
Autophagy Inhibition with Chloroquine Increased Pro-Apoptotic Potential of New Aziridine-Hydrazide Hydrazone Derivatives against Glioblastoma Cells
 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. In Vitro Models of Glioblastoma and Non-Neoplastic Cells (NHA)
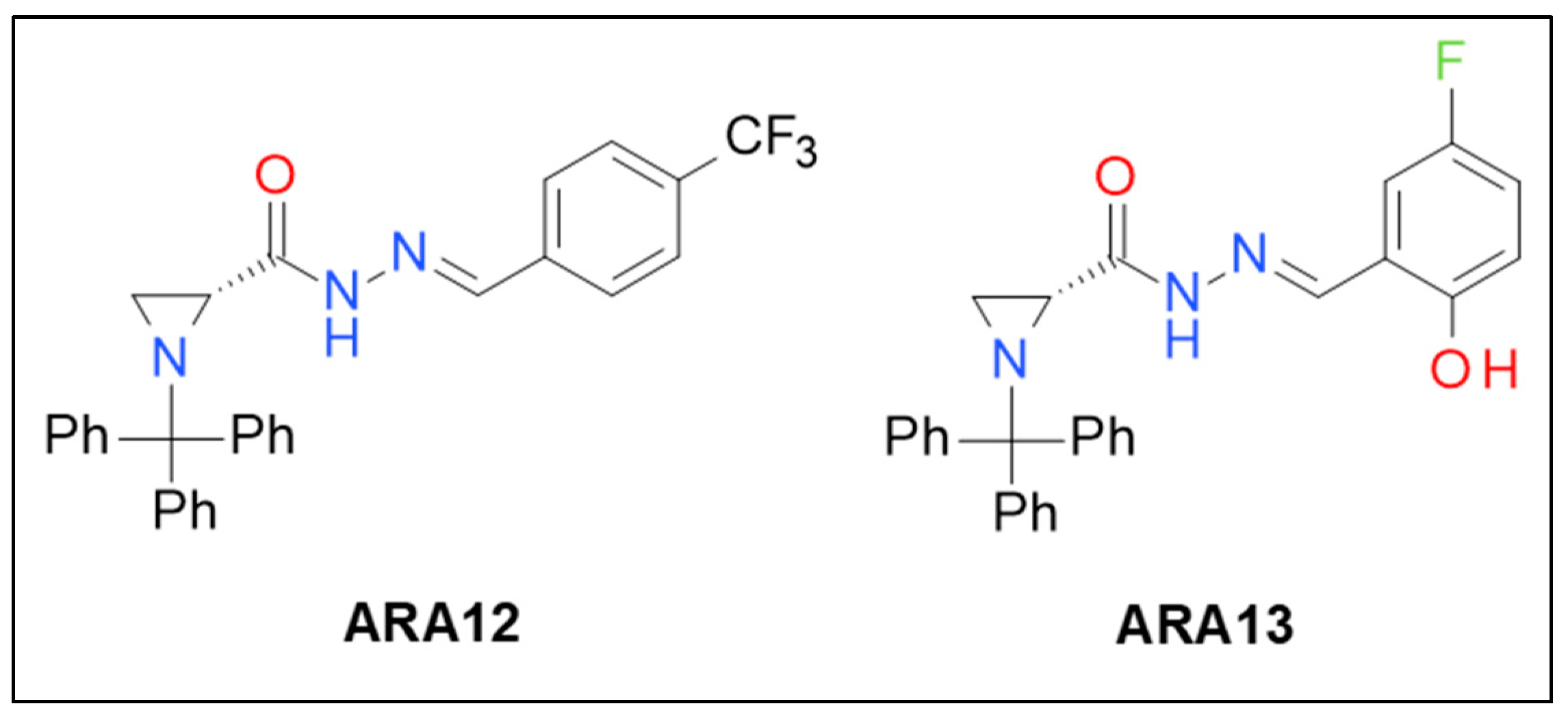
2.2. Synthesis of Tested Compounds
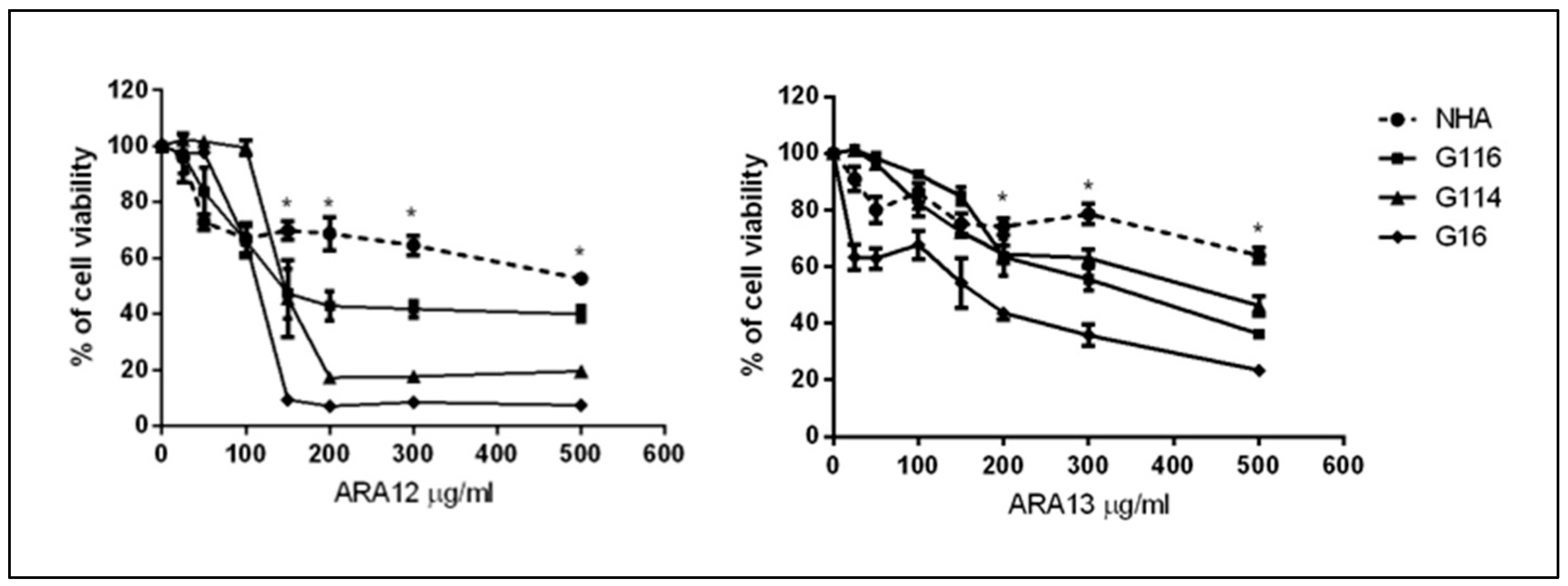
2.3. Drug Cytotoxicity Assay
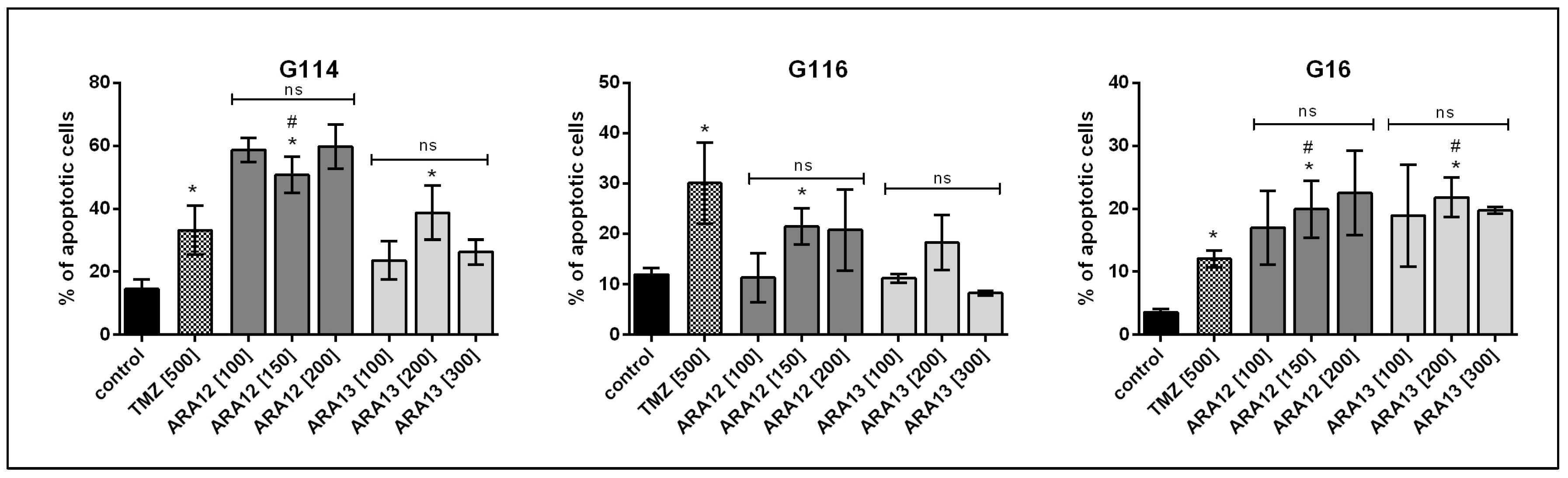
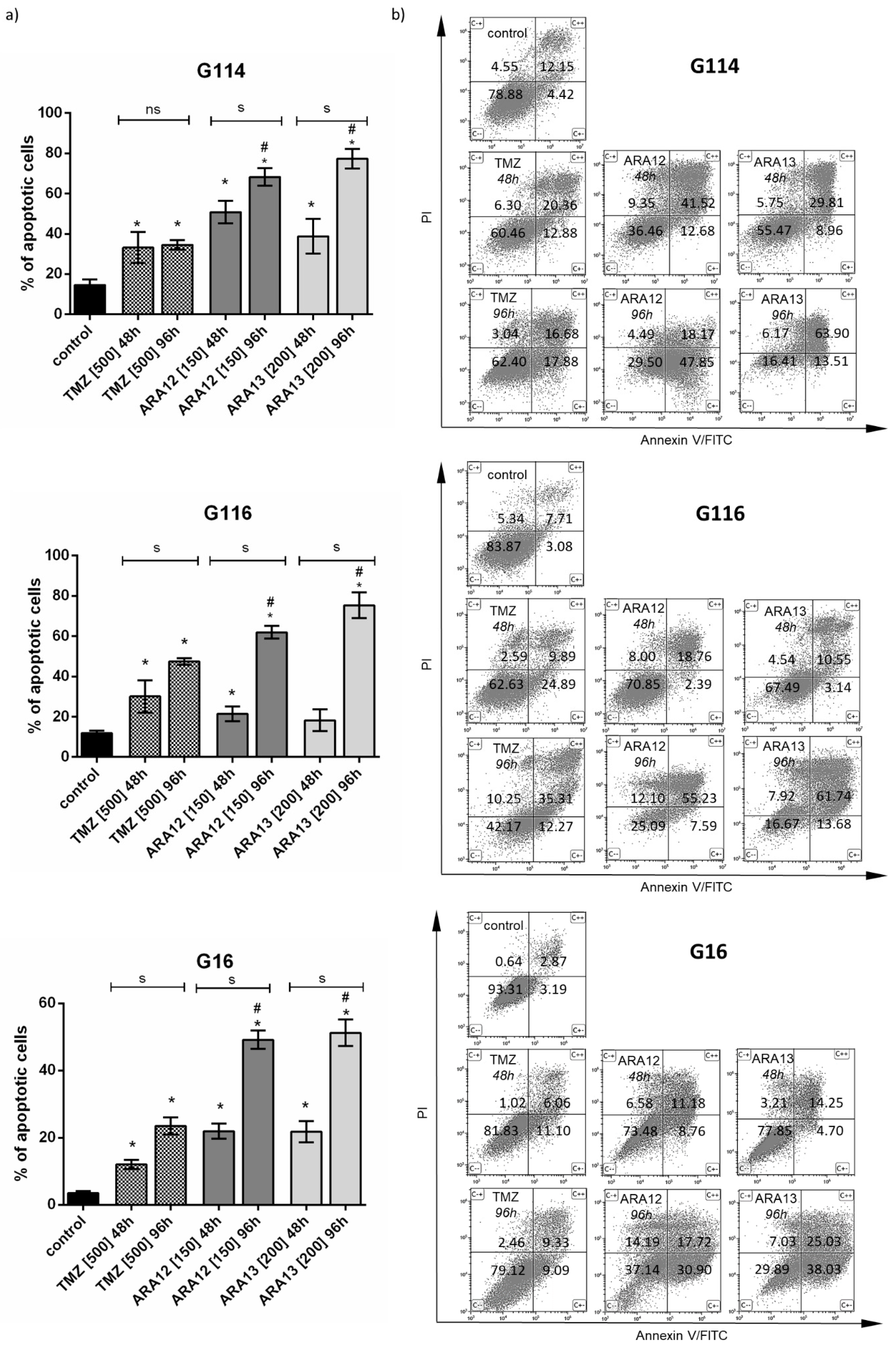
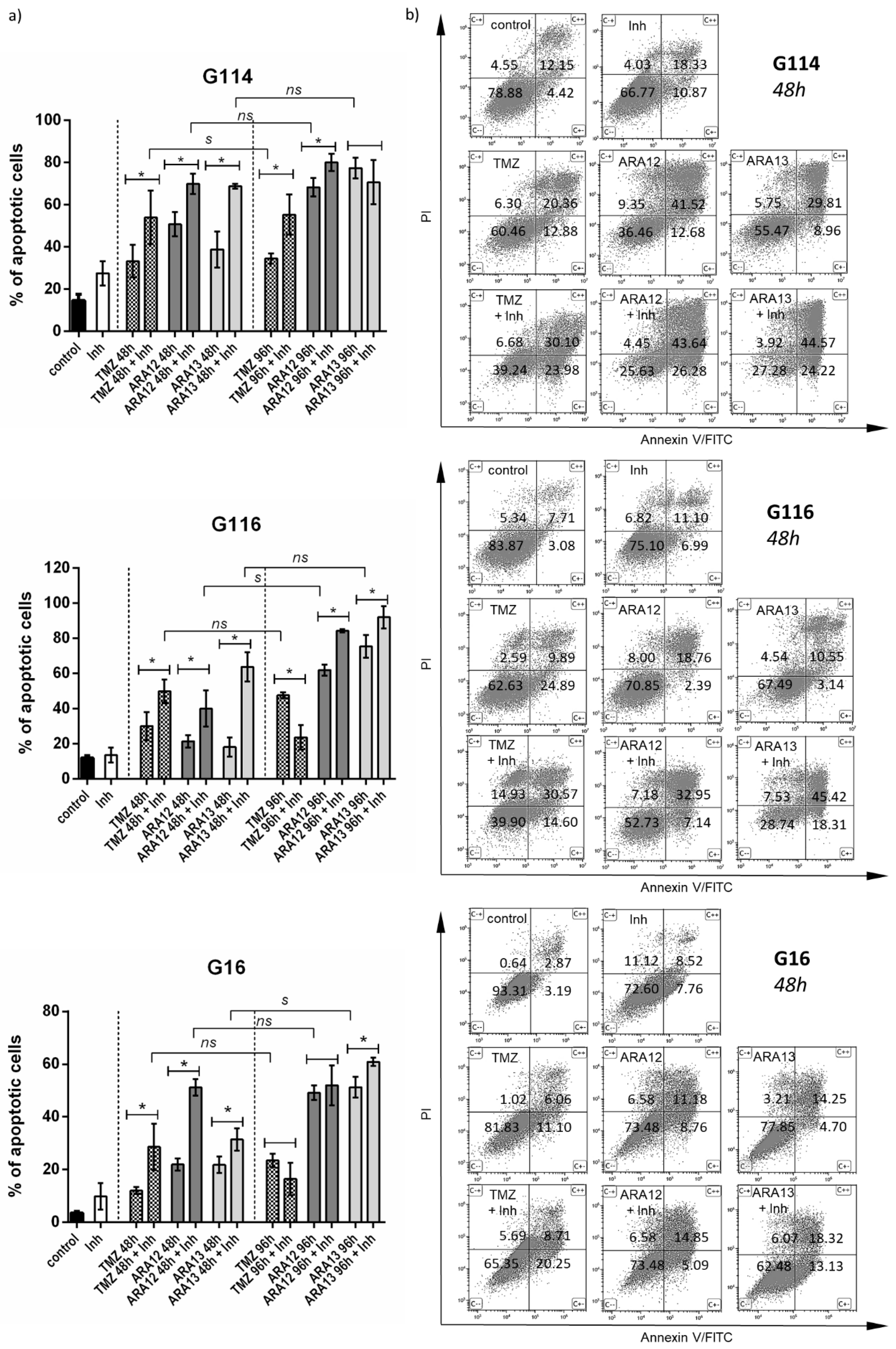
2.4. Assay for Cell Apoptosis by Annexin V/PI Double Staining
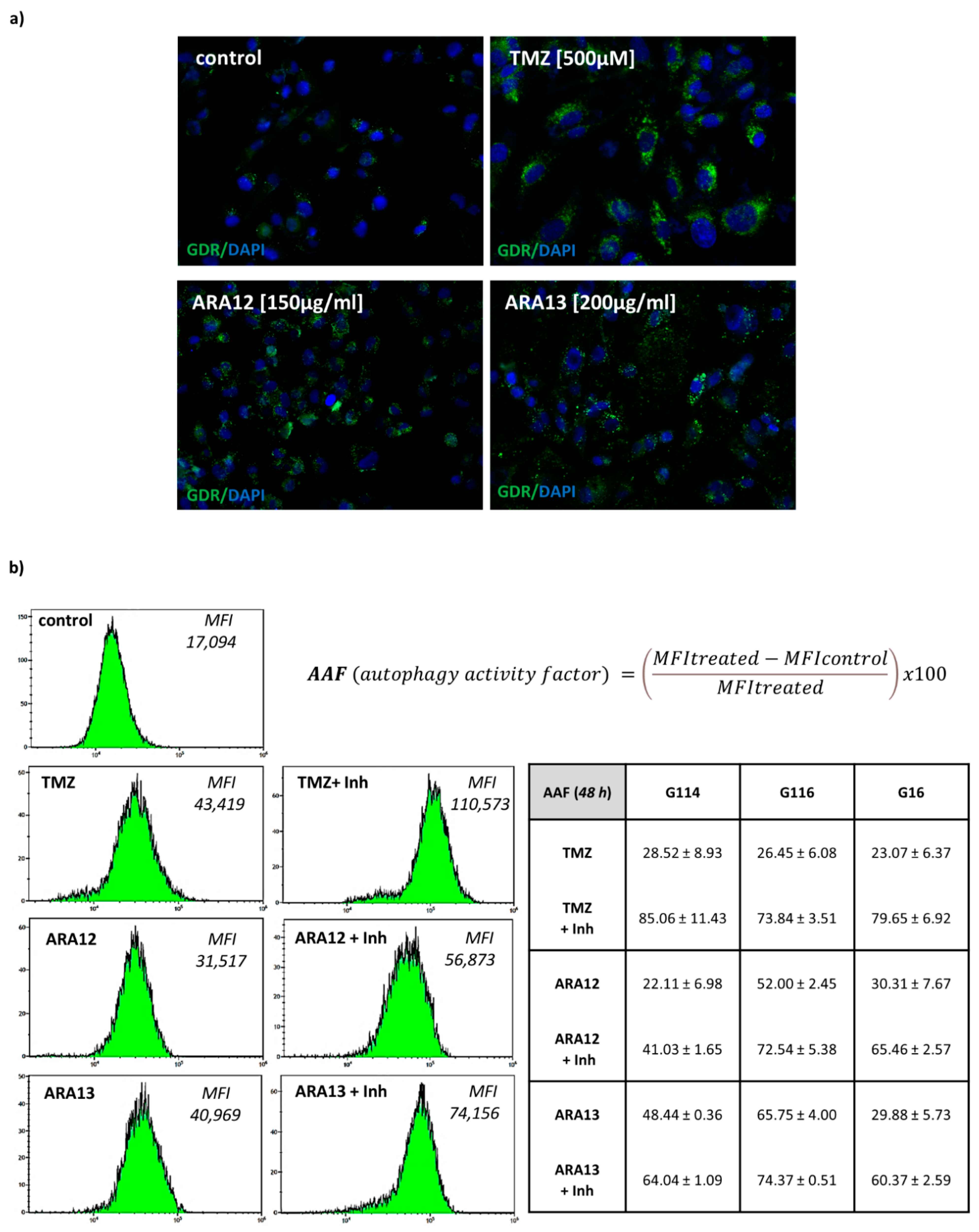
2.5. Assay for Autophagy Detection by FACS with the Use of Autophagy Detection Kit
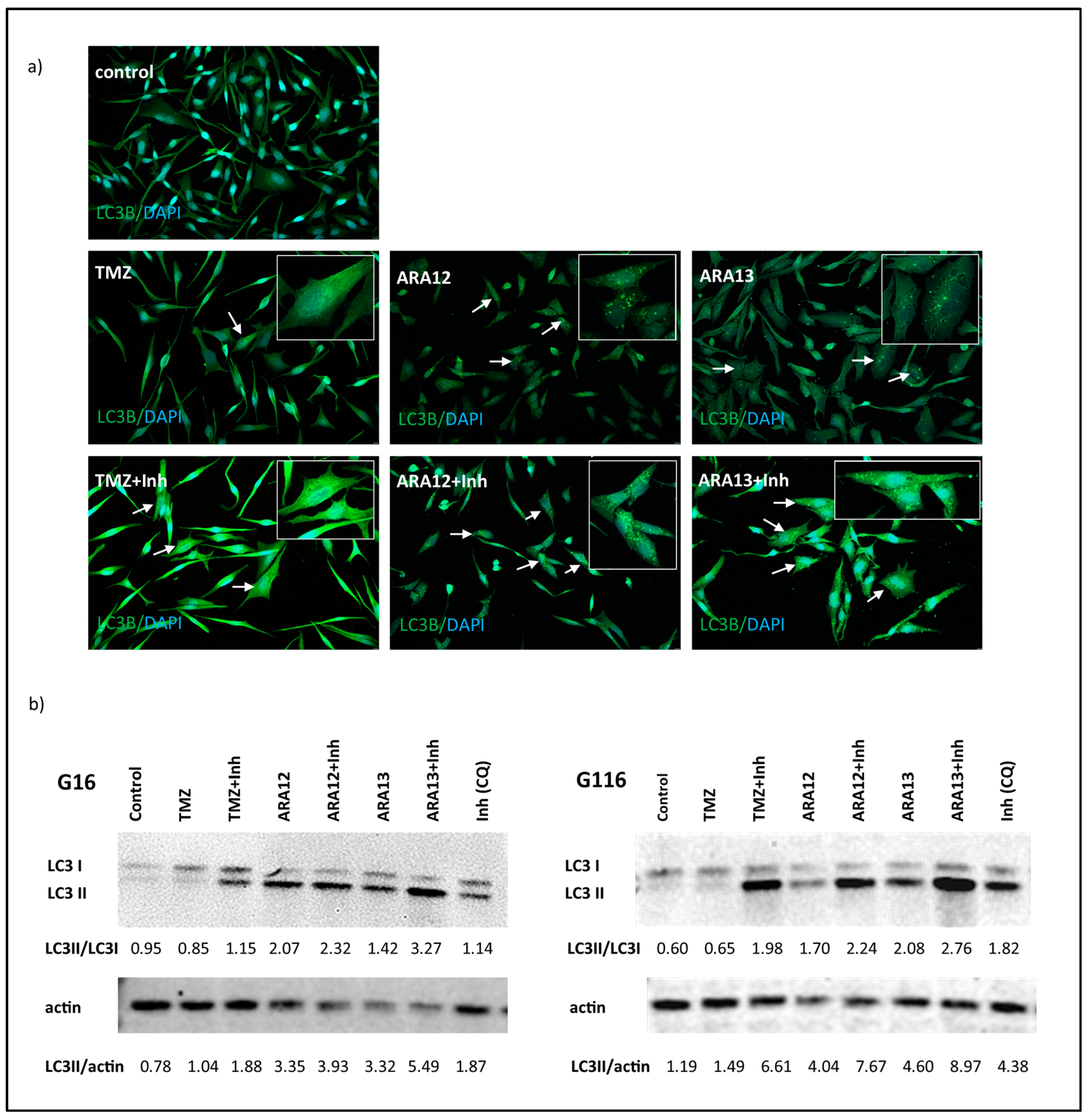
2.6. Immunofluorescence Analysis
2.7. Western Blot Analysis
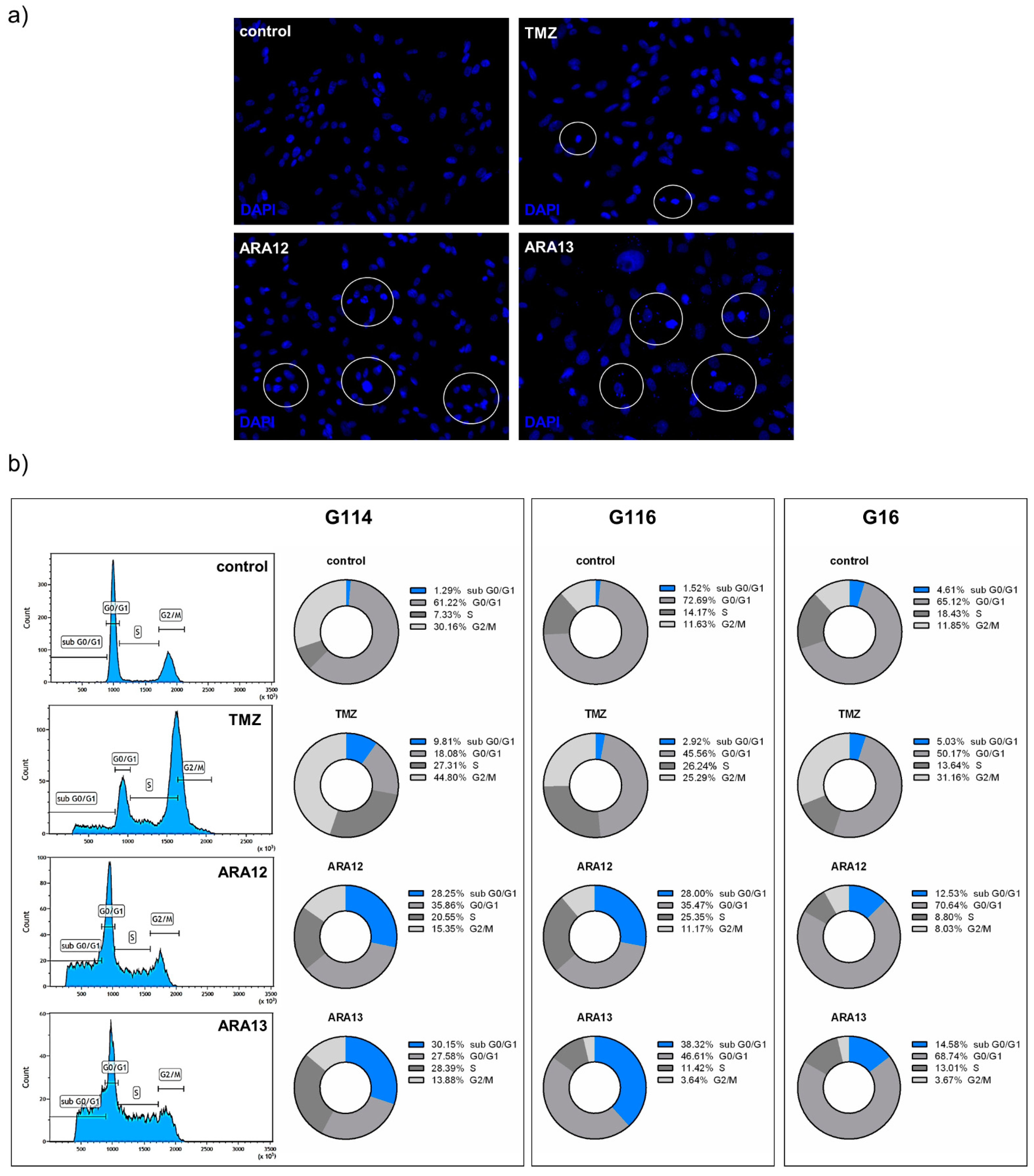
2.8. Detection of subG0/G1 Phase
2.9. Statistical Analysis
3. Results
3.1. Synthesis of New Aziridine–Hydrazide Hydrazone Derivatives and Initial Screening of Their Anti-Tumor Activity
3.2. Newly Synthesized Aziridine–Hydrazide Hydrazone Derivatives Present Proapoptotic Potential against GB Cells Higher than TMZ
3.3. Inhibition of Treatment-Induced Autophagy Facilitate Apoptotic Cell Death of GB Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Delgado-López, P.D.; Corrales-García, E.M. Survival in Glioblastoma: A Review on the Impact of Treatment Modalities. Clin. Transl. Oncol. 2016, 18, 1062–1071. [Google Scholar] [CrossRef]
- Osuka, S.; Van Meir, E.G. Overcoming Therapeutic Resistance in Glioblastoma: The Way Forward. J. Clin. Investig. 2017, 127, 415–426. [Google Scholar] [CrossRef] [Green Version]
- Vollmann-Zwerenz, A.; Leidgens, V.; Feliciello, G.; Klein, C.A.; Hau, P. Tumor Cell Invasion in Glioblastoma. Int. J. Mol. Sci. 2020, 21, 1932. [Google Scholar] [CrossRef] [Green Version]
- Ferri, A.; Stagni, V.; Barilà, D. Targeting the DNA Damage Response to Overcome Cancer Drug Resistance in Glioblastoma. Int. J. Mol. Sci. 2020, 21, 4910. [Google Scholar] [CrossRef]
- Santamaría, P.G.; Moreno-Bueno, G.; Cano, A. Contribution of Epithelial Plasticity to Therapy Resistance. J. Clin. Med. 2019, 8, 676. [Google Scholar] [CrossRef] [Green Version]
- Chien, C.-H.; Hsueh, W.-T.; Chuang, J.-Y.; Chang, K.-Y. Role of Autophagy in Therapeutic Resistance of Glioblastoma. J. Cancer Metastasis Treat. 2019, 5, 66. [Google Scholar] [CrossRef] [Green Version]
- Saleh, T.; Bloukh, S.; Carpenter, V.J.; Alwohoush, E.; Bakeer, J.; Darwish, S.; Azab, B.; Gewirtz, D.A. Therapy-Induced Senescence: An “Old” Friend Becomes the Enemy. Cancers 2020, 12, 822. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Kohli, J.; Demaria, M. Senescent Cells in Cancer Therapy: Friends or Foes? Trends Cancer 2020, 6, 838–857. [Google Scholar] [CrossRef]
- Singh, N.; Miner, A.; Hennis, L.; Mittal, S. Mechanisms of Temozolomide Resistance in Glioblastoma—A Comprehensive Review. Cancer Drug Resist. 2021, 4, 17–43. [Google Scholar] [CrossRef]
- Dvorakova, K.; Payne, C.M.; Tome, M.E.; Briehl, M.M.; McClure, T.; Dorr, R.T. Induction of Oxidative Stress and Apoptosis in Myeloma Cells by the Aziridine-Containing Agent Imexon. Biochem. Pharm. 2000, 60, 749–758. [Google Scholar] [CrossRef]
- Ismail, F.M.D.; Levitsky, D.O.; Dembitsky, V.M. Aziridine Alkaloids as Potential Therapeutic Agents. Eur. J. Med. Chem. 2009, 44, 3373–3387. [Google Scholar] [CrossRef] [PubMed]
- Dembitsky, V.M.; Terent’ev, A.O.; Levitsky, D.O. Aziridine Alkaloids: Origin, Chemistry and Activity. In Natural Products: Phytochemistry, Botany and Metabolism of Alkaloids, Phenolics and Terpenes; Ramawat, K.G., Mérillon, J.-M., Eds.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 977–1006. ISBN 978-3-642-22144-6. [Google Scholar]
- Labib, M.B.; Philoppes, J.N.; Lamie, P.F.; Ahmed, E.R. Azole-Hydrazone Derivatives: Design, Synthesis, in Vitro Biological Evaluation, Dual EGFR/HER2 Inhibitory Activity, Cell Cycle Analysis and Molecular Docking Study as Anticancer Agents. Bioorg. Chem. 2018, 76, 67–80. [Google Scholar] [CrossRef] [PubMed]
- Sreenivasulu, R.; Reddy, K.T.; Sujitha, P.; Kumar, C.G.; Raju, R.R. Synthesis, Antiproliferative and Apoptosis Induction Potential Activities of Novel Bis(Indolyl)Hydrazide-Hydrazone Derivatives. Bioorg. Med. Chem. 2019, 27, 1043–1055. [Google Scholar] [CrossRef]
- Altıntop, M.D.; Özdemir, A.; Turan-Zitouni, G.; Ilgın, S.; Atlı, Ö.; İşcan, G.; Kaplancıklı, Z.A. Synthesis and Biological Evaluation of Some Hydrazone Derivatives as New Anticandidal and Anticancer Agents. Eur. J. Med. Chem. 2012, 58, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Rollas, S.; Küçükgüzel, S.G. Biological Activities of Hydrazone Derivatives. Molecules 2007, 12, 1910–1939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, N.; Ranjana, R.; Kumari, M.; Kumar, B. A Review on Biological Activities of Hydrazone Derivatives. Int. J. Pharm. Clin. Res. 2016, 6, 162–166. [Google Scholar]
- de Oliveira Carneiro Brum, J.; França, T.C.C.; LaPlante, S.R.; Villar, J.D.F. Synthesis and Biological Activity of Hydrazones and Derivatives: A Review. Mini Rev. Med. Chem. 2020, 20, 342–368. [Google Scholar] [CrossRef]
- Kowalczyk, A.; Pieczonka, A.M.; Rachwalski, M.; Leśniak, S.; Stączek, P. Synthesis and Evaluation of Biological Activities of Aziridine Derivatives of Urea and Thiourea. Molecules 2018, 23, 45. [Google Scholar] [CrossRef] [Green Version]
- Pieczonka, A.M.; Strzelczyk, A.; Sadowska, B.; Mlostoń, G.; Stączek, P. Synthesis and Evaluation of Antimicrobial Activity of Hydrazones Derived from 3-Oxido-1H-Imidazole-4-Carbohydrazides. Eur. J. Med. Chem. 2013, 64, 389–395. [Google Scholar] [CrossRef]
- Thota, S.; Rodrigues, D.A.; Pinheiro, P.d.S.M.; Lima, L.M.; Fraga, C.A.M.; Barreiro, E.J. N-Acylhydrazones as Drugs. Bioorg. Med. Chem. Lett. 2018, 28, 2797–2806. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A Summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Witusik-Perkowska, M.; Zakrzewska, M.; Szybka, M.; Papierz, W.; Jaskolski, D.J.; Liberski, P.P.; Sikorska, B. Astrocytoma-Associated Antigens—IL13Rα2, Fra-1, and EphA2 as Potential Markers to Monitor the Status of Tumour-Derived Cell Cultures in Vitro. Cancer Cell Int. 2014, 14, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witusik-Perkowska, M.; Zakrzewska, M.; Sikorska, B.; Papierz, W.; Jaskolski, D.J.; Szemraj, J.; Liberski, P.P. Glioblastoma-Derived Cells in Vitro Unveil the Spectrum of Drug Resistance Capability—Comparative Study of Tumour Chemosensitivity in Different Culture Systems. Biosci. Rep. 2017, 37, BSR20170058. [Google Scholar] [CrossRef] [Green Version]
- Leśniak, S.; Pieczonka, A.M.; Jarzyński, S.; Justyna, K.; Rachwalski, M. Synthesis and Evaluation of the Catalytic Properties of Semicarbazides Derived from N-Triphenylmethyl-Aziridine-2-Carbohydrazides. Tetrahedron Asymmetry 2013, 24, 1341–1344. [Google Scholar] [CrossRef]
- Chan, L.L.-Y.; Shen, D.; Wilkinson, A.R.; Patton, W.; Lai, N.; Chan, E.; Kuksin, D.; Lin, B.; Qiu, J. A Novel Image-Based Cytometry Method for Autophagy Detection in Living Cells. Autophagy 2012, 8, 1371–1382. [Google Scholar] [CrossRef] [Green Version]
- Loos, B.; du Toit, A.; Hofmeyr, J.-H.S. Defining and Measuring Autophagosome Flux—Concept and Reality. Autophagy 2014, 10, 2087–2096. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T. How to Interpret LC3 Immunoblotting. Autophagy 2007, 3, 542–545. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the Use and Interpretation of Assays for Monitoring Autophagy (4th Edition)1. Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef]
- Gupta, M.; Lee, H.J.; Barden, C.J.; Weaver, D.F. The Blood–Brain Barrier (BBB) Score. J. Med. Chem. 2019, 62, 9824–9836. [Google Scholar] [CrossRef] [PubMed]
- Solitro, A.R.; MacKeigan, J.P. Leaving the Lysosome behind: Novel Developments in Autophagy Inhibition. Future Med. Chem. 2016, 8, 73–86. [Google Scholar] [CrossRef] [Green Version]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.-J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine Inhibits Autophagic Flux by Decreasing Autophagosome-Lysosome Fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.; Klockow, J.L.; Zhang, M.; Lafortune, F.; Chang, E.; Jin, L.; Wu, Y.; Daldrup-Link, H.E. Glioblastoma Multiforme (GBM): An Overview of Current Therapies and Mechanisms of Resistance. Pharmacol. Res. 2021, 171, 105780. [Google Scholar] [CrossRef] [PubMed]
- Szklener, K.; Mazurek, M.; Wieteska, M.; Wacławska, M.; Bilski, M.; Mańdziuk, S. New Directions in the Therapy of Glioblastoma. Cancers 2022, 14, 5377. [Google Scholar] [CrossRef] [PubMed]
- Pawlowska, E.; Szczepanska, J.; Szatkowska, M.; Blasiak, J. An Interplay between Senescence, Apoptosis and Autophagy in Glioblastoma Multiforme—Role in Pathogenesis and Therapeutic Perspective. Int. J. Mol. Sci. 2018, 19, 889. [Google Scholar] [CrossRef] [Green Version]
- Beltzig, L.; Schwarzenbach, C.; Leukel, P.; Frauenknecht, K.B.M.; Sommer, C.; Tancredi, A.; Hegi, M.E.; Christmann, M.; Kaina, B. Senescence Is the Main Trait Induced by Temozolomide in Glioblastoma Cells. Cancers 2022, 14, 2233. [Google Scholar] [CrossRef]
- Pérez-Hernández, M.; Arias, A.; Martínez-García, D.; Pérez-Tomás, R.; Quesada, R.; Soto-Cerrato, V. Targeting Autophagy for Cancer Treatment and Tumor Chemosensitization. Cancers 2019, 11, 1599. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, J.; Li, K.; Deng, L.; Wang, H. Combination of an Autophagy Inducer and an Autophagy Inhibitor: A Smarter Strategy Emerging in Cancer Therapy. Front. Pharmacol. 2020, 11, 408. [Google Scholar] [CrossRef] [Green Version]
- Espinos, E.; Lai, R.; Giuriato, S. The Dual Role of Autophagy in Crizotinib-Treated ALK+ ALCL: From the Lymphoma Cells Drug Resistance to Their Demise. Cells 2021, 10, 2517. [Google Scholar] [CrossRef]
- Golden, E.B.; Cho, H.-Y.; Jahanian, A.; Hofman, F.M.; Louie, S.G.; Schönthal, A.H.; Chen, T.C. Chloroquine Enhances Temozolomide Cytotoxicity in Malignant Gliomas by Blocking Autophagy. Neurosurg. Focus 2014, 37, E12. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.; Xu, Z.; Dai, S.; Qian, L.; Sun, L.; Gong, Z. Targeting Autophagy to Sensitive Glioma to Temozolomide Treatment. J. Exp. Clin. Cancer Res. 2016, 35, 23. [Google Scholar] [CrossRef] [Green Version]
- Peterson, Q.P.; Hsu, D.C.; Goode, D.R.; Novotny, C.J.; Totten, R.K.; Hergenrother, P.J. Procaspase-3 Activation as an Anti-Cancer Strategy: Structure-Activity Relationship of Procaspase-Activating Compound 1 (PAC-1) and Its Cellular Co-Localization with Caspase-3. J. Med. Chem. 2009, 52, 5721–5731. [Google Scholar] [CrossRef] [Green Version]
- Peterson, Q.P.; Goode, D.R.; West, D.C.; Ramsey, K.N.; Lee, J.; Hergenrother, P.J. PAC-1 Activates Procaspase-3 in Vitro through Relief of Zinc-Mediated Inhibition. J. Mol. Biol. 2009, 388, 144–158. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Che, X.; Zheng, Q.; Wu, A.; Pan, K.; Shao, A.; Wu, Q.; Zhang, J.; Hong, Y. Caspases: A Molecular Switch Node in the Crosstalk between Autophagy and Apoptosis. Int. J. Biol. Sci. 2014, 10, 1072–1083. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.W.; Kim, H.-K.; Lee, N.-H.; Yi, H.-Y.; Kim, H.-S.; Hong, S.H.; Hong, Y.-K.; Joe, Y.A. The Synergistic Effect of Combination Temozolomide and Chloroquine Treatment Is Dependent on Autophagy Formation and P53 Status in Glioma Cells. Cancer Lett. 2015, 360, 195–204. [Google Scholar] [CrossRef]
- Tsapras, P.; Nezis, I.P. Caspase Involvement in Autophagy. Cell Death Differ. 2017, 24, 1369–1379. [Google Scholar] [CrossRef] [Green Version]
- Inoue, M.; Sumii, Y.; Shibata, N. Contribution of Organofluorine Compounds to Pharmaceuticals. ACS Omega 2020, 5, 10633–10640. [Google Scholar] [CrossRef]
- Dréan, A.; Goldwirt, L.; Verreault, M.; Canney, M.; Schmitt, C.; Guehennec, J.; Delattre, J.-Y.; Carpentier, A.; Idbaih, A. Blood-Brain Barrier, Cytotoxic Chemotherapies and Glioblastoma. Expert Rev. Neurother. 2016, 16, 1285–1300. [Google Scholar] [CrossRef]
- Jain, R.K. Antiangiogenesis Strategies Revisited: From Starving Tumors to Alleviating Hypoxia. Cancer Cell 2014, 26, 605–622. [Google Scholar] [CrossRef] [Green Version]
- Wolff, A.; Antfolk, M.; Brodin, B.; Tenje, M. In Vitro Blood–Brain Barrier Models—An Overview of Established Models and New Microfluidic Approaches. J. Pharm. Sci. 2015, 104, 2727–2746. [Google Scholar] [CrossRef]
- Foo, C.Y.; Munir, N.; Kumaria, A.; Akhtar, Q.; Bullock, C.J.; Narayanan, A.; Fu, R.Z. Medical Device Advances in the Treatment of Glioblastoma. Cancers 2022, 14, 5341. [Google Scholar] [CrossRef]
- Kunwar, S.; Chang, S.; Westphal, M.; Vogelbaum, M.; Sampson, J.; Barnett, G.; Shaffrey, M.; Ram, Z.; Piepmeier, J.; Prados, M.; et al. Phase III Randomized Trial of CED of IL13-PE38QQR vs Gliadel Wafers for Recurrent Glioblastoma†. Neuro Oncol. 2010, 12, 871–881. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, M.H.; Canney, M.; Carpentier, A.; Idbaih, A. Overcoming the Blood Brain Barrier in Glioblastoma: Status and Future Perspective. Rev. Neurol. 2023, 179, 430–436. [Google Scholar] [CrossRef]
- Prados, M.D.; Schold SC JR, S.C.; Fine, H.A.; Jaeckle, K.; Hochberg, F.; Mechtler, L.; Fetell, M.R.; Phuphanich, S.; Feun, L.; Janus, T.J.; et al. A Randomized, Double-Blind, Placebo-Controlled, Phase 2 Study of RMP-7 in Combination with Carboplatin Administered Intravenously for the Treatment of Recurrent Malignant Glioma. Neuro Oncol. 2003, 5, 96–103. [Google Scholar] [CrossRef] [Green Version]
- Vézina, A.; Manglani, M.; Morris, D.; Foster, B.; McCord, M.; Song, H.; Zhang, M.; Davis, D.; Zhang, W.; Bills, J.; et al. Adenosine A2A Receptor Activation Enhances Blood–Tumor Barrier Permeability in a Rodent Glioma Model. Mol. Cancer Res. 2021, 19, 2081–2095. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.R.; Hernandez, Y.; Miyasaki, K.F.; Kwon, E.J. Engineered Nanomaterials That Exploit Blood-Brain Barrier Dysfunction for Delivery to the Brain. Adv. Drug Deliv. Rev. 2023, 197, 114820. [Google Scholar] [CrossRef] [PubMed]
- Mehrabian, A.; Mashreghi, M.; Dadpour, S.; Badiee, A.; Arabi, L.; Hoda Alavizadeh, S.; Alia Moosavian, S.; Reza Jaafari, M. Nanocarriers Call the Last Shot in the Treatment of Brain Cancers. Technol. Cancer Res. Treat. 2022, 21, 15330338221080974. [Google Scholar] [CrossRef] [PubMed]
- Paolino, D.; Cosco, D.; Molinaro, R.; Celia, C.; Fresta, M. Supramolecular Devices to Improve the Treatment of Brain Diseases. Drug Discov. Today 2011, 16, 311–324. [Google Scholar] [CrossRef]
- Wong, K.H.; Riaz, M.K.; Xie, Y.; Zhang, X.; Liu, Q.; Chen, H.; Bian, Z.; Chen, X.; Lu, A.; Yang, Z. Review of Current Strategies for Delivering Alzheimer’s Disease Drugs across the Blood-Brain Barrier. Int. J. Mol. Sci. 2019, 20, 381. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Smith, Q.R.; Liu, X. Brain Penetrating Peptides and Peptide–Drug Conjugates to Overcome the Blood–Brain Barrier and Target CNS Diseases. WIREs Nanomed. Nanobiotechnol. 2021, 13, e1695. [Google Scholar] [CrossRef]
- Wykosky, J.; Gibo, D.M.; Stanton, C.; Debinski, W. Interleukin-13 Receptor Alpha 2, EphA2, and Fos-Related Antigen 1 as Molecular Denominators of High-Grade Astrocytomas and Specific Targets for Combinatorial Therapy. Clin. Cancer Res. 2008, 14, 199–208. [Google Scholar] [CrossRef] [Green Version]
- Pandya, H.; Gibo, D.M.; Garg, S.; Kridel, S.; Debinski, W. An Interleukin 13 Receptor α 2–Specific Peptide Homes to Human Glioblastoma Multiforme Xenografts. Neuro Oncol. 2012, 14, 6–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witusik-Perkowska, M.; Zakrzewska, M.; Jaskolski, D.J.; Liberski, P.P.; Szemraj, J. Artificial Microenvironment of in Vitro Glioblastoma Cell Cultures Changes Profile of MiRNAs Related to Tumor Drug Resistance. OncoTargets Ther. 2019, 12, 3905–3918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, H.; Yang, Z.; Zhang, S.; Cao, S.; Shen, S.; Pang, Z.; Jiang, X. Ligand Modified Nanoparticles Increases Cell Uptake, Alters Endocytosis and Elevates Glioma Distribution and Internalization. Sci. Rep. 2013, 3, 2534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madhankumar, A.B.; Slagle-Webb, B.; Mintz, A.; Sheehan, J.M.; Connor, J.R. Interleukin-13 Receptor-Targeted Nanovesicles Are a Potential Therapy for Glioblastoma Multiforme. Mol. Cancer Ther. 2006, 5, 3162–3169. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Lv, L.; Shi, H.; Hua, Y.; Lv, W.; Wang, X.; Xin, H.; Xu, Q. PEGylated Polyamidoamine Dendrimer Conjugated with Tumor Homing Peptide as a Potential Targeted Delivery System for Glioma. Colloids Surf. B Biointerfaces 2016, 147, 242–249. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
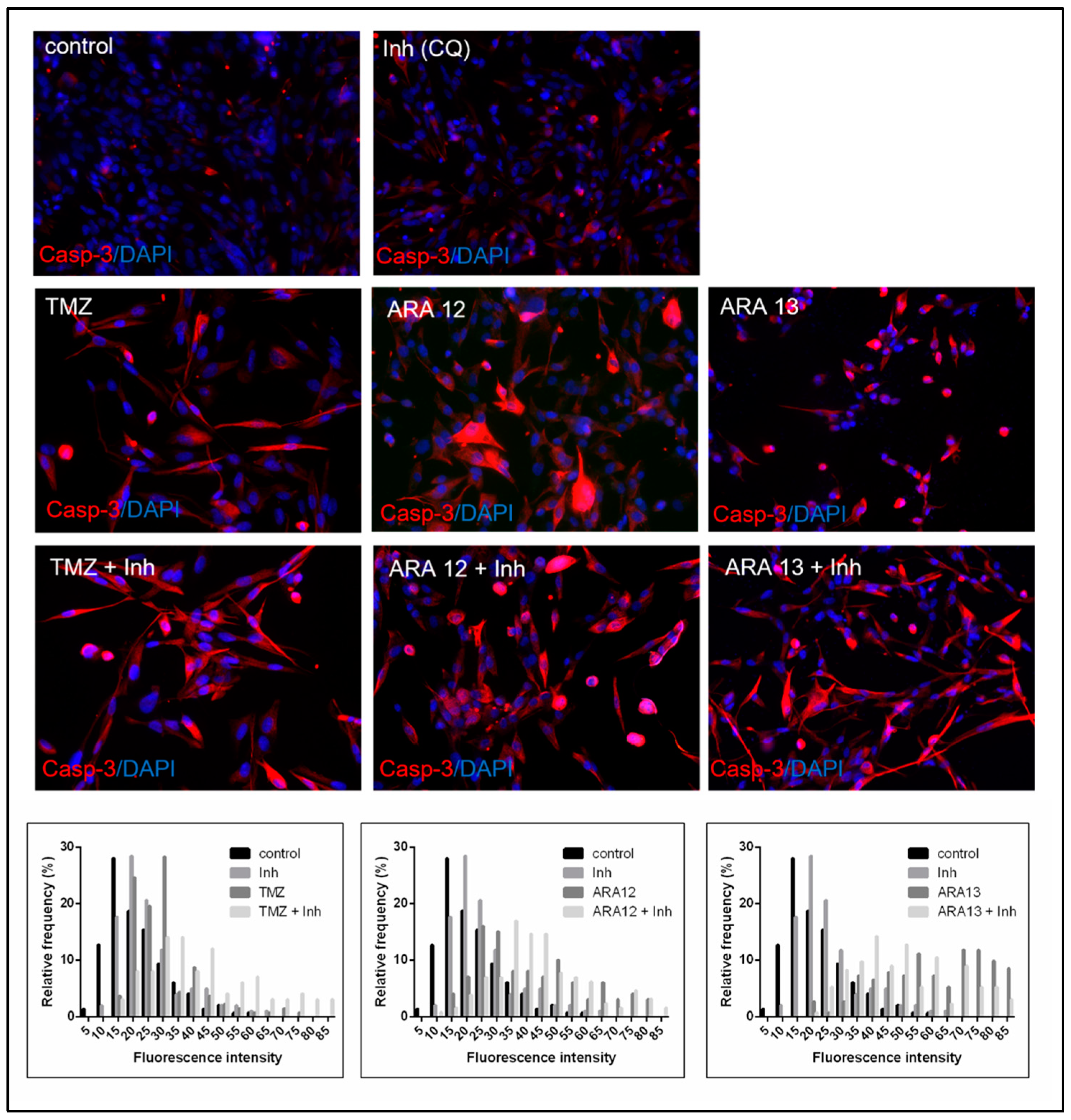{kind=link}
| Treatment Mode | G16 | G116 | G114 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Early Apopt. | Late Apopt. | Viable Cells (%) | Early Apopt. | Late Apopt. | Viable Cells (%) | Early Apopt | Late Apopt. | Viable Cells (%) | |
| (combined/single) | (combined/single) | (combined/single) | |||||||
| untreated | - | - | 93.31 ± 2.69 | - | - | 83.87 ± 4.99 | - | - | 78.88 ± 4.81 |
| TMZ | ↑ (1.83) | ↑ (1.62) | 82.62 ± 6.32 | (0.58) | ↑↑ (3.09) | 62.63 ± 8.85 | ↑ (1.68) | ↑ (1.58) | 60.46 ± 3.59 |
| TMZ + Inh | 65.35 ± 8.65 * | 39.90 ±13.41 * | 39.24 ± 10.54 * | ||||||
| ARA12 | ↑↑↑ (8.50) | (0.53) | 73.48 ± 2.55 | ↑↑ (2.98) | ↑ (1.75) | 70.85 ± 3.88 | ↑↑ (2.07) | (1.05) | 36.46 ± 6.44 |
| ARA12 + Inh | 43.31 ± 2.90 * | 52.73 ± 7.18 * | 25.63 ± 6.10 * | ||||||
| ARA13 | ↑↑ (2.79) | (1.26) | 77.85 ± 6.28 | ↑↑↑ (5.83) | ↑↑↑ (4.30) | 67.49 ± 5.01 | ↑↑ (2.70) | ↑ (1.50) | 55.47 ± 9.96 |
| ARA13 + Inh | 62.48 ± 5.69 * | 28.74 ± 7.53 * | 27.28 ± 1.49 * | ||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Witusik-Perkowska, M.; Głowacka, P.; Pieczonka, A.M.; Świderska, E.; Pudlarz, A.; Rachwalski, M.; Szymańska, J.; Zakrzewska, M.; Jaskólski, D.J.; Szemraj, J. Autophagy Inhibition with Chloroquine Increased Pro-Apoptotic Potential of New Aziridine-Hydrazide Hydrazone Derivatives against Glioblastoma Cells. Cells 2023, 12, 1906. https://doi.org/10.3390/cells12141906
Witusik-Perkowska M, Głowacka P, Pieczonka AM, Świderska E, Pudlarz A, Rachwalski M, Szymańska J, Zakrzewska M, Jaskólski DJ, Szemraj J. Autophagy Inhibition with Chloroquine Increased Pro-Apoptotic Potential of New Aziridine-Hydrazide Hydrazone Derivatives against Glioblastoma Cells. Cells. 2023; 12(14):1906. https://doi.org/10.3390/cells12141906
Chicago/Turabian StyleWitusik-Perkowska, Monika, Pola Głowacka, Adam M. Pieczonka, Ewa Świderska, Agnieszka Pudlarz, Michał Rachwalski, Julia Szymańska, Magdalena Zakrzewska, Dariusz J. Jaskólski, and Janusz Szemraj. 2023. "Autophagy Inhibition with Chloroquine Increased Pro-Apoptotic Potential of New Aziridine-Hydrazide Hydrazone Derivatives against Glioblastoma Cells" Cells 12, no. 14: 1906. https://doi.org/10.3390/cells12141906