
TRPV1 Channels Are New Players in the Reticulum–Mitochondria Ca2+ Coupling in a Rat Cardiomyoblast Cell Line
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Cell Line
2.3. Cell Transfection
2.4. RNA Extraction and RT-PCR
2.5. Subcellular Fractionation
2.6. Western Blotting
2.7. Immunostaining
2.8. Proximity Ligation Assay (PLA) and Confocal Imaging
2.9. Wide-Field Calcium Imaging and Image Analysis
2.10. Electron Microscopy
2.11. Electrophysiology
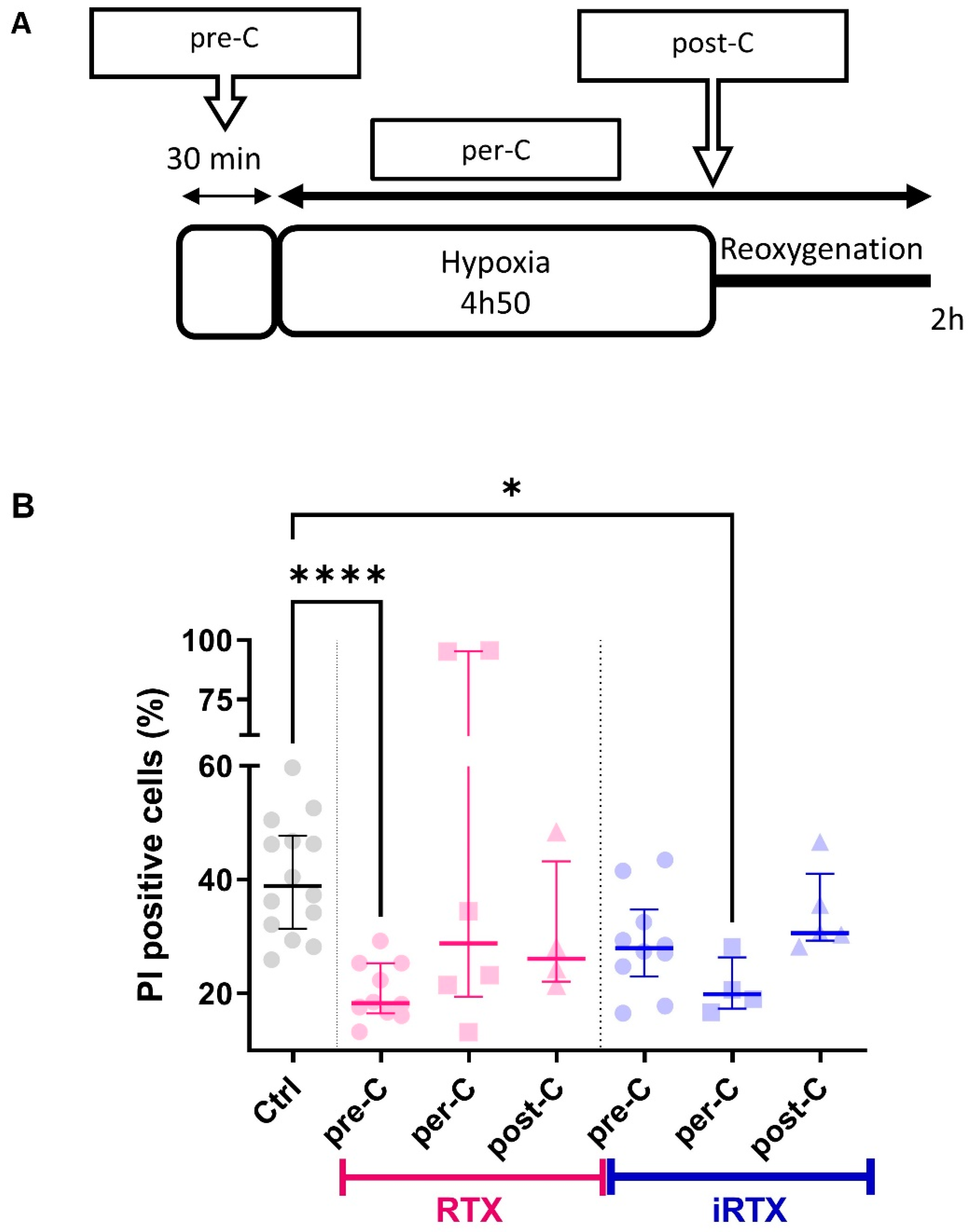
2.12. Hypoxia–Reoxygenation Sequence and Conditioning Strategies
2.13. Statistical Analysis
3. Results and Discussion
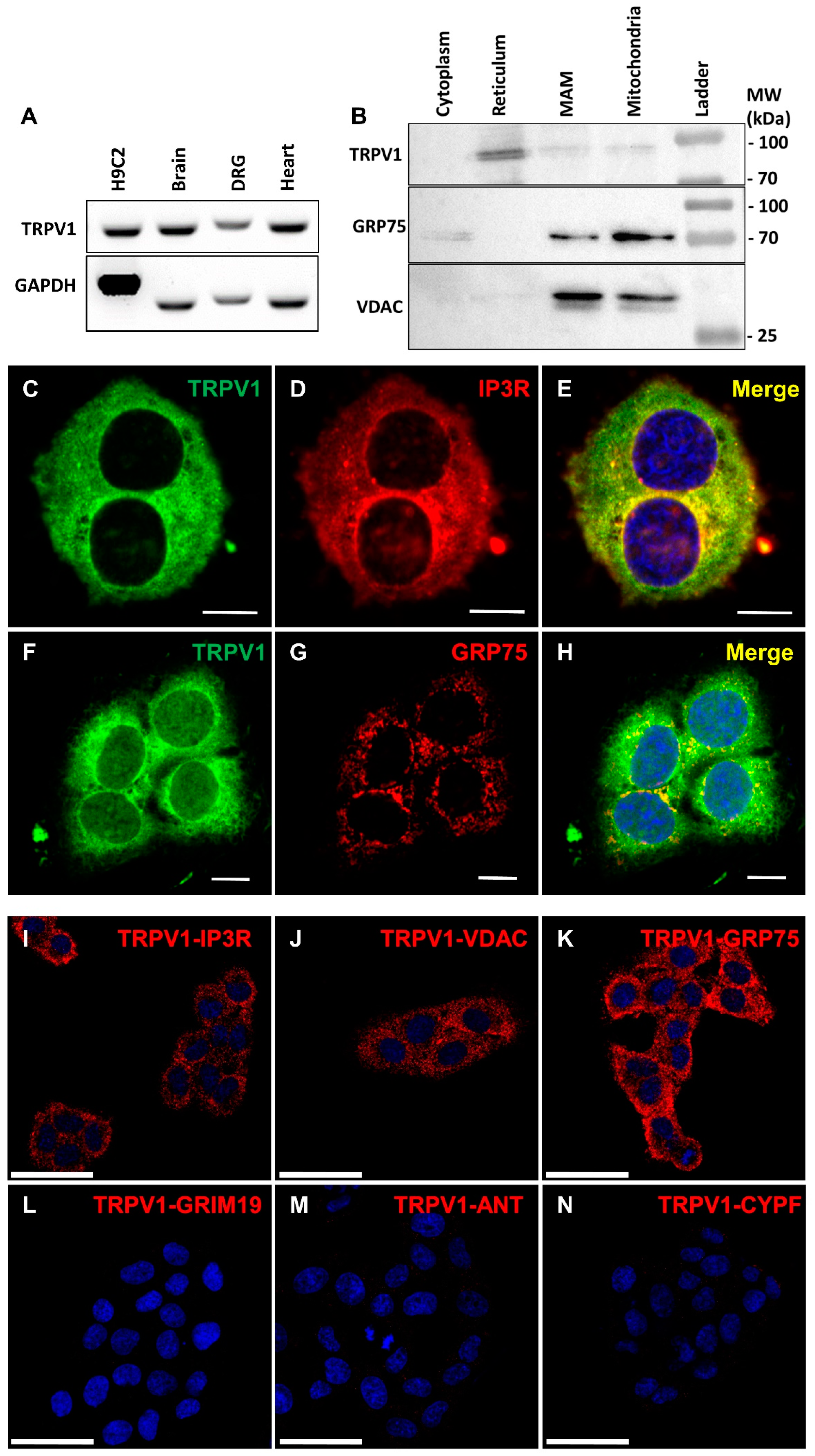
3.1. TRPV1 Channels Are Localized in the Endoplasmic Reticulum Membrane, including at MAMs
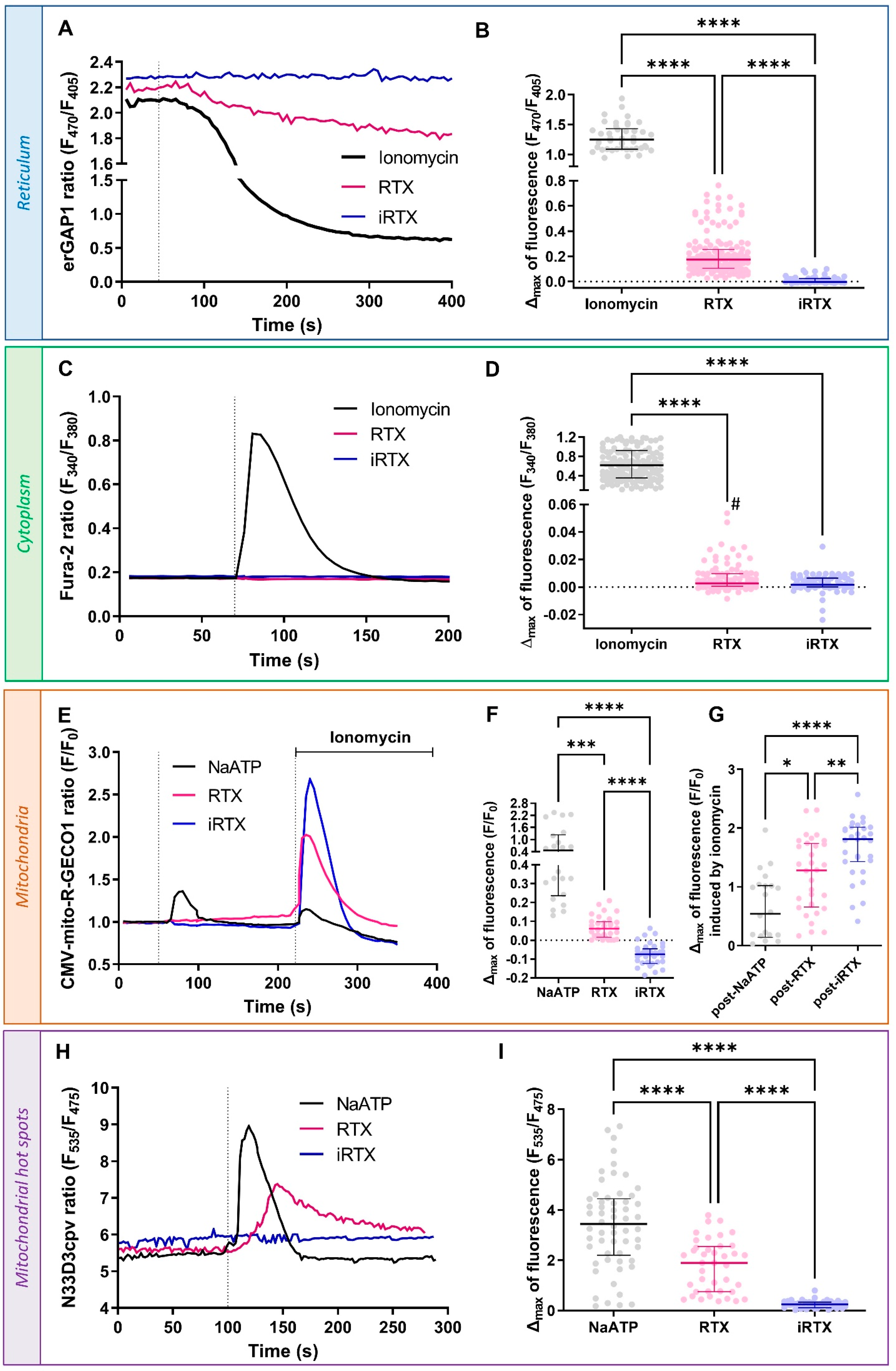
3.2. Acute Effect of TRPV1 Modulation on Ca2+ Homeostasis
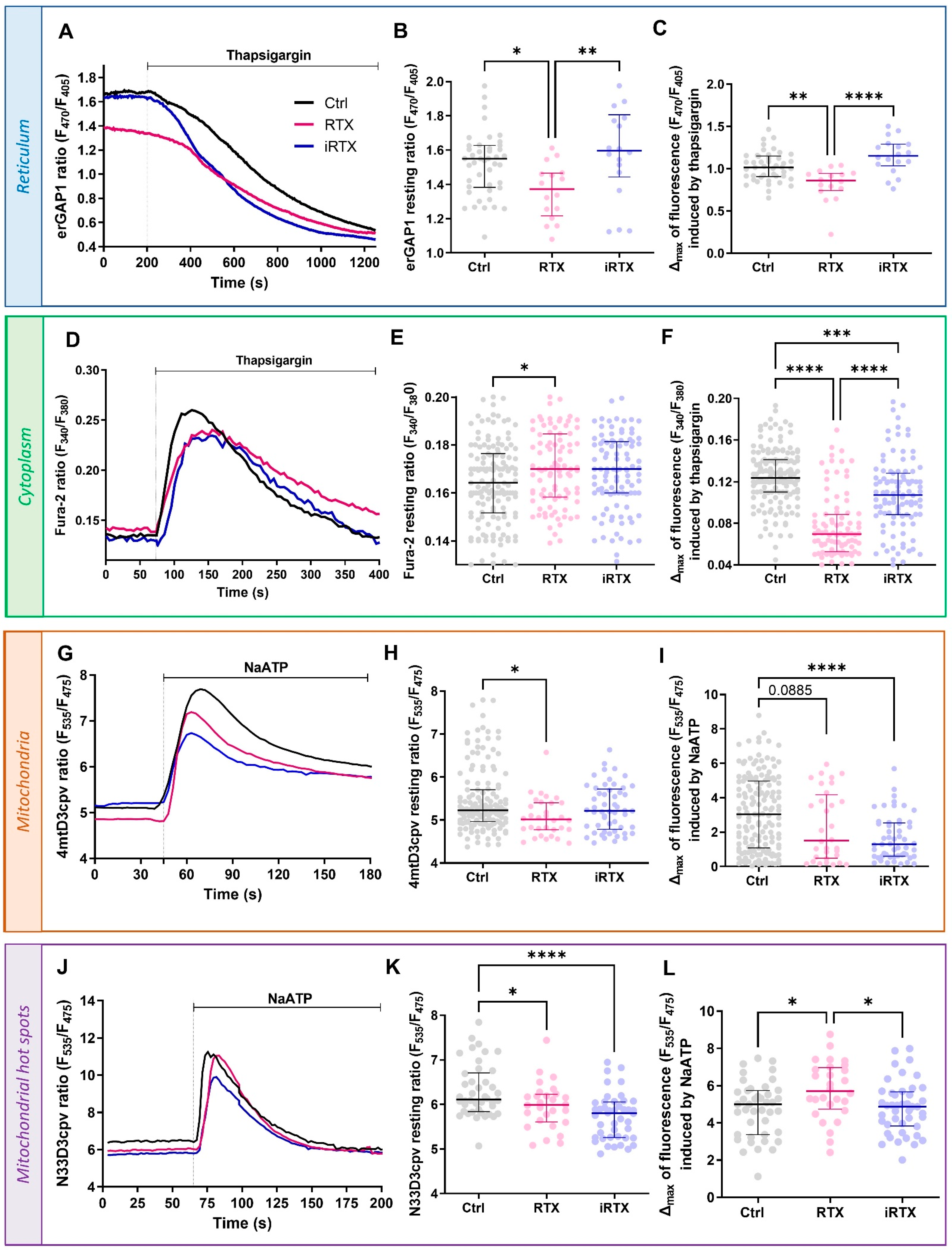
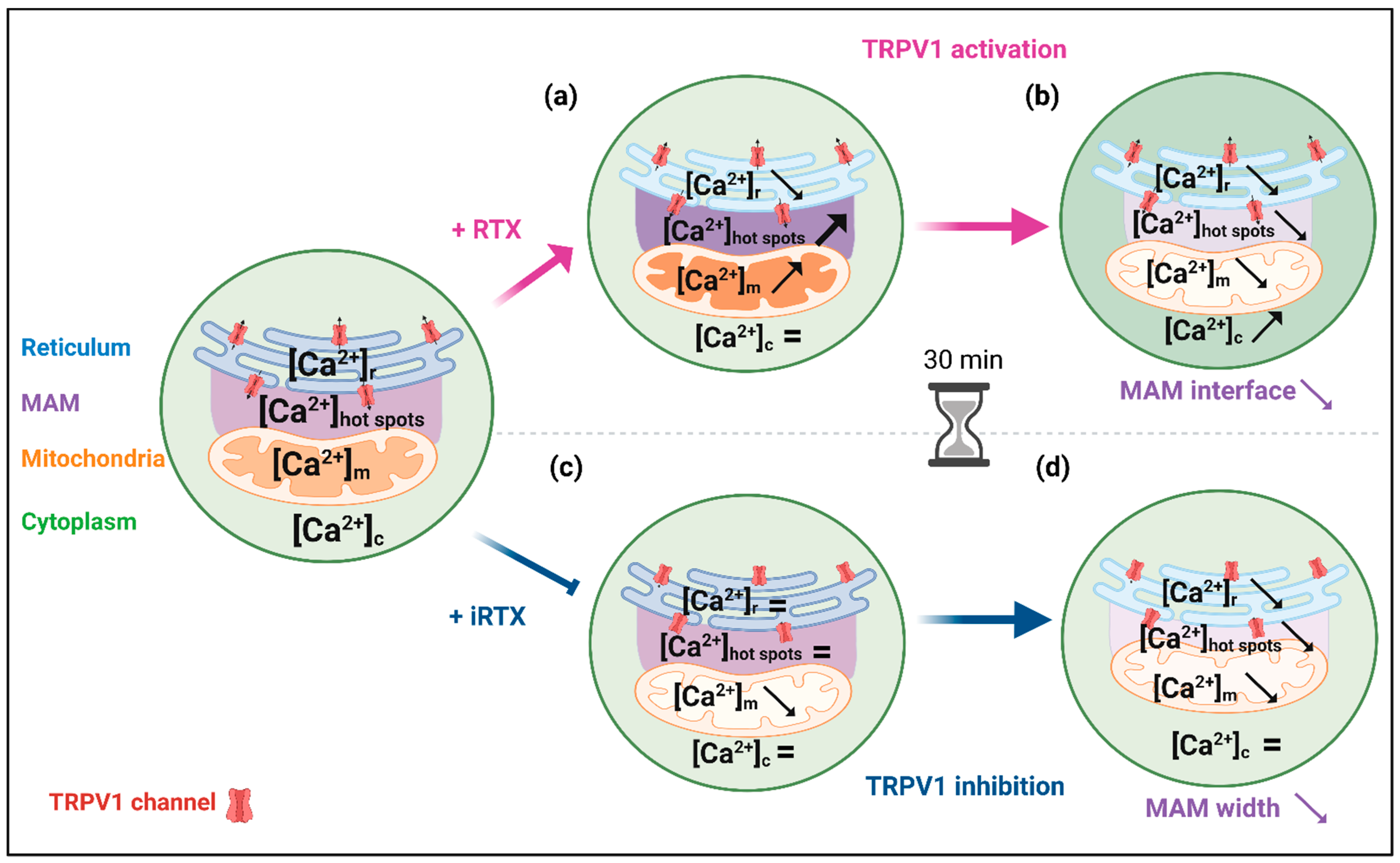
3.3. Effects of a 30 min Prolonged Modulation of TRPV1 on Ca2+ Homeostasis
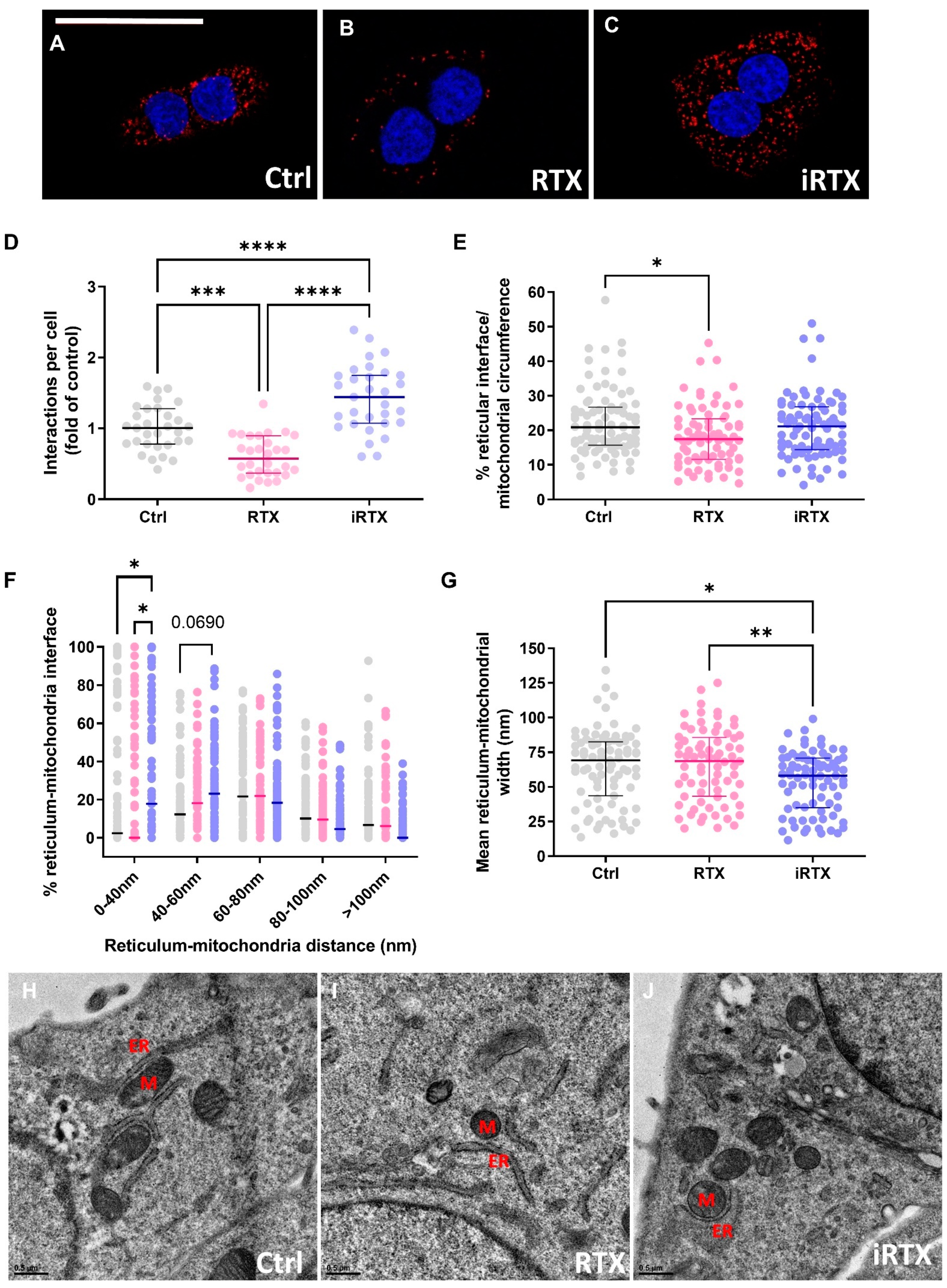
3.4. A 30 Min Prolonged Modulation of TRPV1 Rearranges Reticulum–Mitochondria Interactions
3.5. TRPV1 Pharmacological Modulation Protects SV40-Transformed H9c2 Cells Submitted to In Vitro H/R
4. Conclusions
5. Limitations of the Study
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tominaga, M.; Caterina, M.J.; Malmberg, A.B.; Rosen, T.A.; Gilbert, H.; Skinner, K.; Raumann, B.E.; Basbaum, A.I.; Julius, D. The Cloned Capsaicin Receptor Integrates Multiple Pain-Producing Stimuli. Neuron 1998, 21, 531–543. [Google Scholar] [CrossRef] [PubMed]
- Caterina, M.J.; Leffler, A.; Malmberg, A.B.; Martin, W.J.; Trafton, J.; Petersen-Zeitz, K.R.; Koltzenburg, M.; Basbaum, A.I.; Julius, D. Impaired Nociception and Pain Sensation in Mice Lacking the Capsaicin Receptor. Science 2000, 288, 306–313. [Google Scholar] [CrossRef]
- Caterina, M.J.; Schumacher, M.A.; Tominaga, M.; Rosen, T.A.; Levine, J.D.; Julius, D. The Capsaicin Receptor: A Heat-Activated Ion Channel in the Pain Pathway. Nature 1997, 389, 816–824. [Google Scholar] [CrossRef]
- Edwards, J.G. TRPV1 in the Central Nervous System: Synaptic Plasticity, Function, and Pharmacological Implications. Prog. Drug Res. Fortschritte Arzneimittelforschung Prog. Rech. Pharm. 2014, 68, 77–104. [Google Scholar] [CrossRef]
- Inoue, K.; Koizumi, S.; Fuziwara, S.; Denda, S.; Inoue, K.; Denda, M. Functional Vanilloid Receptors in Cultured Normal Human Epidermal Keratinocytes. Biochem. Biophys. Res. Commun. 2002, 291, 124–129. [Google Scholar] [CrossRef] [PubMed]
- Bielefeldt, K.; Davis, B.M. Differential Effects of ASIC3 and TRPV1 Deletion on Gastroesophageal Sensation in Mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G130–G138. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.L.; Yan Liu, D.; Ma, L.Q.; Luo, Z.D.; Cao, T.B.; Zhong, J.; Yan, Z.C.; Wang, L.J.; Zhao, Z.G.; Zhu, S.J.; et al. Activation of Transient Receptor Potential Vanilloid Type-1 Channel Prevents Adipogenesis and Obesity. Circ. Res. 2007, 100, 1063–1070. [Google Scholar] [CrossRef]
- Vanden Abeele, F.; Lotteau, S.; Ducreux, S.; Dubois, C.; Monnier, N.; Hanna, A.; Gkika, D.; Romestaing, C.; Noyer, L.; Flourakis, M.; et al. TRPV1 Variants Impair Intracellular Ca2+ Signaling and May Confer Susceptibility to Malignant Hyperthermia. Genet. Med. 2019, 21, 441–450. [Google Scholar] [CrossRef]
- Lotteau, S.; Ducreux, S.; Romestaing, C.; Legrand, C.; Coppenolle, F.V. Characterization of Functional TRPV1 Channels in the Sarcoplasmic Reticulum of Mouse Skeletal Muscle. PLoS ONE 2013, 8, e58673. [Google Scholar] [CrossRef]
- Gorbunov, A.S.; Maslov, L.N.; Jaggi, A.S.; Singh, N.; De Petrocellis, L.; Boshchenko, A.A.; Roohbakhsh, A.; Bezuglov, V.V.; Oeltgen, P.R. Physiological and Pathological Role of TRPV1, TRPV2 and TRPV4 Channels in Heart. Curr. Cardiol. Rev. 2019, 15, 244–251. [Google Scholar] [CrossRef]
- Gouin, O.; L’Herondelle, K.; Lebonvallet, N.; Le Gall-Ianotto, C.; Sakka, M.; Buhé, V.; Plée-Gautier, E.; Carré, J.-L.; Lefeuvre, L.; Misery, L.; et al. TRPV1 and TRPA1 in Cutaneous Neurogenic and Chronic Inflammation: Pro-Inflammatory Response Induced by Their Activation and Their Sensitization. Protein Cell 2017, 8, 644–661. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Chen, C.; Chiang, C.; Xiao, T.; Chen, Y.; Zhao, Y.; Zheng, D. The Impact of TRPV1 on Cancer Pathogenesis and Therapy: A Systematic Review. Int. J. Biol. Sci. 2021, 17, 2034–2049. [Google Scholar] [CrossRef] [PubMed]
- Duitama, M.; Vargas-López, V.; Casas, Z.; Albarracin, S.L.; Sutachan, J.-J.; Torres, Y.P. TRP Channels Role in Pain Associated with Neurodegenerative Diseases. Front. Neurosci. 2020, 14, 782. [Google Scholar] [CrossRef] [PubMed]
- Kong, W.-L.; Peng, Y.-Y.; Peng, B.-W. Modulation of Neuroinflammation: Role and Therapeutic Potential of TRPV1 in the Neuro-Immune Axis. Brain. Behav. Immun. 2017, 64, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.S.; Buckley, C.L.; Stokes, A.J. Successful TRPV1 antagonist treatment for cardiac hypertrophy and heart failure in mice. Channels Austin 2013, 7, 17–22. [Google Scholar] [CrossRef]
- Assas, B.M.; Wakid, M.H.; Zakai, H.A.; Miyan, J.A.; Pennock, J.L. Transient Receptor Potential Vanilloid 1 Expression and Function in Splenic Dendritic Cells: A Potential Role in Immune Homeostasis. Immunology 2016, 147, 292–304. [Google Scholar] [CrossRef]
- Majhi, R.K.; Sahoo, S.S.; Yadav, M.; Pratheek, B.M.; Chattopadhyay, S.; Goswami, C. Functional Expression of TRPV Channels in T Cells and Their Implications in Immune Regulation. FEBS J. 2015, 282, 2661–2681. [Google Scholar] [CrossRef]
- Fernandes, E.; Fernandes, M.; Keeble, J. The Functions of TRPA1 and TRPV1: Moving Away from Sensory Nerves. Br. J. Pharmacol. 2012, 166, 510–521. [Google Scholar] [CrossRef]
- Szallasi, A.; Blumberg, P.M.; Annicelli, L.L.; Krause, J.E.; Cortright, D.N. The Cloned Rat Vanilloid Receptor VR1 Mediates Both R-Type Binding and C-Type Calcium Response in Dorsal Root Ganglion Neurons. Mol. Pharmacol. 1999, 56, 581–587. [Google Scholar] [CrossRef]
- Olah, Z.; Szabo, T.; Karai, L.; Hough, C.; Fields, R.D.; Caudle, R.M.; Blumberg, P.M.; Iadarola, M.J. Ligand-Induced Dynamic Membrane Changes and Cell Deletion Conferred by Vanilloid Receptor 1. J. Biol. Chem. 2001, 276, 11021–11030. [Google Scholar] [CrossRef]
- Xin, H.; Tanaka, H.; Yamaguchi, M.; Takemori, S.; Nakamura, A.; Kohama, K. Vanilloid Receptor Expressed in the Sarcoplasmic Reticulum of Rat Skeletal Muscle. Biochem. Biophys. Res. Commun. 2005, 332, 756–762. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Han, J.; Zhao, W.; Zhang, Y.; Wang, S.; Ye, L.; Liu, T.; Zheng, L. TRPV1 Activation Exacerbates Hypoxia/Reoxygenation-Induced Apoptosis in H9C2 Cells via Calcium Overload and Mitochondrial Dysfunction. Int. J. Mol. Sci. 2014, 15, 18362–18380. [Google Scholar] [CrossRef] [PubMed]
- Hurt, C.M.; Lu, Y.; Stary, C.M.; Piplani, H.; Small, B.A.; Urban, T.J.; Qvit, N.; Gross, G.J.; Mochly-Rosen, D.; Gross, E.R. Transient Receptor Potential Vanilloid 1 Regulates Mitochondrial Membrane Potential and Myocardial Reperfusion Injury. J. Am. Heart Assoc. 2016, 5, e003774. [Google Scholar] [CrossRef] [PubMed]
- Bosson, C.; Rendu, J.; Pelletier, L.; Abriat, A.; Chatagnon, A.; Brocard, J.; Brocard, J.; Figarella-Branger, D.; Ducreux, S.; van Coppenolle, F.; et al. Variations in the TRPV1 Gene Are Associated to Exertional Heat Stroke. J. Sci. Med. Sport 2020, 23, 1021–1027. [Google Scholar] [CrossRef]
- Mekahli, D.; Bultynck, G.; Parys, J.B.; De Smedt, H.; Missiaen, L. Endoplasmic-Reticulum Calcium Depletion and Disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a004317. [Google Scholar] [CrossRef]
- Schwaller, B. The Regulation of a Cell’s Ca2+ Signaling Toolkit: The Ca2+ Homeostasome. Adv. Exp. Med. Biol. 2012, 740, 1–25. [Google Scholar] [CrossRef]
- Marchi, S.; Patergnani, S.; Missiroli, S.; Morciano, G.; Rimessi, A.; Wieckowski, M.R.; Giorgi, C.; Pinton, P. Mitochondrial and Endoplasmic Reticulum Calcium Homeostasis and Cell Death. Cell Calcium 2018, 69, 62–72. [Google Scholar] [CrossRef]
- Gong, Y.; Lin, J.; Ma, Z.; Yu, M.; Wang, M.; Lai, D.; Fu, G. Mitochondria-Associated Membrane-Modulated Ca2+ Transfer: A Potential Treatment Target in Cardiac Ischemia Reperfusion Injury and Heart Failure. Life Sci. 2021, 278, 119511. [Google Scholar] [CrossRef]
- van Vliet, A.R.; Verfaillie, T.; Agostinis, P. New Functions of Mitochondria Associated Membranes in Cellular Signaling. Biochim. Biophys. Acta BBA—Mol. Cell Res. 2014, 1843, 2253–2262. [Google Scholar] [CrossRef]
- Szabadkai, G.; Bianchi, K.; Várnai, P.; De Stefani, D.; Wieckowski, M.R.; Cavagna, D.; Nagy, A.I.; Balla, T.; Rizzuto, R. Chaperone-Mediated Coupling of Endoplasmic Reticulum and Mitochondrial Ca2+ Channels. J. Cell Biol. 2006, 175, 901–911. [Google Scholar] [CrossRef]
- Wei, X.; Wei, X.; Lu, Z.; Li, L.; Hu, Y.; Sun, F.; Jiang, Y.; Ma, H.; Zheng, H.; Yang, G.; et al. Activation of TRPV1 Channel Antagonizes Diabetic Nephropathy through Inhibiting Endoplasmic Reticulum-Mitochondria Contact in Podocytes. Metabolism 2020, 105, 154182. [Google Scholar] [CrossRef] [PubMed]
- Knoell, J.; Chillappagari, S.; Knudsen, L.; Korfei, M.; Dartsch, R.; Jonigk, D.; Kuehnel, M.P.; Hoetzenecker, K.; Guenther, A.; Mahavadi, P. PACS2–TRPV1 Axis Is Required for ER–Mitochondrial Tethering during ER Stress and Lung Fibrosis. Cell. Mol. Life Sci. 2022, 79, 151. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, X.; Xu, X.; Qu, X.; Yang, Y. Transient Receptor Potential Vanilloid Type 1 Protects Against Pressure Overload–Induced Cardiac Hypertrophy by Promoting Mitochondria-Associated Endoplasmic Reticulum Membranes. J. Cardiovasc. Pharmacol. 2022, 80, 430. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Yan, Z.; Zhu, Z. Mitochondria-Associated Endoplasmic Reticulum Membranes in Cardiovascular Diseases. Front. Cell Dev. Biol. 2020, 8, 604240. [Google Scholar] [CrossRef]
- Dia, M.; Gomez, L.; Thibault, H.; Tessier, N.; Leon, C.; Chouabe, C.; Ducreux, S.; Gallo-Bona, N.; Tubbs, E.; Bendridi, N.; et al. Reduced Reticulum–Mitochondria Ca2+ Transfer Is an Early and Reversible Trigger of Mitochondrial Dysfunctions in Diabetic Cardiomyopathy. Basic Res. Cardiol. 2020, 115, 74. [Google Scholar] [CrossRef]
- Tubbs, E.; Chanon, S.; Robert, M.; Bendridi, N.; Bidaux, G.; Chauvin, M.-A.; Ji-Cao, J.; Durand, C.; Gauvrit-Ramette, D.; Vidal, H.; et al. Disruption of Mitochondria-Associated Endoplasmic Reticulum Membrane (MAM) Integrity Contributes to Muscle Insulin Resistance in Mice and Humans. Diabetes 2018, 67, 636–650. [Google Scholar] [CrossRef]
- Paillard, M.; Tubbs, E.; Thiebaut, P.-A.; Gomez, L.; Fauconnier, J.; Da Silva, C.C.; Teixeira, G.; Mewton, N.; Belaidi, E.; Durand, A.; et al. Depressing Mitochondria-Reticulum Interactions Protects Cardiomyocytes from Lethal Hypoxia-Reoxygenation Injury. Circulation 2013, 128, 1555–1565. [Google Scholar] [CrossRef]
- Zhong, B.; Wang, D.H. TRPV1 Gene Knockout Impairs Preconditioning Protection against Myocardial Injury in Isolated Perfused Hearts in Mice. Am. J. Physiol.-Heart Circ. Physiol. 2007, 293, H1791–H1798. [Google Scholar] [CrossRef]
- Randhawa, P.K.; Jaggi, A.S. Investigating the Involvement of TRPV1 Ion Channels in Remote Hind Limb Preconditioning-Induced Cardioprotection in Rats. Naunyn. Schmiedebergs Arch. Pharmacol. 2017, 390, 117–126. [Google Scholar] [CrossRef]
- Ren, J.-Y.; Song, J.-X.; Lu, M.-Y.; Chen, H. Cardioprotection by Ischemic Postconditioning Is Lost in Isolated Perfused Heart from Diabetic Rats: Involvement of Transient Receptor Potential Vanilloid 1, Calcitonin Gene-Related Peptide and Substance P. Regul. Pept. 2011, 169, 49–57. [Google Scholar] [CrossRef]
- Gao, Y.; Song, J.; Chen, H.; Cao, C.; Lee, C. TRPV1 Activation Is Involved in the Cardioprotection of Remote Limb Ischemic Postconditioning in Ischemia-Reperfusion Injury Rats. Biochem. Biophys. Res. Commun. 2015, 463, 1034–1039. [Google Scholar] [CrossRef]
- Yang, D.; Luo, Z.; Ma, S.; Wong, W.T.; Ma, L.; Zhong, J.; He, H.; Zhao, Z.; Cao, T.; Yan, Z.; et al. Activation of TRPV1 by Dietary Capsaicin Improves Endothelium-Dependent Vasorelaxation and Prevents Hypertension. Cell Metab. 2010, 12, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Munjuluri, S.; Wilkerson, D.A.; Sooch, G.; Chen, X.; White, F.A.; Obukhov, A.G. Capsaicin and TRPV1 Channels in the Cardiovascular System: The Role of Inflammation. Cells 2021, 11, 18. [Google Scholar] [CrossRef] [PubMed]
- Bizouarne, N.; Denis, V.; Legrand, A.; Monsigny, M.; Kieda, C. A SV-40 Immortalized Murine Endothelial Cell Line from Peripheral Lymph Node High Endothelium Expresses a New Alpha-L-Fucose Binding Protein. Biol. Cell 1993, 79, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Bochaton, T.; Crola-Da-Silva, C.; Pillot, B.; Villedieu, C.; Ferreras, L.; Alam, M.R.; Thibault, H.; Strina, M.; Gharib, A.; Ovize, M.; et al. Inhibition of Myocardial Reperfusion Injury by Ischemic Postconditioning Requires Sirtuin 3-Mediated Deacetylation of Cyclophilin D. J. Mol. Cell. Cardiol. 2015, 84, 61–69. [Google Scholar] [CrossRef]
- Gouriou, Y.; Alam, M.R.; Harhous, Z.; Crola Da Silva, C.; Baetz, D.B.; Badawi, S.; Lefai, E.; Rieusset, J.; Durand, A.; Harisseh, R.; et al. ANT2-Mediated ATP Import into Mitochondria Protects against Hypoxia Lethal Injury. Cells 2020, 9, 2542. [Google Scholar] [CrossRef]
- Delcroix, V.; Mauduit, O.; Tessier, N.; Montillaud, A.; Lesluyes, T.; Ducret, T.; Chibon, F.; Van Coppenolle, F.; Ducreux, S.; Vacher, P. The Role of the Anti-Aging Protein Klotho in IGF-1 Signaling and Reticular Calcium Leak: Impact on the Chemosensitivity of Dedifferentiated Liposarcomas. Cancers 2018, 10, 439. [Google Scholar] [CrossRef]
- Wu, J.; Liu, L.; Matsuda, T.; Zhao, Y.; Rebane, A.; Drobizhev, M.; Chang, Y.-F.; Araki, S.; Arai, Y.; March, K.; et al. Improved Orange and Red Ca2+ Indicators and Photophysical Considerations for Optogenetic Applications. ACS Chem. Neurosci. 2013, 4, 963–972. [Google Scholar] [CrossRef]
- Palmer, A.E.; Giacomello, M.; Kortemme, T.; Hires, S.A.; Lev-Ram, V.; Baker, D.; Tsien, R.Y. Ca2+ Indicators Based on Computationally Redesigned Calmodulin-Peptide Pairs. Chem. Biol. 2006, 13, 521–530. [Google Scholar] [CrossRef]
- Rodriguez-Garcia, A.; Rojo-Ruiz, J.; Navas-Navarro, P.; Aulestia, F.J.; Gallego-Sandin, S.; Garcia-Sancho, J.; Alonso, M.T. GAP, an Aequorin-Based Fluorescent Indicator for Imaging Ca2+ in Organelles. Proc. Natl. Acad. Sci. USA 2014, 111, 2584–2589. [Google Scholar] [CrossRef]
- Wieckowski, M.R.; Giorgi, C.; Lebiedzinska, M.; Duszynski, J.; Pinton, P. Isolation of Mitochondria-Associated Membranes and Mitochondria from Animal Tissues and Cells. Nat. Protoc. 2009, 4, 1582–1590. [Google Scholar] [CrossRef] [PubMed]
- Gouriou, Y.; Gonnot, F.; Wehbi, M.; Brun, C.; Gomez, L.; Bidaux, G. High-Sensitivity Calcium Biosensor on the Mitochondrial Surface Reveals That IP3R Channels Participate in the Reticular Ca2+ Leak towards Mitochondria. PLoS ONE 2023, 18, e0285670. [Google Scholar] [CrossRef] [PubMed]
- Bartok, A.; Weaver, D.; Golenár, T.; Nichtova, Z.; Katona, M.; Bánsághi, S.; Alzayady, K.J.; Thomas, V.K.; Ando, H.; Mikoshiba, K.; et al. IP3 Receptor Isoforms Differently Regulate ER-Mitochondrial Contacts and Local Calcium Transfer. Nat. Commun. 2019, 10, 3726. [Google Scholar] [CrossRef] [PubMed]
- Hamill, O.P.; Marty, A.; Neher, E.; Sakmann, B.; Sigworth, F.J. Improved Patch-Clamp Techniques for High-Resolution Current Recording from Cells and Cell-Free Membrane Patches. Pflugers Arch. 1981, 391, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.J.; Yellon, D.M. Ischaemic Conditioning and Reperfusion Injury. Nat. Rev. Cardiol. 2016, 13, 193–209. [Google Scholar] [CrossRef]
- Jahnel, R.; Dreger, M.; Gillen, C.; Bender, O.; Kurreck, J.; Hucho, F. Biochemical Characterization of the Vanilloid Receptor 1 Expressed in a Dorsal Root Ganglia Derived Cell Line. Eur. J. Biochem. 2001, 268, 5489–5496. [Google Scholar] [CrossRef]
- Chou, M.Z.; Mtui, T.; Gao, Y.-D.; Kohler, M.; Middleton, R.E. Resiniferatoxin Binds to the Capsaicin Receptor (TRPV1) near the Extracellular Side of the S4 Transmembrane Domain. Biochemistry 2004, 43, 2501–2511. [Google Scholar] [CrossRef]
- Wahl, P.; Foged, C.; Tullin, S.; Thomsen, C. Iodo-Resiniferatoxin, a New Potent Vanilloid Receptor Antagonist. Mol. Pharmacol. 2001, 59, 9–15. [Google Scholar] [CrossRef]
- Bertero, E.; Maack, C. Calcium Signaling and Reactive Oxygen Species in Mitochondria. Circ. Res. 2018, 122, 1460–1478. [Google Scholar] [CrossRef]
- Marchi, S.; Bittremieux, M.; Missiroli, S.; Morganti, C.; Patergnani, S.; Sbano, L.; Rimessi, A.; Kerkhofs, M.; Parys, J.B.; Bultynck, G.; et al. Endoplasmic Reticulum-Mitochondria Communication Through Ca2+ Signaling: The Importance of Mitochondria-Associated Membranes (MAMs). In Organelle Contact Sites: From Molecular Mechanism to Disease; Tagaya, M., Simmen, T., Eds.; Advances in Experimental Medicine and Biology; Springer: Singapore, 2017; pp. 49–67. ISBN 978-981-10-4567-7. [Google Scholar]
- Márta, K.; Hasan, P.; Rodríguez-Prados, M.; Paillard, M.; Hajnóczky, G. Pharmacological Inhibition of the Mitochondrial Ca2+ Uniporter: Relevance for Pathophysiology and Human Therapy. J. Mol. Cell. Cardiol. 2021, 151, 135–144. [Google Scholar] [CrossRef]
- Lax, A.; Soler, F.; Fernández-Belda, F. Intracellular Ca2+ Pools and Fluxes in Cardiac Muscle-Derived H9c2 Cells. J. Bioenerg. Biomembr. 2005, 37, 249–259. [Google Scholar] [CrossRef]
- Csordás, G.; Várnai, P.; Golenár, T.; Roy, S.; Purkins, G.; Schneider, T.G.; Balla, T.; Hajnóczky, G. Imaging Interorganelle Contacts and Local Calcium Dynamics at the ER-Mitochondrial Interface. Mol. Cell 2010, 39, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Giacomello, M.; Drago, I.; Bortolozzi, M.; Scorzeto, M.; Gianelle, A.; Pizzo, P.; Pozzan, T. Ca2+ Hot Spots on the Mitochondrial Surface Are Generated by Ca2+ Mobilization from Stores, but Not by Activation of Store-Operated Ca2+ Channels. Mol. Cell 2010, 38, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Wacquier, B.; Combettes, L.; Dupont, G. Cytoplasmic and Mitochondrial Calcium Signaling: A Two-Way Relationship. Cold Spring Harb. Perspect. Biol. 2019, 11, a035139. [Google Scholar] [CrossRef] [PubMed]
- Turner, H.; Fleig, A.; Stokes, A.; Kinet, J.-P.; Penner, R. Discrimination of Intracellular Calcium Store Subcompartments Using TRPV1 (Transient Receptor Potential Channel, Vanilloid Subfamily Member 1) Release Channel Activity. Biochem. J. 2003, 371, 341–350. [Google Scholar] [CrossRef]
- Wisnoskey, B.J.; Sinkins, W.G.; Schilling, W.P. Activation of Vanilloid Receptor Type I in the Endoplasmic Reticulum Fails to Activate Store-Operated Ca2+ Entry. Biochem. J. 2003, 372, 517–528. [Google Scholar] [CrossRef]
- Bassani, J.W.; Bassani, R.A.; Bers, D.M. Relaxation in Rabbit and Rat Cardiac Cells: Species-Dependent Differences in Cellular Mechanisms. J. Physiol. 1994, 476, 279–293. [Google Scholar] [CrossRef]
- Giamogante, F.; Barazzuol, L.; Brini, M.; Calì, T. ER–Mitochondria Contact Sites Reporters: Strengths and Weaknesses of the Available Approaches. Int. J. Mol. Sci. 2020, 21, 8157. [Google Scholar] [CrossRef]
- Giacomello, M.; Pellegrini, L. The Coming of Age of the Mitochondria–ER Contact: A Matter of Thickness. Cell Death Differ. 2016, 23, 1417–1427. [Google Scholar] [CrossRef]
- Cho, E.; Woo, Y.; Suh, Y.; Suh, B.K.; Kim, S.J.; Nhung, T.T.M.; Yoo, J.Y.; Nghi, T.D.; Lee, S.B.; Mun, D.J.; et al. Ratiometric Measurement of MAM Ca2+ Dynamics Using a Modified CalfluxVTN. Nat. Commun. 2023, 14, 3586. [Google Scholar] [CrossRef]
- Goswami, C.; Hucho, T.B.; Hucho, F. Identification and Characterisation of Novel Tubulin-Binding Motifs Located within the C-Terminus of TRPV1. J. Neurochem. 2007, 101, 250–262. [Google Scholar] [CrossRef] [PubMed]
- Carreá, M.; Andreá, N.; Carles, G.; Borghi, H.; Brichese, L.; Briand, C.; Braguer, D. Tubulin Is an Inherent Component of Mitochondrial Membranes That Interacts with the Voltage-Dependent Anion Channel. J. Biol. Chem. 2002, 277, 33664–33669. [Google Scholar] [CrossRef] [PubMed]
- Goswami, C.; Dreger, M.; Otto, H.; Schwappach, B.; Hucho, F. Rapid Disassembly of Dynamic Microtubules upon Activation of the Capsaicin Receptor TRPV1. J. Neurochem. 2006, 96, 254–266. [Google Scholar] [CrossRef] [PubMed]
- Birsa, N.; Norkett, R.; Higgs, N.; Lopez-Domenech, G.; Kittler, J.T. Mitochondrial Trafficking in Neurons and the Role of the Miro Family of GTPase Proteins. Biochem. Soc. Trans. 2013, 41, 1525–1531. [Google Scholar] [CrossRef] [PubMed]
- Gamper, N.; Shapiro, M.S. Regulation of Ion Transport Proteins by Membrane Phosphoinositides. Nat. Rev. Neurosci. 2007, 8, 921–934. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.-D.; Li, M.-M.; Xu, G.; Zhang, E.-L.; Chen, J.; Sun, B.; Chen, D.-W.; Gao, Y.-Q. Targeting Mitochondria-Associated Membranes as a Potential Therapy against Endothelial Injury Induced by Hypoxia. J. Cell. Biochem. 2019, 120, 18967–18978. [Google Scholar] [CrossRef]
- Lemos, F.O.; Bultynck, G.; Parys, J.B. A Comprehensive Overview of the Complex World of the Endo- and Sarcoplasmic Reticulum Ca2+-Leak Channels. Biochim. Biophys. Acta BBA—Mol. Cell Res. 2021, 1868, 119020. [Google Scholar] [CrossRef]
- Makarewich, C.A.; Zhang, H.; Davis, J.; Correll, R.N.; Trappanese, D.M.; Hoffman, N.E.; Troupes, C.D.; Berretta, R.M.; Kubo, H.; Madesh, M.; et al. Transient Receptor Potential Channels Contribute to Pathological Structural and Functional Remodeling After Myocardial Infarction. Circ. Res. 2014, 115, 567–580. [Google Scholar] [CrossRef]
- Al-Mawla, R.; Ducrozet, M.; Tessier, N.; Païta, L.; Pillot, B.; Gouriou, Y.; Villedieu, C.; Harhous, Z.; Paccalet, A.; Crola Da Silva, C.; et al. Acute Induction of Translocon-Mediated Ca2+ Leak Protects Cardiomyocytes Against Ischemia/Reperfusion Injury. Cells 2020, 9, 1319. [Google Scholar] [CrossRef]
- Bauer, T.M.; Murphy, E. Role of Mitochondrial Calcium and the Permeability Transition Pore in Regulating Cell Death. Circ Res 2020, 126, 280–293. [Google Scholar] [CrossRef]
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthuis, R.J. Cell Biology of Ischemia/Reperfusion Injury. Int. Rev. Cell Mol. Biol. 2012, 298, 229–317. [Google Scholar] [CrossRef] [PubMed]
- Consolini, A.E.; Ragone, M.I.; Bonazzola, P.; Colareda, G.A. Mitochondrial Bioenergetics During Ischemia and Reperfusion. In Mitochondrial Dynamics in Cardiovascular Medicine; Santulli, G., Ed.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2017; pp. 141–167. ISBN 978-3-319-55330-6. [Google Scholar]
- Sala-Vila, A.; Navarro-Lérida, I.; Sánchez-Alvarez, M.; Bosch, M.; Calvo, C.; López, J.A.; Calvo, E.; Ferguson, C.; Giacomello, M.; Serafini, A.; et al. Interplay between Hepatic Mitochondria-Associated Membranes, Lipid Metabolism and Caveolin-1 in Mice. Sci. Rep. 2016, 6, 27351. [Google Scholar] [CrossRef] [PubMed]
- Bidaux, G.; Gordienko, D.; Shapovalov, G.; Farfariello, V.; Borowiec, A.-S.; Iamshanova, O.; Lemonnier, L.; Gueguinou, M.; Guibon, R.; Fromont, G.; et al. 4TM-TRPM8 Channels Are New Gatekeepers of the ER-Mitochondria Ca2+ Transfer. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 981–994. [Google Scholar] [CrossRef]
- Acharya, T.K.; Kumar, A.; Kumar, S.; Goswami, C. TRPV4 Interacts with MFN2 and Facilitates Endoplasmic Reticulum-Mitochondrial Contact Points for Ca2+-Buffering. Life Sci. 2022, 310, 121112. [Google Scholar] [CrossRef] [PubMed]
- Bagur, R.; Hajnóczky, G. Intracellular Ca2+ Sensing: Its Role in Calcium Homeostasis and Signaling. Mol. Cell 2017, 66, 780–788. [Google Scholar] [CrossRef]
- Chen, J.; Li, L.; Bai, X.; Xiao, L.; Shangguan, J.; Zhang, W.; Zhang, X.; Wang, S.; Liu, G. Inhibition of Autophagy Prevents Panax Notoginseng Saponins (PNS) Protection on Cardiac Myocytes Against Endoplasmic Reticulum (ER) Stress-Induced Mitochondrial Injury, Ca2+ Homeostasis and Associated Apoptosis. Front. Pharmacol. 2021, 12, 620812. [Google Scholar] [CrossRef]
- Grings, M.; Seminotti, B.; Karunanidhi, A.; Ghaloul-Gonzalez, L.; Mohsen, A.-W.; Wipf, P.; Palmfeldt, J.; Vockley, J.; Leipnitz, G. ETHE1 and MOCS1 Deficiencies: Disruption of Mitochondrial Bioenergetics, Dynamics, Redox Homeostasis and Endoplasmic Reticulum-Mitochondria Crosstalk in Patient Fibroblasts. Sci. Rep. 2019, 9, 12651. [Google Scholar] [CrossRef]
- Biczo, G.; Vegh, E.T.; Shalbueva, N.; Mareninova, O.A.; Elperin, J.; Lotshaw, E.; Gretler, S.; Lugea, A.; Malla, S.R.; Dawson, D.; et al. Mitochondrial Dysfunction, Through Impaired Autophagy, Leads to Endoplasmic Reticulum Stress, Deregulated Lipid Metabolism, and Pancreatitis in Animal Models. Gastroenterology 2018, 154, 689–703. [Google Scholar] [CrossRef]
- Zhou, H.; Wang, S.; Hu, S.; Chen, Y.; Ren, J. ER-Mitochondria Microdomains in Cardiac Ischemia-Reperfusion Injury: A Fresh Perspective. Front. Physiol. 2018, 9, 755. [Google Scholar] [CrossRef]
- Rieusset, J. Mitochondria-Associated Membranes (MAMs): An Emerging Platform Connecting Energy and Immune Sensing to Metabolic Flexibility. Biochem. Biophys. Res. Commun. 2018, 500, 35–44. [Google Scholar] [CrossRef]
- Ouyang, M.; Zhang, Q.; Shu, J.; Wang, Z.; Fan, J.; Yu, K.; Lei, L.; Li, Y.; Wang, Q. Capsaicin Ameliorates the Loosening of Mitochondria-Associated Endoplasmic Reticulum Membranes and Improves Cognitive Function in Rats With Chronic Cerebral Hypoperfusion. Front. Cell. Neurosci. 2022, 16, 822702. [Google Scholar] [CrossRef] [PubMed]
- Kerkhofs, M.; Giorgi, C.; Marchi, S.; Seitaj, B.; Parys, J.B.; Pinton, P.; Bultynck, G.; Bittremieux, M. Alterations in Ca2+ Signalling via ER-Mitochondria Contact Site Remodelling in Cancer. Adv. Exp. Med. Biol. 2017, 997, 225–254. [Google Scholar] [CrossRef] [PubMed]
- Kimes, B.W.; Brandt, B.L. Properties of a Clonal Muscle Cell Line from Rat Heart. Exp. Cell Res. 1976, 98, 367–381. [Google Scholar] [CrossRef] [PubMed]
- Onódi, Z.; Visnovitz, T.; Kiss, B.; Hambalkó, S.; Koncz, A.; Ágg, B.; Váradi, B.; Tóth, V.É.; Nagy, R.N.; Gergely, T.G.; et al. Systematic Transcriptomic and Phenotypic Characterization of Human and Murine Cardiac Myocyte Cell Lines and Primary Cardiomyocytes Reveals Serious Limitations and Low Resemblances to Adult Cardiac Phenotype. J. Mol. Cell. Cardiol. 2022, 165, 19–30. [Google Scholar] [CrossRef]
- Kuznetsov, A.V.; Javadov, S.; Sickinger, S.; Frotschnig, S.; Grimm, M. H9c2 and HL-1 Cells Demonstrate Distinct Features of Energy Metabolism, Mitochondrial Function and Sensitivity to Hypoxia-Reoxygenation. Biochim. Biophys. Acta 2015, 1853, 276–284. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular Mechanisms of Cell Death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primers | |
|---|---|
| Mouse TRPV1 | Forward 5′-GCTCTCATGGGCGAGACTGTC-3′ |
| Reverse 5′-CGGAAGGCCTTCCTCATGCAC-3′ | |
| Rat TRPV1 | Forward 5′-GCTCTCATGGGTGAGACCGTC-3′ |
| Reverse 5′-CGGAAGGCCTTCCTCATGCAC-3′ | |
| Mouse and rat GAPDH | Forward 5′-GGCTGGCATTGCTCTCAA-3′ |
| Reverse 5′-GCTGTAGCCGTATTCATTGTC-3′ | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tessier, N.; Ducrozet, M.; Dia, M.; Badawi, S.; Chouabe, C.; Crola Da Silva, C.; Ovize, M.; Bidaux, G.; Van Coppenolle, F.; Ducreux, S. TRPV1 Channels Are New Players in the Reticulum–Mitochondria Ca2+ Coupling in a Rat Cardiomyoblast Cell Line. Cells 2023, 12, 2322. https://doi.org/10.3390/cells12182322
Tessier N, Ducrozet M, Dia M, Badawi S, Chouabe C, Crola Da Silva C, Ovize M, Bidaux G, Van Coppenolle F, Ducreux S. TRPV1 Channels Are New Players in the Reticulum–Mitochondria Ca2+ Coupling in a Rat Cardiomyoblast Cell Line. Cells. 2023; 12(18):2322. https://doi.org/10.3390/cells12182322
Chicago/Turabian StyleTessier, Nolwenn, Mallory Ducrozet, Maya Dia, Sally Badawi, Christophe Chouabe, Claire Crola Da Silva, Michel Ovize, Gabriel Bidaux, Fabien Van Coppenolle, and Sylvie Ducreux. 2023. "TRPV1 Channels Are New Players in the Reticulum–Mitochondria Ca2+ Coupling in a Rat Cardiomyoblast Cell Line" Cells 12, no. 18: 2322. https://doi.org/10.3390/cells12182322