
Chimeric Antigen Receptor T Cells in Glioblastoma—Current Concepts and Promising Future

Abstract
:1. Introduction
2. Preclinical Studies
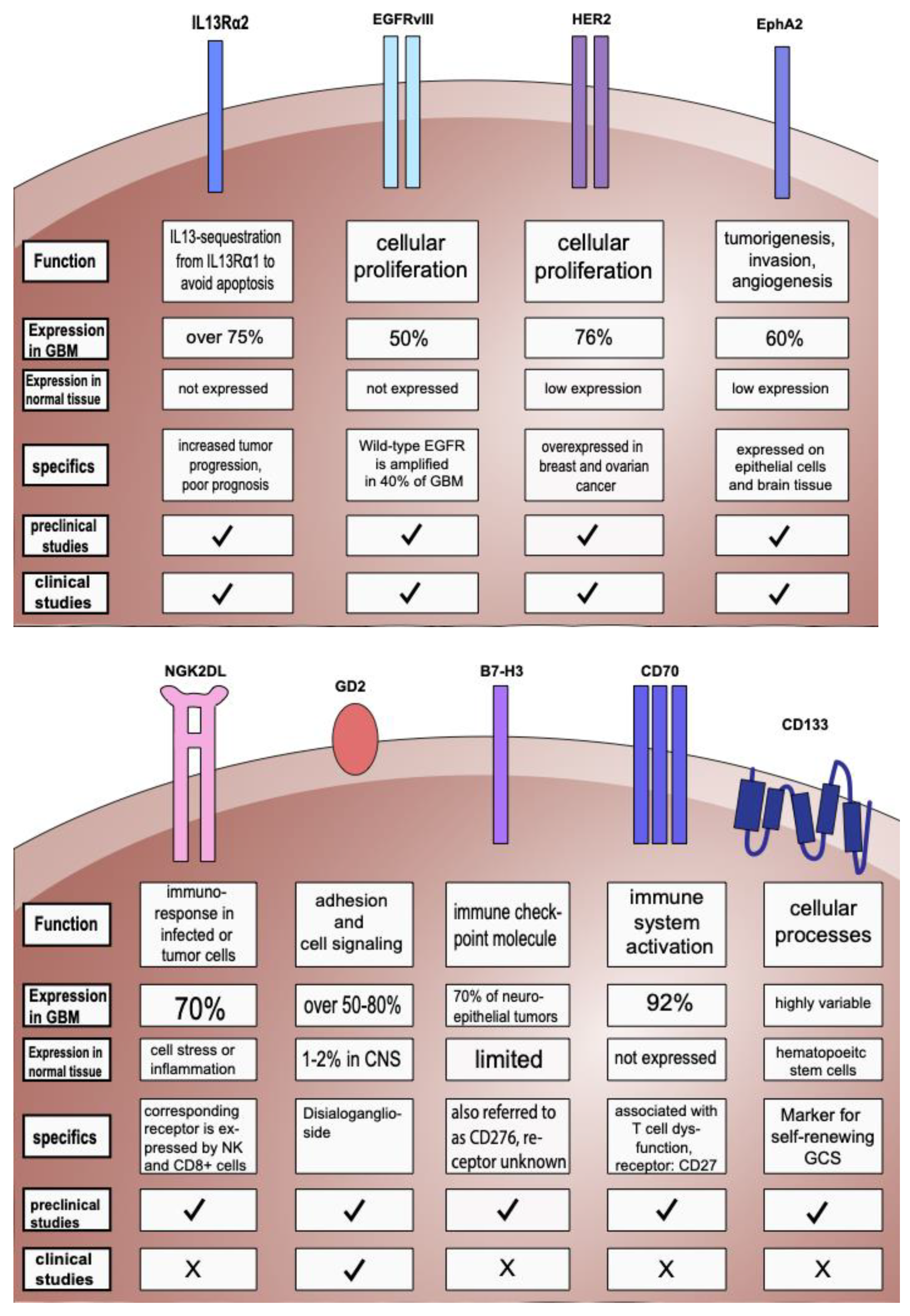
2.1. Targeting IL-13Rα2
2.2. Targeting EGFR and EGFRvIII
2.3. Targeting HER2 and Combinatorial Targets
2.4. Targeting EphA2
2.5. Targeting NKG2D Ligands (NKG2DLs)
2.6. Targeting GD2
2.7. Other Targets
3. Clinical Studies
3.1. Targeting IL-13Rα2
3.2. Targeting EGFRvIII
3.3. Targeting HER2
3.4. Targeting EphA2
3.5. Targeting GD2
4. Challenges
4.1. GBM-Related Challenges
4.1.1. Neuroanatomical Barriers
4.1.2. Tumor Microenvironment and Immunosuppression
4.1.3. Heterogeneity and Antigen Loss
4.2. CAR-T Cell Toxicity
5. Outlook
5.1. Breaking down Neuroanatomical Barriers
5.2. Adjusting the TME
5.3. Conquer Heterogeneity and Antigen Loss
5.4. Targeting Cancer Stem Cells
5.5. Management of Toxicity
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ou, A.; Alfred Yung, W.K.; Majd, N. Molecular Mechanisms of Treatment Resistance in Glioblastoma. Int. J. Mol. Sci. 2021, 22, 351. [Google Scholar] [CrossRef]
- Choi, B.D.; Maus, M.V.; June, C.H.; Sampson, J.H. Immunotherapy for Glioblastoma: Adoptive T-Cell Strategies. Clin. Cancer Res. 2019, 25, 2042–2048. [Google Scholar] [CrossRef] [PubMed]
- Berger, M.S.; Berger, G. Handbook of Clinical Neurology; Berger, M.S., Weller, E.S., Eds.; Elsevier: Amsterdam, The Netherlands, 2016; Volume 134, pp. 2–446. [Google Scholar]
- Alexander, B.M.; Cloughesy, T.F. Adult glioblastoma. J. Clin. Oncol. 2017, 35, 2402–2409. [Google Scholar] [CrossRef] [PubMed]
- Dymova, M.A.; Kuligina, E.V.; Richter, V.A. Molecular Mechanisms of Drug Resistance in Glioblastoma. Int. J. Mol. Sci. 2021, 22, 6385. [Google Scholar] [CrossRef] [PubMed]
- Bianco, J.; Bastiancich, C.; Jankovski, A.; des Rieux, A.; Préat, V.; Danhier, F. On Glioblastoma and the Search for a Cure: Where Do We Stand? Cell. Mol. Life Sci. 2017, 74, 2451–2466. [Google Scholar] [CrossRef]
- Majd, N.; Dasgupta, P.; de Groot, J. Immunotherapy for Neuro-Oncology. Adv. Exp. Med. Biol. 2020, 1244, 183–203. [Google Scholar]
- Desai, K.; Hubben, A.; Ahluwalia, M. The Role of Checkpoint Inhibitors in Glioblastoma. Target Oncol. 2019, 14, 375–394. [Google Scholar] [CrossRef]
- Lan, X.; Jörg, D.J.; Cavalli, F.M.G.; Richards, L.M.; Nguyen, L.V.; Vanner, R.J.; Guilhamon, P.; Lee, L.; Kushida, M.M.; Pellacani, D.; et al. Fate Mapping of Human Glioblastoma Reveals an Invariant Stem Cell Hierarchy. Nature 2017, 549, 227–232. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-Cell RNA-Seq Highlights Intratumoral Heterogeneity in Primary Glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [Green Version]
- Tirosh, I.; Suvà, M.L. Dissecting Human Gliomas by Single-Cell RNA Sequencing. Neuro Oncol. 2018, 20, 37–43. [Google Scholar] [CrossRef]
- Chaligne, R.; Gaiti, F.; Silverbush, D.; Schiffman, J.S.; Weisman, H.R.; Kluegel, L.; Gritsch, S.; Deochand, S.D.; Gonzalez Castro, L.N.; Richman, A.R.; et al. Epigenetic Encoding, Heritability and Plasticity of Glioma Transcriptional Cell States. Nat. Genet. 2021, 53, 1469–1479. [Google Scholar] [CrossRef]
- Fedele, M.; Cerchia, L.; Pegoraro, S.; Sgarra, R.; Manfioletti, G. Proneural-Mesenchymal Transition: Phenotypic Plasticity to Acquire Multitherapy Resistance in Glioblastoma. Int. J. Mol. Sci. 2019, 20, 2746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeo, E.C.F.; Brown, M.P.; Gargett, T.; Ebert, L.M. The Role of Cytokines and Chemokines in Shaping the Immune Microenvironment of Glioblastoma: Implications for Immunotherapy. Cells 2021, 10, 607. [Google Scholar] [CrossRef]
- Jansen, J.A.; Omuro, A.; Lucca, L.E. T Cell Dysfunction in Glioblastoma: A Barrier and an Opportunity for the Development of Successful Immunotherapies. Curr. Opin. Neurol. 2021, 34, 827–833. [Google Scholar] [CrossRef] [PubMed]
- Labanieh, L.; Majzner, R.G.; Mackall, C.L. Programming CAR-T Cells to Kill Cancer. Nat. Biomed. Eng. 2018, 2, 377–391. [Google Scholar] [CrossRef] [PubMed]
- Oved, J.H.; Barrett, D.M.; Teachey, D.T. Cellular Therapy: Immune-Related Complications. Immunol. Rev. 2019, 290, 114–126. [Google Scholar] [CrossRef]
- Mohanty, R.; Chowdhury, C.R.; Arega, S.; Sen, P.; Ganguly, P.; Ganguly, N. CAR T Cell Therapy: A New Era for Cancer Treatment (Review). Oncol. Rep. 2019, 42, 2183–2195. [Google Scholar] [CrossRef]
- Cruz-Ramos, M.; García-Foncillas, J. CAR-T Cell and Personalized Medicine. Adv. Exp. Med. Biol. 2019, 1168, 131–145. [Google Scholar]
- Nair, R.; Westin, J. CAR T-Cells. Adv. Exp. Med. Biol. 2020, 1244, 215–233. [Google Scholar]
- Benmebarek, M.R.; Karches, C.H.; Cadilha, B.L.; Lesch, S.; Endres, S.; Kobold, S. Killing Mechanisms of Chimeric Antigen Receptor (CAR) T Cells. Int. J. Mol. Sci. 2019, 20, 1283. [Google Scholar] [CrossRef] [Green Version]
- Sahebjam, S.; Sharabi, A.; Lim, M.; Kesarwani, P.; Chinnaiyan, P. Immunotherapy and Radiation in Glioblastoma. J. Neurooncol. 2017, 134, 531–539. [Google Scholar] [CrossRef]
- Sterner, R.C.; Sterner, R.M. CAR-T Cell Therapy: Current Limitations and Potential Strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef] [PubMed]
- Prinzing, B.L.; Gottschalk, S.M.; Krenciute, G. CAR T-Cell Therapy for Glioblastoma: Ready for the next Round of Clinical Testing? Expert Rev. Anticancer 2018, 18, 451–461. [Google Scholar] [CrossRef]
- Rafiq, S.; Hackett, C.S.; Brentjens, R.J. Engineering Strategies to Overcome the Current Roadblocks in CAR T Cell Therapy. Nat. Rev. Clin. Oncol. 2020, 17, 147–167. [Google Scholar] [CrossRef]
- Schaft, N. The Landscape of Car-t Cell Clinical Trials against Solid Tumors—A Comprehensive Overview. Cancers 2020, 12, 2567. [Google Scholar] [CrossRef] [PubMed]
- Land, C.A.; Musich, P.R.; Haydar, D.; Krenciute, G.; Xie, Q. Chimeric Antigen Receptor T-Cell Therapy in Glioblastoma: Charging the T Cells to Fight. J. Transl. Med. 2020, 18, 428. [Google Scholar] [CrossRef]
- Brown, C.E.; Aguilar, B.; Starr, R.; Yang, X.; Chang, W.C.; Weng, L.; Chang, B.; Sarkissian, A.; Brito, A.; Sanchez, J.F.; et al. Optimization of IL13Rα2-Targeted Chimeric Antigen Receptor T Cells for Improved Anti-Tumor Efficacy against Glioblastoma. Mol. Ther. 2018, 26, 31–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Aguilar, B.; Starr, R.; Alizadeh, D.; Brito, A.; Sarkissian, A.; Ostberg, J.R.; Forman, S.J.; Brown, C.E. Glioblastoma-Targeted CD4+ CAR T Cells Mediate Superior Antitumor Activity. JCI Insight 2018, 3, e99048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegde, M.; Mukherjee, M.; Grada, Z.; Pignata, A.; Landi, D.; Navai, S.A.; Wakefield, A.; Fousek, K.; Bielamowicz, K.; Chow, K.K.H.; et al. Tandem CAR T Cells Targeting HER2 and IL13Rα2 Mitigate Tumor Antigen Escape. J. Clin. Investig. 2016, 126, 3036–3052. [Google Scholar] [CrossRef] [Green Version]
- Bielamowicz, K.; Fousek, K.; Byrd, T.T.; Samaha, H.; Mukherjee, M.; Aware, N.; Wu, M.F.; Orange, J.S.; Sumazin, P.; Man, T.K.; et al. Trivalent CAR T Cells Overcome Interpatient Antigenic Variability in Glioblastoma. Neuro Oncol. 2018, 20, 506–518. [Google Scholar] [CrossRef] [Green Version]
- Krenciute, G.; Prinzing, B.L.; Yi, Z.; Wu, M.-F.; Liu, H.; Dotti, G.; Balyasnikova, I.V.; Gottschalk, S. Transgenic Expression of IL15 Improves Antiglioma Activity of IL13Rα2-CAR T Cells but Results in Antigen Loss Variants. Cancer Immunol. Res. 2017, 5, 571–581. [Google Scholar] [CrossRef] [Green Version]
- Sampson, J.H.; Choi, B.D.; Sanchez-Perez, L.; Suryadevara, C.M.; Snyder, D.J.; Flores, C.T.; Schmittling, R.J.; Nair, S.K.; Reap, E.A.; Norberg, P.K.; et al. EGFRvIII MCAR-Modified T-Cell Therapy Cures Mice with Established Intracerebral Glioma and Generates Host Immunity against Tumor-Antigen Loss. Clin. Cancer Res. 2014, 20, 972–984. [Google Scholar] [CrossRef] [Green Version]
- Choe, J.H.; Watchmaker, P.B.; Simic, M.S.; Gilbert, R.D.; Li, A.W.; Krasnow, N.A.; Downey, K.M.; Yu, W.; Carrera, D.A.; Celli, A.; et al. SynNotch-CAR T Cells Overcome Challenges of Specificity, Heterogeneity, and Persistence in Treating Glioblastoma. Sci. Transl. Med. 2021, 13, eabe7378. [Google Scholar] [CrossRef]
- Jiang, H.; Gao, H.; Kong, J.; Song, B.; Wang, P.; Shi, B.; Wang, H.; Li, Z. Selective Targeting of Glioblastoma with EGFRvIII/EGFR Bitargeted Chimeric Antigen Receptor T Cell. Cancer Immunol. Res. 2018, 6, 1314–1326. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, E.R.; D’Souza, R.R.; Klampatsa, A. Armored CAR T-Cells: The Next Chapter in T-Cell Cancer Immunotherapy. Biologics 2021, 15, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.D.; Yu, X.; Castano, A.P.; Bouffard, A.A.; Schmidts, A.; Larson, R.C.; Bailey, S.R.; Boroughs, A.C.; Frigault, M.J.; Leick, M.B.; et al. CAR-T Cells Secreting BiTEs Circumvent Antigen Escape without Detectable Toxicity. Nat. Biotechnol. 2019, 37, 1049–1058. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Yin, J. Prospective Approaches to Enhancing CAR T Cell Therapy for Glioblastoma. Front. Immunol. 2022, 13, 1008751. [Google Scholar] [CrossRef]
- Agliardi, G.; Liuzzi, A.R.; Hotblack, A.; De Feo, D.; Núñez, N.; Stowe, C.L.; Friebel, E.; Nannini, F.; Rindlisbacher, L.; Roberts, T.A.; et al. Intratumoral IL-12 Delivery Empowers CAR-T Cell Immunotherapy in a Pre-Clinical Model of Glioblastoma. Nat. Commun. 2021, 12, 444. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Ren, J.; Amoozgar, Z.; Lee, S.; Datta, M.; Roberge, S.; Duquette, M.; Fukumura, D.; Jain, R.K. Anti-VEGF Therapy Improves EGFR-VIII-CAR-T Cell Delivery and Efficacy in Syngeneic Glioblastoma Models in Mice. J. Immunother. Cancer 2023, 11, e005583. [Google Scholar] [CrossRef]
- Muhammad, N.; Wang, R.; Li, W.; Zhang, Z.; Chang, Y.; Hu, Y.; Zhao, J.; Zheng, X.; Mao, Q.; Xia, H. A Novel TanCAR Targeting IL13Rα2 and EphA2 for Enhanced Glioblastoma Therapy. Mol. Oncolytics 2022, 24, 729–741. [Google Scholar] [CrossRef] [PubMed]
- An, Z.; Hu, Y.; Bai, Y.; Zhang, C.; Xu, C.; Kang, X.; Yang, S.; Li, W.; Zhong, X. Antitumor Activity of the Third Generation EphA2 CAR-T Cells against Glioblastoma Is Associated with Interferon Gamma Induced PD-L1. Oncoimmunology 2021, 10, 1960728. [Google Scholar] [CrossRef]
- Larson, R.C.; Kann, M.C.; Bailey, S.R.; Haradhvala, N.J.; Llopis, P.M.; Bouffard, A.A.; Scarfó, I.; Leick, M.B.; Grauwet, K.; Berger, T.R.; et al. CAR T Cell Killing Requires the IFNγR Pathway in Solid but Not Liquid Tumours. Nature 2022, 604, 563–570. [Google Scholar] [CrossRef]
- Zingoni, A.; Molfetta, R.; Fionda, C.; Soriani, A.; Paolini, R.; Cippitelli, M.; Cerboni, C.; Santoni, A. NKG2D and Its Ligands: “One for All, All for One”. Front. Immunol. 2018, 9, 746. [Google Scholar] [CrossRef]
- Yang, D.; Sun, B.; Dai, H.; Li, W.; Shi, L.; Zhang, P.; Li, S.; Zhao, X. T Cells Expressing NKG2D Chimeric Antigen Receptors Efficiently Eliminate Glioblastoma and Cancer Stem Cells. J. Immunother. Cancer 2019, 7, 93. [Google Scholar] [CrossRef] [Green Version]
- Weiss, T.; Weller, M.; Guckenberger, M.; Sentman, C.L.; Roth, P. NKG2D-Based CAR T Cells and Radiotherapy Exert Synergistic Efficacy in Glioblastoma. Cancer Res. 2018, 78, 1031–1043. [Google Scholar] [CrossRef] [Green Version]
- Prapa, M.; Chiavelli, C.; Golinelli, G.; Grisendi, G.; Bestagno, M.; Di Tinco, R.; Dall’Ora, M.; Neri, G.; Candini, O.; Spano, C.; et al. GD2 CAR T Cells against Human Glioblastoma. NPJ Precis Oncol. 2021, 5, 93. [Google Scholar] [CrossRef]
- Gargett, T.; Ebert, L.M.; Truong, N.T.H.; Kollis, P.M.; Sedivakova, K.; Yu, W.; Yeo, E.C.F.; Wittwer, N.L.; Gliddon, B.L.; Tea, M.N.; et al. GD2-Targeting CAR-T Cells Enhanced by Transgenic IL-15 Expression Are an Effective and Clinically Feasible Therapy for Glioblastoma. J. Immunother. Cancer 2022, 10, 9. [Google Scholar] [CrossRef] [PubMed]
- De Billy, E.; Pellegrino, M.; Orlando, D.; Pericoli, G.; Ferretti, R.; Businaro, P.; Ajmone-Cat, M.A.; Rossi, S.; Petrilli, L.L.; Maestro, N.; et al. Dual IGF1R/IR Inhibitors in Combination with GD2-CAR T-Cells Display a Potent Anti-Tumor Activity in Diffuse Midline Glioma H3K27M-Mutant. Neuro Oncol. 2022, 24, 1150–1163. [Google Scholar] [CrossRef]
- Tang, X.; Zhao, S.; Zhang, Y.; Wang, Y.; Zhang, Z.; Yang, M.; Zhu, Y.; Zhang, G.; Guo, G.; Tong, A.; et al. B7-H3 as a Novel CAR-T Therapeutic Target for Glioblastoma. Mol. Oncolytics 2019, 14, 279–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; Zhang, Z.; Zhong, K.; Wang, Z.; Yang, N.; Tang, X.; Li, H.; Lu, Q.; Wu, Z.; Yuan, B.; et al. CXCL11-Armed Oncolytic Adenoviruses Enhance CAR-T Cell Therapeutic Efficacy and Reprogram Tumor Microenvironment in Glioblastoma. Mol. Ther. 2023, 31, 134–153. [Google Scholar] [CrossRef] [PubMed]
- Sauer, T.; Parikh, K.; Sharma, S.; Omer, B.; Sedloev, D.; Chen, Q.; Angenendt, L.; Schliemann, C.; Schmitt, M.; Müller-Tidow, C.; et al. CD70-Specific CAR T Cells Have Potent Activity against Acute Myeloid Leukemia without HSC Toxicity. Blood 2021, 138, 318–330. [Google Scholar] [CrossRef]
- Jin, L.; Ge, H.; Long, Y.; Yang, C.; Chang, Y.E.; Mu, L.; Sayour, E.J.; De Leon, G.; Wang, Q.J.; Yang, J.C.; et al. CD70, a Novel Target of CAR T-Cell Therapy for Gliomas. Neuro Oncol. 2018, 20, 55–65. [Google Scholar] [CrossRef] [Green Version]
- Seyfrid, M.; Maich, W.T.; Shaikh, V.M.; Tatari, N.; Upreti, D.; Piyasena, D.; Subapanditha, M.; Savage, N.; McKenna, D.; Mikolajewicz, N.; et al. CD70 as an Actionable Immunotherapeutic Target in Recurrent Glioblastoma and Its Microenvironment. J. Immunother. Cancer 2022, 10, e003289. [Google Scholar] [CrossRef]
- Zhu, G.; Zhang, J.; Zhang, Q.; Jin, G.; Su, X.; Liu, S.; Liu, F. Enhancement of CD70-Specific CAR T Treatment by IFN-γ Released from OHSV-1-Infected Glioblastoma. Cancer Immunol. Immunother. 2022, 71, 2433–2448. [Google Scholar] [CrossRef] [PubMed]
- Vora, P.; Venugopal, C.; Salim, S.K.; Tatari, N.; Bakhshinyan, D.; Singh, M.; Seyfrid, M.; Upreti, D.; Rentas, S.; Wong, N.; et al. The Rational Development of CD133-Targeting Immunotherapies for Glioblastoma. Cell Stem Cell 2020, 26, 832–844.e6. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.E.; Badie, B.; Barish, M.E.; Weng, L.; Ostberg, J.R.; Chang, W.C.; Naranjo, A.; Starr, R.; Wagner, J.; Wright, C.; et al. Bioactivity and Safety of IL13Rα2-Redirected Chimeric Antigen Receptor CD8+ T Cells in Patients with Recurrent Glioblastoma. Clin. Cancer Res. 2015, 21, 4062–4072. [Google Scholar] [CrossRef] [Green Version]
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N. Engl. J. Med. 2016, 375, 2561–2569. [Google Scholar] [CrossRef]
- Brown, C.E.; Rodriguez, A.; Palmer, J.; Ostberg, J.R.; Naranjo, A.; Wagner, J.R.; Aguilar, B.; Starr, R.; Weng, L.; Synold, T.W.; et al. Off-the-Shelf, Steroid-Resistant, IL13Rα2-Specific CAR T Cells for Treatment of Glioblastoma. Neuro Oncol. 2022, 24, 1318–1330. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A Single Dose of Peripherally Infused EGFRvIII-Directed CAR T Cells Mediates Antigen Loss and Induces Adaptive Resistance in Patients with Recurrent Glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goff, S.L.; Morgan, R.A.; Yang, J.C.; Sherry, R.M.; Robbins, P.F.; Restifo, N.P.; Feldman, S.A.; Lu, Y.C.; Lu, L.; Zheng, Z.; et al. Pilot Trial of Adoptive Transfer of Chimeric Antigen Receptor-Transduced t Cells Targeting Egfrviii in Patients with Glioblastoma. J. Immunother. 2019, 42, 126–135. [Google Scholar] [CrossRef]
- Ahmed, N.; Brawley, V.; Hegde, M.; Bielamowicz, K.; Wakefield, A.; Ghazi, A.; Ashoori, A.; Diouf, O.; Gerken, C.; Landi, D.; et al. Autologous HER2 CMV Bispecific CAR T Cells Are Safe and Demonstrate Clinical Benefit for Glioblastoma in a Phase I Trial. J. Immunother. Cancer 2015, 3, O11. [Google Scholar] [CrossRef] [Green Version]
- Lin, Q.; Ba, T.; Ho, J.; Chen, D.; Cheng, Y.; Wang, L.; Xu, G.; Xu, L.; Zhou, Y.; Wei, Y.; et al. First-in-Human Trial of EphA2-Redirected CAR T-Cells in Patients With Recurrent Glioblastoma: A Preliminary Report of Three Cases at the Starting Dose. Front. Oncol. 2021, 11, 694941. [Google Scholar] [CrossRef]
- Majzner, R.G.; Ramakrishna, S.; Yeom, K.W.; Patel, S.; Chinnasamy, H.; Schultz, L.M.; Richards, R.M.; Jiang, L.; Barsan, V.; Mancusi, R.; et al. GD2-CAR T Cell Therapy for H3K27M-Mutated Diffuse Midline Gliomas. Nature 2022, 603, 934–941. [Google Scholar] [CrossRef]
- Liu, Z.; Zhou, J.; Yang, X.; Liu, Y.; Zou, C.; Lv, W.; Chen, C.; Cheng, K.K.; Chen, T.; Chang, L.J.; et al. Safety and Antitumor Activity of GD2-Specific 4SCAR-T Cells in Patients with Glioblastoma. Mol. Cancer 2023, 22, 3. [Google Scholar] [CrossRef] [PubMed]
- Adhikaree, J.; Moreno-Vicente, J.; Kaur, A.P.; Jackson, A.M.; Patel, P.M. Resistance Mechanisms and Barriers to Successful Immunotherapy for Treating Glioblastoma. Cells 2020, 9, 263. [Google Scholar] [CrossRef] [Green Version]
- Chuang, D.F.; Lin, X. Targeted Therapies for the Treatment of Glioblastoma in Adults. Curr. Oncol. Rep. 2019, 21, 61. [Google Scholar] [CrossRef]
- Haddad, A.F.; Young, J.S.; Amara, D.; Berger, M.S.; Raleigh, D.R.; Aghi, M.K.; Butowski, N.A. Mouse Models of Glioblastoma for the Evaluation of Novel Therapeutic Strategies. Neurooncol. Adv. 2021, 3, vdab100. [Google Scholar] [CrossRef] [PubMed]
- Huszthy, P.C.; Daphu, I.; Niclou, S.P.; Stieber, D.; Nigro, J.M.; Sakariassen, P.O.; Miletic, H.; Thorsen, F.; Bjerkvig, R. In Vivo Models of Primary Brain Tumors: Pitfalls and Perspectives. Neuro Oncol. 2012, 14, 979–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbott, N.J.; Patabendige, A.A.K.; Dolman, D.E.M.; Yusof, S.R.; Begley, D.J. Structure and Function of the Blood–Brain Barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef]
- Chuntova, P.; Downey, K.M.; Hegde, B.; Almeida, N.D.; Okada, H. Genetically Engineered T-Cells for Malignant Glioma: Overcoming the Barriers to Effective Immunotherapy. Front. Immunol. 2019, 10, 3062. [Google Scholar] [CrossRef] [Green Version]
- Davies, D.C. Blood-Brain Barrier Breakdown in Septic Encephalopathy and Brain Tumours*. J Anat. 2002, 200, 639–646. [Google Scholar] [CrossRef]
- Donnadieu, E.; Dupré, L.; Pinho, L.G.; Cotta-de-Almeida, V. Surmounting the Obstacles That Impede Effective CAR T Cell Trafficking to Solid Tumors. J. Leukoc. Biol. 2020, 108, 1067–1079. [Google Scholar] [CrossRef] [PubMed]
- Marx, S.; Godicelj, A.; Wucherpfennig, K.W. A Conceptual Framework for Inducing T Cell-Mediated Immunity Against Glioblastoma. Semin. Immunopathol. 2022, 44, 697–707. [Google Scholar] [CrossRef] [PubMed]
- Bagley, S.J.; Desai, A.S.; Linette, G.P.; June, C.H.; O’Rourke, D.M. CAR T-Cell Therapy for Glioblastoma: Recent Clinical Advances and Future Challenges. Neuro Oncol. 2018, 20, 1429–1438. [Google Scholar] [CrossRef] [Green Version]
- Mormino, A.; Garofalo, S. Dialogue among Lymphocytes and Microglia in Glioblastoma Microenvironment. Cancers 2022, 14, 2632. [Google Scholar] [CrossRef]
- Kang, X.; Zheng, Y.; Hong, W.; Chen, X.; Li, H.; Huang, B.; Huang, Z.; Tang, H.; Geng, W. Recent Advances in Immune Cell Therapy for Glioblastoma. Front. Immunol. 2020, 11, 544563. [Google Scholar] [CrossRef] [PubMed]
- Oelkrug, C.; Ramage, J.M. Enhancement of T Cell Recruitment and Infiltration into Tumours. Clin. Exp. Immunol. 2014, 178, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Xue, S.; Hu, M.; Iyer, V.; Yu, J. Blocking the PD-1/PD-L1 Pathway in Glioma: A Potential New Treatment Strategy. J. Hematol. Oncol. 2017, 10, 81. [Google Scholar] [CrossRef] [Green Version]
- Zhu, G.; Zhang, Q.; Zhang, J.; Liu, F. Targeting Tumor-Associated Antigen: A Promising CAR-T Therapeutic Strategy for Glioblastoma Treatment. Front. Pharm. 2021, 12, 661606. [Google Scholar] [CrossRef]
- Lemoine, J.; Ruella, M.; Houot, R. Born to Survive: How Cancer Cells Resist CAR T Cell Therapy. J. Hematol. Oncol. 2021, 14, 199. [Google Scholar] [CrossRef] [PubMed]
- Salinas, R.D.; Durgin, J.S.; O’Rourke, D.M. Potential of Glioblastoma-Targeted Chimeric Antigen Receptor (CAR) T-Cell Therapy. CNS Drugs 2020, 34, 127–145. [Google Scholar] [CrossRef]
- Simula, L.; Ollivier, E.; Icard, P.; Donnadieu, E. Immune Checkpoint Proteins, Metabolism and Adhesion Molecules: Overlooked Determinants of CAR T-Cell Migration? Cells 2022, 11, 1854. [Google Scholar] [CrossRef] [PubMed]
- Karachi, A.; Dastmalchi, F.; Nazarian, S.; Huang, J.; Sayour, E.J.; Jin, L.; Yang, C.; Mitchell, D.A.; Rahman, M. Optimizing T Cell-Based Therapy for Glioblastoma. Front. Immunol. 2021, 12, 705580. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 Human Cancer Genomes Reveals the Landscape of Tumor Mutational Burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef] [Green Version]
- Müller, S.; Kohanbash, G.; Liu, S.J.; Alvarado, B.; Carrera, D.; Bhaduri, A.; Watchmaker, P.B.; Yagnik, G.; Di Lullo, E.; Malatesta, M.; et al. Single-Cell Profiling of Human Gliomas Reveals Macrophage Ontogeny as a Basis for Regional Differences in Macrophage Activation in the Tumor Microenvironment. Genome Biol. 2017, 18, 234. [Google Scholar] [CrossRef] [Green Version]
- Qazi, M.A.; Vora, P.; Venugopal, C.; Sidhu, S.S.; Moffat, J.; Swanton, C.; Singh, S.K. Intratumoral Heterogeneity: Pathways to Treatment Resistance and Relapse in Human Glioblastoma. Ann. Oncol. 2017, 28, 1448–1456. [Google Scholar] [CrossRef]
- Akhoundi, M.; Mohammadi, M.; Sahraei, S.S.; Sheykhhasan, M.; Fayazi, N. CAR T Cell Therapy as a Promising Approach in Cancer Immunotherapy: Challenges and Opportunities. Cell. Oncol. 2021, 44, 495–523. [Google Scholar] [CrossRef]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [Green Version]
- Rubin, D.B.; Danish, H.H.; Ali, A.B.; Li, K.; Larose, S.; Monk, A.D.; Cote, D.J.; Spendley, L.; Kim, A.H.; Robertson, M.S.; et al. Neurological Toxicities Associated with Chimeric Antigen Receptor T-Cell Therapy. Brain 2019, 142, 1334–1348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majzner, R.G.; Mackall, C.L. Clinical Lessons Learned from the First Leg of the CAR T Cell Journey. Nat. Med. 2019, 25, 1341–1355. [Google Scholar] [CrossRef]
- Liu, L.; Qu, Y.; Cheng, L.; Yoon, C.W.; He, P.; Monther, A.; Guo, T.; Chittle, S.; Wang, Y. Engineering Chimeric Antigen Receptor T Cells for Solid Tumour Therapy. Clin. Transl. Med. 2022, 12, e1141. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Tao, H.; Karachi, A.; Long, Y.; Hou, A.Y.; Na, M.; Dyson, K.A.; Grippin, A.J.; Deleyrolle, L.P.; Zhang, W.; et al. CXCR1- or CXCR2-Modified CAR T Cells Co-Opt IL-8 for Maximal Antitumor Efficacy in Solid Tumors. Nat. Commun. 2019, 10, 4016. [Google Scholar] [CrossRef] [Green Version]
- Qin, Y.; Xu, G. Enhancing CAR T-Cell Therapies against Solid Tumors: Mechanisms and Reversion of Resistance. Front. Immunol. 2022, 13, 1053120. [Google Scholar] [CrossRef]
- Sridhar, P.; Petrocca, F. Regional Delivery of Chimeric Antigen Receptor (CAR) T-Cells for Cancer Therapy. Cancers 2017, 9, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitanza, N.A.; Johnson, A.J.; Wilson, A.L.; Brown, C.; Yokoyama, J.K.; Künkele, A.; Chang, C.A.; Rawlings-Rhea, S.; Huang, W.; Seidel, K.; et al. Locoregional Infusion of HER2-Specific CAR T Cells in Children and Young Adults with Recurrent or Refractory CNS Tumors: An Interim Analysis. Nat. Med. 2021, 27, 1544–1552. [Google Scholar] [CrossRef]
- Antonucci, L.; Canciani, G.; Mastronuzzi, A.; Carai, A.; Del Baldo, G.; Del Bufalo, F. CAR-T Therapy for Pediatric High-Grade Gliomas: Peculiarities, Current Investigations and Future Strategies. Front. Immunol. 2022, 13, 2066. [Google Scholar] [CrossRef]
- Vitanza, N.A.; Ronsley, R.; Choe, M.; Henson, C.; Breedt, M.; Barrios-Anderson, A.; Wein, A.; Brown, C.; Beebe, A.; Kong, A.; et al. Locoregional CAR T Cells for Children with CNS Tumors: Clinical Procedure and Catheter Safety. Neoplasia 2023, 36, 100870. [Google Scholar] [CrossRef] [PubMed]
- Razeghian, E.; Nasution, M.K.M.; Rahman, H.S.; Gardanova, Z.R.; Abdelbasset, W.K.; Aravindhan, S.; Bokov, D.O.; Suksatan, W.; Nakhaei, P.; Shariatzadeh, S.; et al. A Deep Insight into CRISPR/Cas9 Application in CAR-T Cell-Based Tumor Immunotherapies. Stem Cell Res. 2021, 12, 428. [Google Scholar] [CrossRef] [PubMed]
- Jafarzadeh, L.; Masoumi, E.; Fallah-Mehrjardi, K.; Mirzaei, H.R.; Hadjati, J. Prolonged Persistence of Chimeric Antigen Receptor (CAR) T Cell in Adoptive Cancer Immunotherapy: Challenges and Ways Forward. Front. Immunol. 2020, 11, 702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietrobon, V.; Todd, L.A.; Goswami, A.; Stefanson, O.; Yang, Z.; Marincola, F. Improving CAR T-Cell Persistence. Int. J. Mol. Sci. 2021, 22, 10828. [Google Scholar] [CrossRef] [PubMed]
- John, L.B.; Devaud, C.; Duong, C.P.M.; Yong, C.S.; Beavis, P.A.; Haynes, N.M.; Chow, M.T.; Smyth, M.J.; Kershaw, M.H.; Darcy, P.K. Anti-PD-1 Antibody Therapy Potently Enhances the Eradication of Established Tumors By Gene-Modified T Cells. Clin. Cancer Res. 2013, 19, 5636–5646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, Y.; Boesteanu, A.C.; Binder, Z.A.; Xu, C.; Reid, R.A.; Rodriguez, J.L.; Cook, D.R.; Thokala, R.; Blouch, K.; McGettigan-Croce, B.; et al. Checkpoint Blockade Reverses Anergy in IL-13Rα2 Humanized ScFv-Based CAR T Cells to Treat Murine and Canine Gliomas. Mol. Oncolytics 2018, 11, 20–38. [Google Scholar] [CrossRef] [Green Version]
- Shen, S.; Chen, L.; Liu, J.; Yang, L.; Zhang, M.; Wang, L.; Zhang, R.; Uemura, Y.; Wu, Q.; Yu, X.; et al. Current State and Future of Co-Inhibitory Immune Checkpoints for the Treatment of Glioblastoma. Cancer Biol. Med. 2020, 17, 555–568. [Google Scholar] [CrossRef]
- Rafiq, S.; Yeku, O.O.; Jackson, H.J.; Purdon, T.J.; van Leeuwen, D.G.; Drakes, D.J.; Song, M.; Miele, M.M.; Li, Z.; Wang, P.; et al. Targeted Delivery of a PD-1-Blocking ScFv by CAR-T Cells Enhances Anti-Tumor Efficacy in Vivo. Nat. Biotechnol. 2018, 36, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Rupp, L.J.; Schumann, K.; Roybal, K.T.; Gate, R.E.; Ye, C.J.; Lim, W.A.; Marson, A. CRISPR/Cas9-Mediated PD-1 Disruption Enhances Anti-Tumor Efficacy of Human Chimeric Antigen Receptor T Cells. Sci. Rep. 2017, 7, 737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Wu, H.; Chen, G.; Wei, X.; Wang, C.; Zhou, S.; Huang, A.; Zhang, Z.; Zhan, C.; Wu, Y.; et al. Arming Anti-EGFRvIII CAR-T With TGFβ Trap Improves Antitumor Efficacy in Glioma Mouse Models. Front. Oncol. 2020, 10, 1117. [Google Scholar] [CrossRef]
- Choi, B.D.; Yu, X.; Castano, A.P.; Darr, H.; Henderson, D.B.; Bouffard, A.A.; Larson, R.C.; Scarfò, I.; Bailey, S.R.; Gerhard, G.M.; et al. CRISPR-Cas9 Disruption of PD-1 Enhances Activity of Universal EGFRvIII CAR T Cells in a Preclinical Model of Human Glioblastoma. J. Immunother. Cancer 2019, 7, 304. [Google Scholar] [CrossRef]
- Liu, X.; Ranganathan, R.; Jiang, S.; Fang, C.; Sun, J.; Kim, S.; Newick, K.; Lo, A.; June, C.H.; Zhao, Y.; et al. A Chimeric Switch-Receptor Targeting PD1 Augments the Efficacy of Second-Generation CAR T Cells in Advanced Solid Tumors. Cancer Res. 2016, 76, 1578–1590. [Google Scholar] [CrossRef] [Green Version]
- Tahmasebi, S.; Elahi, R.; Khosh, E.; Esmaeilzadeh, A. Programmable and Multi-Targeted CARs: A New Breakthrough in Cancer CAR-T Cell Therapy. Clin. Transl. Oncol. 2021, 23, 1003–1019. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, A.R.; Owens, M.N.; Geyer, C.R. Modular Chimeric Antigen Receptor Systems for Universal CAR T Cell Retargeting. Int. J. Mol. Sci. 2020, 21, 7222. [Google Scholar] [CrossRef]
- Gisina, A.; Kholodenko, I.; Kim, Y.; Abakumov, M.; Lupatov, A.; Yarygin, K. Glioma Stem Cells: Novel Data Obtained by Single-Cell Sequencing. Int. J. Mol. Sci. 2022, 23, 14224. [Google Scholar] [CrossRef] [PubMed]
- Maggs, L.; Cattaneo, G.; Dal, A.E.; Moghaddam, A.S.; Ferrone, S. CAR T Cell-Based Immunotherapy for the Treatment of Glioblastoma. Front. Neurosci. 2021, 15, 662064. [Google Scholar] [CrossRef] [PubMed]
- Qin, V.M.; Haynes, N.M.; D’Souza, C.; Neeson, P.J.; Zhu, J.J. CAR-T Plus Radiotherapy: A Promising Combination for Immunosuppressive Tumors. Front. Immunol. 2022, 12, 813832. [Google Scholar] [CrossRef]
- Xu, J.; Wang, Y.; Shi, J.; Liu, J.; Li, Q.; Chen, L. Combination Therapy: A Feasibility Strategy for Car-t Cell Therapy in the Treatment of Solid Tumors (Review). Oncol. Lett. 2018, 16, 2063–2070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labanieh, L.; Majzner, R.G.; Klysz, D.; Sotillo, E.; Fisher, C.J.; Vilches-Moure, J.G.; Pacheco, K.Z.B.; Malipatlolla, M.; Xu, P.; Hui, J.H.; et al. Enhanced Safety and Efficacy of Protease-Regulated CAR-T Cell Receptors. Cell 2022, 185, 1745–1763.e22. [Google Scholar] [CrossRef] [PubMed]
- Debinski, W.; Obiri, N.I.; Powers, S.K.; Pastan, I.; Puri, R.K. Human Glioma Cells Overexpress Receptors for Interleukin 13 and Are Extremely Sensitive to a Novel Chimeric Protein Composed of Interleukin 13 and Pseudomonas Exotoxin. Clin. Cancer Res. 1995, 1, 1253–1258. [Google Scholar]
- Kawakami, M.; Kawakami, K.; Takahashi, S.; Abe, M.; Puri, R.K. Analysis of Interleukin-13 Receptorα2 Expression in Human Pediatric Brain Tumors. Cancer 2004, 101, 1036–1042. [Google Scholar] [CrossRef] [PubMed]
- Thaci, B.; Brown, C.E.; Binello, E.; Werbaneth, K.; Sampath, P.; Sengupta, S. Significance of Interleukin-13 Receptor Alpha 2-Targeted Glioblastoma Therapy. Neuro Oncol. 2014, 16, 1304–1312. [Google Scholar] [CrossRef] [Green Version]
- Ohgaki, H.; Kleihues, P. The Definition of Primary and Secondary Glioblastoma. Clin. Cancer Res. 2013, 19, 764–772. [Google Scholar] [CrossRef] [Green Version]
- Song, E.Z.; Wang, X.; Philipson, B.I.; Zhang, Q.; Thokala, R.; Zhang, L.; Assenmacher, C.A.; Binder, Z.A.; Ming, G.; O’Rourke, D.M.; et al. The IAP Antagonist Birinapant Enhances Chimeric Antigen Receptor T Cell Therapy for Glioblastoma by Overcoming Antigen Heterogeneity. Mol. Oncolytics 2022, 27, 288–304. [Google Scholar] [CrossRef]
- Zhu, H.; You, Y.; Shen, Z.; Shi, L. EGFRvIII-CAR-T Cells with PD-1 Knockout Have Improved Anti-Glioma Activity. Pathol. Oncol. Res. 2020, 26, 2135–2141. [Google Scholar] [CrossRef] [PubMed]
- Duan, S.; Guo, W.; Xu, Z.; He, Y.; Liang, C.; Mo, Y.; Wang, Y.; Xiong, F.; Guo, C.; Li, Y.; et al. Natural Killer Group 2D Receptor and Its Ligands in Cancer Immune Escape. Mol. Cancer 2019, 18, 29. [Google Scholar] [CrossRef]
- Huang, W.-C.; Easom, N.J.; Tang, X.-Z.; Gill, U.S.; Singh, H.; Robertson, F.; Chang, C.; Trowsdale, J.; Davidson, B.R.; Rosenberg, W.M.; et al. T Cells Infiltrating Diseased Liver Express Ligands for the NKG2D Stress Surveillance System. J. Immunol. 2017, 198, 1172–1182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strid, J.; Sobolev, O.; Zafirova, B.; Polic, B.; Hayday, A. The Intraepithelial T Cell Response to NKG2D-Ligands Links Lymphoid Stress Surveillance to Atopy. Science 2011, 334, 1293–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
| Current Challenges | Potential Solutions |
|---|---|
| Limitations of GBM mouse models [68,69] |
|
| Neuroanatomical barriers such as the BBB and insufficient trafficking of CAR-T cells in solid tumors [1,8,70,71,72,73,74] | |
| Immunosuppressive TME [7,8,14,22,36,66,75,76,77,78,79,80,81,82,83] | |
| Intra- and intertumoral heterogeneity and antigen loss [1,4,16,19,21,57,60,67,75,85,86,87], | |
| CAR-T cell toxicity [2,16,17,18,19,20,25,82,88,89,90,91] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kringel, R.; Lamszus, K.; Mohme, M. Chimeric Antigen Receptor T Cells in Glioblastoma—Current Concepts and Promising Future. Cells 2023, 12, 1770. https://doi.org/10.3390/cells12131770
Kringel R, Lamszus K, Mohme M. Chimeric Antigen Receptor T Cells in Glioblastoma—Current Concepts and Promising Future. Cells. 2023; 12(13):1770. https://doi.org/10.3390/cells12131770
Chicago/Turabian StyleKringel, Rebecca, Katrin Lamszus, and Malte Mohme. 2023. "Chimeric Antigen Receptor T Cells in Glioblastoma—Current Concepts and Promising Future" Cells 12, no. 13: 1770. https://doi.org/10.3390/cells12131770