]
- Lynch, M.A. The Multifaceted Profile of Activated Microglia. Mol. Neurobiol. 2009, 40, 139–156. [Google Scholar] [CrossRef]
- Chastain, L.G.; Franklin, T.; Gangisetty, O.; Cabrera, M.A.; Mukherjee, S.; Shrivastava, P.; Jabbar, S.; Sarkar, D.K. Early Life Alcohol Exposure Primes Hypothalamic Microglia to Later-Life Hypersensitivity to Immune Stress: Possible Epigenetic Mechanism. Neuropsychopharmacology 2019, 44, 1579–1588. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Cabrera, M.A.; Boyadjieva, N.I.; Berger, G.; Rousseau, B.; Sarkar, D.K. Alcohol Increases Exosome Release from Microglia to Promote Complement C1q-Induced Cellular Death of Proopiomelanocortin Neurons in the Hypothalamus in a Rat Model of Fetal Alcohol Spectrum Disorders. J. Neurosci. 2020, 40, 7965–7979. [Google Scholar] [CrossRef] [PubMed]
- Boyadjieva, N.I.; Sarkar, D.K. Cyclic Adenosine Monophosphate and Brain-Derived Neurotrophic Factor Decreased Oxidative Stress and Apoptosis in Developing Hypothalamic Neuronal Cells: Role of Microglia. Alcohol. Clin. Exp. Res. 2013, 37, 1370–1379. [Google Scholar] [CrossRef] [PubMed]
- Bekdash, R.; Zhang, C.; Sarkar, D. Fetal Alcohol Programming of Hypothalamic Proopiomelanocortin System by Epigenetic Mechanisms and Later Life Vulnerability to Stress. Alcohol. Clin. Exp. Res. 2014, 38, 2323–2330. [Google Scholar] [CrossRef] [PubMed]
- Boyadjieva, N.I.; Sarkar, D.K. Microglia Play a Role in Ethanol-Induced Oxidative Stress and Apoptosis in Developing Hypothalamic Neurons. Alcohol. Clin. Exp. Res. 2013, 37, 252–262. [Google Scholar] [CrossRef]
- Shrivastava, P.; Cabrera, M.A.; Chastain, L.G.; Boyadjieva, N.I.; Jabbar, S.; Franklin, T.; Sarkar, D.K. Mu-Opioid Receptor and Delta-Opioid Receptor Differentially Regulate Microglial Inflammatory Response to Control Proopiomelanocortin Neuronal Apoptosis in the Hypothalamus: Effects of Neonatal Alcohol. J. Neuroinflamm. 2017, 14, 83. [Google Scholar] [CrossRef]
- Boyadjieva, N.I.; Sarkar, D.K. Role of Microglia in Ethanol’s Apoptotic Action on Hypothalamic Neuronal Cells in Primary Cultures. Alcohol. Clin. Exp. Res. 2010, 34, 1835–1842. [Google Scholar] [CrossRef]
- Lawrimore, C.J.; Crews, F.T. Ethanol, TLR3, and TLR4 Agonists Have Unique Innate Immune Responses in Neuron-Like SH-SY5Y and Microglia-Like BV2. Alcohol. Clin. Exp. Res. 2017, 41, 939–954. [Google Scholar] [CrossRef]
- Tao, L.; Zhang, L.; Gao, R.; Jiang, F.; Cao, J.; Liu, H. Andrographolide Alleviates Acute Brain Injury in a Rat Model of Traumatic Brain Injury: Possible Involvement of Inflammatory Signaling. Front. Neurosci. 2018, 12, 657. [Google Scholar] [CrossRef]
- Cui, W.; Sun, C.; Ma, Y.; Wang, S.; Wang, X.; Zhang, Y. Inhibition of TLR4 Induces M2 Microglial Polarization and Provides Neuroprotection via the NLRP3 Inflammasome in Alzheimer’s Disease. Front. Neurosci. 2020, 14, 444. [Google Scholar] [CrossRef]
- Fernandez-Lizarbe, S.; Montesinos, J.; Guerri, C. Ethanol Induces TLR4/TLR2 Association, Triggering an Inflammatory Response in Microglial Cells. J. Neurochem. 2013, 126, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Alfonso-Loeches, S.; Ureña-Peralta, J.; Morillo-Bargues, M.J.; Gómez-Pinedo, U.; Guerri, C. Ethanol-Induced TLR4/NLRP3 Neuroinflammatory Response in Microglial Cells Promotes Leukocyte Infiltration Across the BBB. Neurochem. Res. 2016, 41, 193–209. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Zhang, D.; Yu, H.; Yuan, H.; Shen, H.; Lan, X.; Liu, H.; Chen, X.; Meng, F.; Wu, X.; et al. Gut Microbiota Regulates Chronic Ethanol Exposure-Induced Depressive-like Behavior through Hippocampal NLRP3-Mediated Neuroinflammation. Mol. Psychiatry 2023, 28, 919–930. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K. Role of MCP-1 and CCR2 in Ethanol-Induced Damage in the Developing Brain. Ph.D. Thesis, University of Kentucky, Lexington, KY, USA, 2019. [Google Scholar] [CrossRef]
- Coccini, T.; Ottonello, M.; Spigno, P.; Malovini, A.; Fiabane, E.; Roda, E.; Signorini, C.; Pistarini, C. Biomarkers for Alcohol Abuse/Withdrawal and Their Association with Clinical Scales and Temptation to Drink. A Prospective Pilot Study during 4-Week Residential Rehabilitation. Alcohol 2021, 94, 43–56. [Google Scholar] [CrossRef]
- Zhang, K.; Wang, H.; Xu, M.; Frank, J.A.; Luo, J. Role of MCP-1 and CCR2 in Ethanol-Induced Neuroinflammation and Neurodegeneration in the Developing Brain. J. Neuroinflamm. 2018, 15, 197. [Google Scholar] [CrossRef]
- Sofroniew, M.V. Astrocyte Reactivity: Subtypes, States, and Functions in CNS Innate Immunity. Trends Immunol. 2020, 41, 758–770. [Google Scholar] [CrossRef]
- Sloan, S.A.; Barres, B.A. Mechanisms of Astrocyte Development and Their Contributions to Neurodevelopmental Disorders. Curr. Opin. Neurobiol. 2014, 27, 75–81. [Google Scholar] [CrossRef]
- Molofsky, A.V.; Krenick, R.; Ullian, E.; Tsai, H.; Deneen, B.; Richardson, W.D.; Barres, B.A.; Rowitch, D.H. Astrocytes and Disease: A Neurodevelopmental Perspective. Genes Dev. 2012, 26, 891–907. [Google Scholar] [CrossRef]
- Jurga, A.M.; Paleczna, M.; Kadluczka, J.; Kuter, K.Z. Beyond the GFAP-Astrocyte Protein Markers in the Brain. Biomolecules 2021, 11, 1361. [Google Scholar] [CrossRef]
- Lawrimore, C.J.; Coleman, L.G.; Crews, F.T. Ethanol Induces Interferon Expression in Neurons via TRAIL: Role of Astrocyte-to-Neuron Signaling. Psychopharmacology 2019, 236, 2881–2897. [Google Scholar] [CrossRef]
- Miller, M.W.; Robertson, S. Prenatal Exposure to Ethanol Alters the Postnatal Development and Transformation of Radial Glia to Astrocytes in the Cortex. J. Comp. Neurol. 1993, 337, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Vesce, S.; Rossi, D.; Brambilla, L.; Volterra, A. Glutamate Release from Astrocytes in Physiological Conditions and in Neurodegenerative Disorders Characterized by Neuroinflammation. Int. Rev. Neurobiol. 2007, 82, 57–71. [Google Scholar]
- Acioglu, C.; Li, L.; Elkabes, S. Contribution of Astrocytes to Neuropathology of Neurodegenerative Diseases. Brain Res. 2021, 1758, 147291. [Google Scholar] [CrossRef] [PubMed]
- Clark, D.P.Q.; Perreau, V.M.; Shultz, S.R.; Brady, R.D.; Lei, E.; Dixit, S.; Taylor, J.M.; Beart, P.M.; Boon, W.C. Inflammation in Traumatic Brain Injury: Roles for Toxic A1 Astrocytes and Microglial–Astrocytic Crosstalk. Neurochem. Res. 2019, 44, 1410–1424. [Google Scholar] [CrossRef]
- Hinkle, J.T.; Dawson, V.L.; Dawson, T.M. The A1 Astrocyte Paradigm: New Avenues for Pharmacological Intervention in Neurodegeneration. Mov. Disord. 2019, 34, 959–969. [Google Scholar] [CrossRef]
- Moore, N.H.; Costa, L.G.; Shaffer, S.A.; Goodlett, D.R.; Guizzetti, M. Shotgun Proteomics Implicates Extracellular Matrix Proteins and Protease Systems in Neuronal Development Induced by Astrocyte Cholinergic Stimulation. J. Neurochem. 2009, 108, 891–908. [Google Scholar] [CrossRef] [PubMed]
- Guizzetti, M.; Zhang, X.; Goeke, C.; Gavin, D.P. Glia and Neurodevelopment: Focus on Fetal Alcohol Spectrum Disorders. Front. Pediatr. 2014, 2, 123. [Google Scholar] [CrossRef]
- Miller, M.W.; Potempa, G. Numbers of Neurons and Glia in Mature Rat Somatosensory Cortex: Effects of Prenatal Exposure to Ethanol. J. Comp. Neurol. 1990, 293, 92–102. [Google Scholar] [CrossRef]
- Pérez-Torrero, E.; Durán, P.; Granados, L.; Gutiérez-Ospina, G.; Cintra, L.; Díaz-Cintra, S. Effects of Acute Prenatal Ethanol Exposure on Bergmann Glia Cells Early Postnatal Development. Brain Res. 1997, 746, 305–308. [Google Scholar] [CrossRef]
- Vallés, S.; Pitarch, J.; Renau-Piqueras, J.; Guerri, C. Ethanol Exposure Affects Glial Fibrillary Acidic Protein Gene Expression and Transcription during Rat Brain Development. J. Neurochem. 2002, 69, 2484–2493. [Google Scholar] [CrossRef]
- Siqueira, M.; Araujo, A.P.B.; Gomes, F.C.A.; Stipursky, J. Ethanol Gestational Exposure Impairs Vascular Development and Endothelial Potential to Control BBB-Associated Astrocyte Function in the Developing Cerebral Cortex. Mol. Neurobiol. 2021, 58, 1755–1768. [Google Scholar] [CrossRef] [PubMed]
- Sarc, L.; Wraber, B.; Lipnik-Stangelj, M. Ethanol and Acetaldehyde Disturb TNF-Alpha and IL-6 Production in Cultured Astrocytes. Hum. Exp. Toxicol. 2011, 30, 1256–1265. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Huo, J.; Yang, M.; Zhang, G.; Wan, S.; Chen, X.; Zhang, B.; Liu, H. ERK1/2 Signalling Pathway Regulates Tubulin-Binding Cofactor B Expression and Affects Astrocyte Process Formation after Acute Foetal Alcohol Exposure. Brain Sci. 2022, 12, 813. [Google Scholar] [CrossRef] [PubMed]
- Blanco, A.M.; Vallés, S.L.; Pascual, M.; Guerri, C. Involvement of TLR4/Type I IL-1 Receptor Signaling in the Induction of Inflammatory Mediators and Cell Death Induced by Ethanol in Cultured Astrocytes. J. Immunol. 2005, 175, 6893–6899. [Google Scholar] [CrossRef]
- Mangan, M.S.J.; Olhava, E.J.; Roush, W.R.; Seidel, H.M.; Glick, G.D.; Latz, E. Targeting the NLRP3 Inflammasome in Inflammatory Diseases. Nat. Rev. Drug Discov. 2018, 17, 588–606. [Google Scholar] [CrossRef]
- Kane, C.J.M.; Drew, P.D. Neuroinflammatory Contribution of Microglia and Astrocytes in Fetal Alcohol Spectrum Disorders. J. Neurosci. Res. 2021, 99, 1973–1985. [Google Scholar] [CrossRef]
- Zou, J.; Crews, F. Inflammasome-IL-1β Signaling Mediates Ethanol Inhibition of Hippocampal Neurogenesis. Front. Neurosci. 2012, 6, 77. [Google Scholar] [CrossRef]
- Jiang, X.; Nardelli, J. Cellular and Molecular Introduction to Brain Development. Neurobiol. Dis. 2016, 92, 3–17. [Google Scholar] [CrossRef]
- Darbinian, N.; Darbinyan, A.; Merabova, N.; Bajwa, A.; Tatevosian, G.; Martirosyan, D.; Zhao, H.; Selzer, M.E.; Goetzl, L. Ethanol-Mediated Alterations in Oligodendrocyte Differentiation in the Developing Brain. Neurobiol. Dis. 2021, 148, 105181. [Google Scholar] [CrossRef]
- Stratoulias, V.; Venero, J.L.; Tremblay, M.-È.; Joseph, B. Microglial Subtypes: Diversity within the Microglial Community. EMBO J. 2019, 38, e101997. [Google Scholar] [CrossRef]
- Arifuzzaman, S.; Das, A.; Kim, S.H.; Yoon, T.; Lee, Y.S.; Jung, K.H.; Chai, Y.G. Selective Inhibition of EZH2 by a Small Molecule Inhibitor Regulates Microglial Gene Expression Essential for Inflammation. Biochem. Pharmacol. 2017, 137, 61–80. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, J.; Jiang, Q.; Zhang, L.; Song, W. Zinc Binding Groups for Histone Deacetylase Inhibitors. J. Enzym. Inhib. Med. Chem. 2018, 33, 714–721. [Google Scholar] [CrossRef]
- Carrillo-Jimenez, A.; Deniz, Ö.; Niklison-Chirou, M.V.; Ruiz, R.; Bezerra-Salomão, K.; Stratoulias, V.; Amouroux, R.; Yip, P.K.; Vilalta, A.; Cheray, M.; et al. TET2 Regulates the Neuroinflammatory Response in Microglia. Cell Rep. 2019, 29, 697–713.e8. [Google Scholar] [CrossRef]
- MacArthur, I.C.; Dawlaty, M.M. TET Enzymes and 5-Hydroxymethylcytosine in Neural Progenitor Cell Biology and Neurodevelopment. Front. Cell Dev. Biol. 2021, 9, 645335. [Google Scholar] [CrossRef]
- Chen, Y.; Ozturk, N.C.; Zhou, F.C. DNA Methylation Program in Developing Hippocampus and Its Alteration by Alcohol. PLoS ONE 2013, 8, e60503. [Google Scholar] [CrossRef]
- Kim, P.; Park, J.H.; Choi, C.S.; Choi, I.; Joo, S.H.; Kim, M.K.; Kim, S.Y.; Kim, K.C.; Park, S.H.; Kwon, K.J.; et al. Effects of Ethanol Exposure during Early Pregnancy in Hyperactive, Inattentive and Impulsive Behaviors and MeCP2 Expression in Rodent Offspring. Neurochem. Res. 2013, 38, 620–631. [Google Scholar] [CrossRef]
- Nagre, N.N.; Subbanna, S.; Shivakumar, M.; Psychoyos, D.; Basavarajappa, B.S. CB1-Receptor Knockout Neonatal Mice Are Protected against Ethanol-Induced Impairments of DNMT1, DNMT3A, and DNA Methylation. J. Neurochem. 2015, 132, 429–442. [Google Scholar] [CrossRef]
- Varadinova, M.; Boyadjieva, N. Epigenetic Mechanisms: A Possible Link between Autism Spectrum Disorders and Fetal Alcohol Spectrum Disorders. Pharmacol. Res. 2015, 102, 71–80. [Google Scholar] [CrossRef]
- Veazey, K.J.; Wang, H.; Bedi, Y.S.; Skiles, W.M.; Chang, R.C.-A.; Golding, M.C. Disconnect between Alcohol-Induced Alterations in Chromatin Structure and Gene Transcription in a Mouse Embryonic Stem Cell Model of Exposure. Alcohol 2017, 60, 121–133. [Google Scholar] [CrossRef]
- Boschen, K.E.; Gong, H.; Murdaugh, L.B.; Parnell, S.E. Knockdown of Mns1 Increases Susceptibility to Craniofacial Defects following Gastrulation-Stage Alcohol Exposure in Mice. Alcohol. Clin. Exp. Res. 2018, 42, 2136–2143. [Google Scholar] [CrossRef]
- Alberry, B.; Laufer, B.I.; Chater-Diehl, E.; Singh, S.M. Epigenetic Impacts of Early Life Stress in Fetal Alcohol Spectrum Disorders Shape the Neurodevelopmental Continuum. Front. Mol. Neurosci. 2021, 14, 671891. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef]
- Augusto-Oliveira, M.; Arrifano, G.P.; Delage, C.I.; Tremblay, M.-È.; Crespo-Lopez, M.E.; Verkhratsky, A. Plasticity of Microglia. Biol. Rev. 2022, 97, 217–250. [Google Scholar] [CrossRef]
- Cheray, M.; Joseph, B. Epigenetics Control Microglia Plasticity. Front. Cell. Neurosci. 2018, 12, 243. [Google Scholar] [CrossRef]
- Martins-Ferreira, R.; Leal, B.; Costa, P.P.; Ballestar, E. Microglial Innate Memory and Epigenetic Reprogramming in Neurological Disorders. Prog. Neurobiol. 2021, 200, 101971. [Google Scholar] [CrossRef]
- Crews, F.T.; Zou, J.; Coleman, L.G., Jr. Extracellular Microvesicles Promote Microglia-Mediated Pro-Inflammatory Responses to Ethanol. J. Neurosci. Res. 2021, 99, 1940–1956. [Google Scholar] [CrossRef]
- Ibáñez, F.; Ureña-Peralta, J.R.; Costa-Alba, P.; Torres, J.-L.; Laso, F.-J.; Marcos, M.; Guerri, C.; Pascual, M. Circulating MicroRNAs in Extracellular Vesicles as Potential Biomarkers of Alcohol-Induced Neuroinflammation in Adolescence: Gender Differences. Int. J. Mol. Sci. 2020, 21, 6730. [Google Scholar] [CrossRef]
- Ibáñez, F.; Montesinos, J.; Ureña-Peralta, J.R.; Guerri, C.; Pascual, M. TLR4 Participates in the Transmission of Ethanol-Induced Neuroinflammation via Astrocyte-Derived Extracellular Vesicles. J. Neuroinflamm. 2019, 16, 136. [Google Scholar] [CrossRef]
- Tseng, A.M.; Chung, D.D.; Pinson, M.R.; Salem, N.A.; Eaves, S.E.; Miranda, R.C. Ethanol Exposure Increases MiR-140 in Extracellular Vesicles: Implications for Fetal Neural Stem Cell Proliferation and Maturation. Alcohol. Clin. Exp. Res. 2019, 43, 1414–1426. [Google Scholar] [CrossRef]
- Chung, D.D.; Mahnke, A.H.; Pinson, M.R.; Salem, N.A.; Lai, M.S.; Collins, N.P.; Hillhouse, A.E.; Miranda, R.C. Sex Differences in the Transcriptome of Extracellular Vesicles Secreted by Fetal Neural Stem Cells and Effects of Chronic Alcohol Exposure. Biol. Sex Differ. 2023, 14, 19. [Google Scholar] [CrossRef]
- Chung, D.D.; Pinson, M.R.; Mahnke, A.H.; Salem, N.A.; Le, K.T.; Payne, E.A.; Lehman, T.E.; Weintraub, S.T.; Miranda, R.C. Dose-Related Shifts in Proteome and Function of Extracellular Vesicles Secreted by Fetal Neural Stem Cells Following Chronic Alcohol Exposure. Heliyon 2022, 8, e11348. [Google Scholar] [CrossRef]
- Di Rocco, G.; Baldari, S.; Pani, G.; Toietta, G. Stem Cells under the Influence of Alcohol: Effects of Ethanol Consumption on Stem/Progenitor Cells. Cell. Mol. Life Sci. 2019, 76, 231–244. [Google Scholar] [CrossRef]
- Bell, C.C.; Chimata, R. Prevalence of Neurodevelopmental Disorders Among Low-Income African Americans at a Clinic on Chicago’s South Side. Psychiatr. Serv. 2015, 66, 539–542. [Google Scholar] [CrossRef]
- CDC. Fetal Alcohol Spectrum Disorders (FASDs). Available online: https://www.cdc.gov/ncbddd/fasd/index.html (accessed on 17 April 2023).
- Derbyshire, E.; Obeid, R. Choline, Neurological Development and Brain Function: A Systematic Review Focusing on the First 1000 Days. Nutrients 2020, 12, 1731. [Google Scholar] [CrossRef]
- Fuglestad, A.J.; Miller, N.C.; Fink, B.A.; Boys, C.J.; Eckerle, J.K.; Georgieff, M.K.; Wozniak, J.R. Neurophysiological Correlates of Memory Change in Children with Fetal Alcohol Spectrum Disorders Treated with Choline. Front. Psychol. 2022, 13, 936019. [Google Scholar] [CrossRef]
- Gimbel, B.A.; Anthony, M.E.; Ernst, A.M.; Roediger, D.J.; de Water, E.; Eckerle, J.K.; Boys, C.J.; Radke, J.P.; Mueller, B.A.; Fuglestad, A.J.; et al. Long-Term Follow-up of a Randomized Controlled Trial of Choline for Neurodevelopment in Fetal Alcohol Spectrum Disorder: Corpus Callosum White Matter Microstructure and Neurocognitive Outcomes. J. Neurodev. Disord. 2022, 14, 59. [Google Scholar] [CrossRef]
- Wozniak, J.R.; Fink, B.A.; Fuglestad, A.J.; Eckerle, J.K.; Boys, C.J.; Sandness, K.E.; Radke, J.P.; Miller, N.C.; Lindgren, C.; Brearley, A.M.; et al. Four-Year Follow-up of a Randomized Controlled Trial of Choline for Neurodevelopment in Fetal Alcohol Spectrum Disorder. J. Neurodev. Disord. 2020, 12, 9. [Google Scholar] [CrossRef]
- Baker, J.A.; Bodnar, T.S.; Breit, K.R.; Weinberg, J.; Thomas, J.D. Choline Supplementation Alters Hippocampal Cytokine Levels in Adolescence and Adulthood in an Animal Model of Fetal Alcohol Spectrum Disorders. Cells 2023, 12, 546. [Google Scholar] [CrossRef]
- Baker, J.A.; Breit, K.R.; Bodnar, T.S.; Weinberg, J.; Thomas, J.D. Choline Supplementation Modifies the Effects of Developmental Alcohol Exposure on Immune Responses in Adult Rats. Nutrients 2022, 14, 2868. [Google Scholar] [CrossRef]
- Grafe, E.L.; Wade, M.M.M.; Hodson, C.E.; Thomas, J.D.; Christie, B.R. Postnatal Choline Supplementation Rescues Deficits in Synaptic Plasticity Following Prenatal Ethanol Exposure. Nutrients 2022, 14, 2004. [Google Scholar] [CrossRef]
- Bottom, R.T.; Abbott, C.W.; Huffman, K.J. Rescue of Ethanol-Induced FASD-like Phenotypes via Prenatal Co-Administration of Choline. Neuropharmacology 2020, 168, 107990. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Drew, P.D. Peroxisome Proliferator-Activated Receptor-γ Agonists Suppress the Production of IL-12 Family Cytokines by Activated Glia. J. Immunol. 2007, 178, 1904–1913. [Google Scholar] [CrossRef] [PubMed]
- Kane, C.J.M.; Phelan, K.D.; Han, L.; Smith, R.R.; Xie, J.; Douglas, J.C.; Drew, P.D. Protection of Neurons and Microglia against Ethanol in a Mouse Model of Fetal Alcohol Spectrum Disorders by Peroxisome Proliferator-Activated Receptor-γ Agonists. Brain Behav. Immun. 2011, 25 (Suppl. S1), S137–S145. [Google Scholar] [CrossRef] [PubMed]
- Patten, A.R.; Sickmann, H.M.; Dyer, R.A.; Innis, S.M.; Christie, B.R. Omega-3 Fatty Acids Can Reverse the Long-Term Deficits in Hippocampal Synaptic Plasticity Caused by Prenatal Ethanol Exposure. Neurosci. Lett. 2013, 551, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Helland, I.B.; Smith, L.; Saarem, K.; Saugstad, O.D.; Drevon, C.A. Maternal Supplementation with Very-Long-Chain n-3 Fatty Acids during Pregnancy and Lactation Augments Children’s IQ at 4 Years of Age. Pediatrics 2003, 111, e39–e44. [Google Scholar] [CrossRef]
- Wellmann, K.A.; George, F.; Brnouti, F.; Mooney, S.M. Docosahexaenoic Acid Partially Ameliorates Deficits in Social Behavior and Ultrasonic Vocalizations Caused by Prenatal Ethanol Exposure. Behav. Brain Res. 2015, 286, 201–211. [Google Scholar] [CrossRef]
- Ren, Z.; Wang, X.; Xu, M.; Frank, J.A.; Luo, J. Minocycline Attenuates Ethanol-Induced Cell Death and Microglial Activation in the Developing Spinal Cord. Alcohol 2019, 79, 25–35. [Google Scholar] [CrossRef]
- Cantacorps, L.; Montagud-Romero, S.; Valverde, O. Curcumin Treatment Attenuates Alcohol-Induced Alterations in a Mouse Model of Foetal Alcohol Spectrum Disorders. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2020, 100, 109899. [Google Scholar] [CrossRef]
- García-Baos, A.; Puig-Reyne, X.; García-Algar, Ó.; Valverde, O. Cannabidiol Attenuates Cognitive Deficits and Neuroinflammation Induced by Early Alcohol Exposure in a Mice Model. Biomed. Pharmacother. 2021, 141, 111813. [Google Scholar] [CrossRef]
- Almeida-Toledano, L.; Andreu-Fernández, V.; Aras-López, R.; García-Algar, Ó.; Martínez, L.; Gómez-Roig, M.D. Epigallocatechin Gallate Ameliorates the Effects of Prenatal Alcohol Exposure in a Fetal Alcohol Spectrum Disorder-Like Mouse Model. Int. J. Mol. Sci. 2021, 22, 715. [Google Scholar] [CrossRef]
- Sabzali, M.; Eidi, A.; Khaksari, M.; Khastar, H. Anti-Inflammatory, Antioxidant, and Antiapoptotic Action of Metformin Attenuates Ethanol Neurotoxicity in the Animal Model of Fetal Alcohol Spectrum Disorders. Neurotox. Res. 2022, 40, 605–613. [Google Scholar] [CrossRef] [PubMed]
- Skorput, A.G.; Lee, S.M.; Yeh, P.W.; Yeh, H.H. The NKCC1 Antagonist Bumetanide Mitigates Interneuronopathy Associated with Ethanol Exposure in Utero. eLife 2019, 8, e48648. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, S.; Page, S.J.; Wang, L.; Ishii, S.; Li, P.; Sasaki, T.; Basha, A.; Salzberg, A.; Quezado, Z.; Imamura, F.; et al. Kcnn2 Blockade Reverses Learning Deficits in a Mouse Model of Fetal Alcohol Spectrum Disorders. Nat. Neurosci. 2020, 23, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Tuazon, J.P.; Castelli, V.; Lee, J.-Y.; Desideri, G.B.; Stuppia, L.; Cimini, A.M.; Borlongan, C.V. Neural Stem Cells. In Stem Cells: Therapeutic Applications; Ratajczak, M.Z., Ed.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2019; pp. 79–91. ISBN 978-3-030-31206-0. [Google Scholar]
- Shirasaka, T.; Ukai, W.; Yoshinaga, T.; Watanabe, K.; Kaneta, H.; Kigawa, Y.; Igarashi, T.; Tateno, M.; Hashimoto, E.; Saito, T. Promising therapy of neural stem cell transplantation for FASD model–Neural network reconstruction and behavior recovery. Nihon Arukoru Yakubutsu Igakkai Zasshi 2011, 46, 576–584. [Google Scholar] [PubMed]
- Yoshinaga, T.; Hashimoto, E.; Ukai, W.; Toki, S.; Saito, S.; Saito, T. Neural Stem Cell Transplantation in a Model of Fetal Alcohol Effects. In Neuropsychiatric Disorders: An Integrative Approach; Gerlach, M., Deckert, J., Double, K., Koutsilieri, E., Eds.; Springer: Vienna, Austria, 2007; pp. 331–337. [Google Scholar]
- Arjona, A.; Boyadjieva, N.; Kuhn, P.; Sarkar, D.K. Fetal Ethanol Exposure Disrupts the Daily Rhythms of Splenic Granzyme B, IFN-γ, and NK Cell Cytotoxicity in Adulthood. Alcohol. Clin. Exp. Res. 2006, 30, 1039–1044. [Google Scholar] [CrossRef] [PubMed]
- Boyadjieva, N.I.; Dokur, M.; Advis, J.P.; Meadows, G.G.; Sarkar, D.K. Beta-Endorphin Modulation of Lymphocyte Proliferation: Effects of Ethanol. Alcohol. Clin. Exp. Res. 2002, 26, 1719–1727. [Google Scholar] [CrossRef]
- Sarkar, D.K.; Boyadjieva, N.I.; Chen, C.P.; Ortigüela, M.; Reuhl, K.; Clement, E.M.; Kuhn, P.; Marano, J. Cyclic adenosine monophosphate differentiated β-endorphin neurons promote immune function and prevent prostate cancer growth. Proc. Natl. Acad. Sci. USA 2008, 105, 9105–9110. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.P.; Boyadjieva, N.I.; Advis, J.P.; Sarkar, D.K. Ethanol Suppression of the Hypothalamic Proopiomelanocortin Level and the Splenic NK Cell Cytolytic Activity Is Associated with a Reduction in the Expression of Proinflammatory Cytokines but Not Anti-Inflammatory Cytokines in Neuroendocrine and Immune Cells. Alcohol. Clin. Exp. Res. 2006, 30, 1925–1932. [Google Scholar] [CrossRef]
- Boyadjieva, N.I.; Ortigüela, M.; Arjona, A.; Cheng, X.; Sarkar, D.K. β-Endorphin Neuronal Cell Transplant Reduces Corticotropin Releasing Hormone Hyperresponse to Lipopolysaccharide and Eliminates Natural Killer Cell Functional Deficiencies in Fetal Alcohol Exposed Rats. Alcohol. Clin. Exp. Res. 2009, 33, 931–937. [Google Scholar] [CrossRef]
- Wynne, O.; Sarkar, D.K. Stress and Neuroendocrine–Immune Interaction: A Therapeutic Role for β-Endorphin. In The Wiley-Blackwell Handbook of Psychoneuroimmunology; John Wiley & Sons, Ltd.: West Sussex, UK, 2013; pp. 198–211. ISBN 978-1-118-31481-4. [Google Scholar]
- Yasuhara, T.; Matsukawa, N.; Hara, K.; Yu, G.; Xu, L.; Maki, M.; Kim, S.U.; Borlongan, C.V. Transplantation of Human Neural Stem Cells Exerts Neuroprotection in a Rat Model of Parkinson’s Disease. J. Neurosci. 2006, 26, 12497–12511. [Google Scholar] [CrossRef] [PubMed]
- Goetzl, L.; Darbinian, N.; Goetzl, E.J. Novel Window on Early Human Neurodevelopment via Fetal Exosomes in Maternal Blood. Ann. Clin. Transl. Neurol. 2016, 3, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Goetzl, L.; Darbinian, N.; Merabova, N. Noninvasive Assessment of Fetal Central Nervous System Insult: Potential Application to Prenatal Diagnosis. Prenat. Diagn. 2019, 39, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Darbinian, N.; Darbinyan, A.; Sinard, J.; Tatevosian, G.; Merabova, N.; D’Amico, F.; Khader, T.; Bajwa, A.; Martirosyan, D.; Gawlinski, A.K.; et al. Molecular Markers in Maternal Blood Exosomes Allow Early Detection of Fetal Alcohol Spectrum Disorders. Int. J. Mol. Sci. 2023, 24, 135. [Google Scholar] [CrossRef] [PubMed]
- Tavanasefat, H.; Li, F.; Koyano, K.; Gourtani, B.K.; Marty, V.; Mulpuri, Y.; Lee, S.H.; Shin, K.-H.; Wong, D.T.W.; Xiao, X.; et al. Molecular Consequences of Fetal Alcohol Exposure on Amniotic Exosomal MiRNAs with Functional Implications for Stem Cell Potency and Differentiation. PLoS ONE 2020, 15, e0242276. [Google Scholar] [CrossRef]
- Balaraman, S.; Lunde, E.R.; Sawant, O.; Cudd, T.A.; Washburn, S.E.; Miranda, R.C. Maternal and Neonatal Plasma MicroRNA Biomarkers for Fetal Alcohol Exposure in an Ovine Model. Alcohol. Clin. Exp. Res. 2014, 38, 1390–1400. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mukherjee, S.; Tarale, P.; Sarkar, D.K. Neuroimmune Interactions in Fetal Alcohol Spectrum Disorders: Potential Therapeutic Targets and Intervention Strategies. Cells 2023, 12, 2323. https://doi.org/10.3390/cells12182323
Mukherjee S, Tarale P, Sarkar DK. Neuroimmune Interactions in Fetal Alcohol Spectrum Disorders: Potential Therapeutic Targets and Intervention Strategies. Cells. 2023; 12(18):2323. https://doi.org/10.3390/cells12182323
Chicago/Turabian StyleMukherjee, Sayani, Prashant Tarale, and Dipak K. Sarkar. 2023. "Neuroimmune Interactions in Fetal Alcohol Spectrum Disorders: Potential Therapeutic Targets and Intervention Strategies" Cells 12, no. 18: 2323. https://doi.org/10.3390/cells12182323