
Propofol Inhibits Glioma Stem Cell Growth and Migration and Their Interaction with Microglia via BDNF-AS and Extracellular Vesicles
 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Experimental Protocols
2.3. GSC and Microglia Cultures
2.4. Neurosphere Formation/Self-Renewal Assay
2.5. In Vitro Limiting Dilution Assay
2.6. Cell Proliferation
2.7. Cell Death Assays
2.8. siRNA Transfection
2.9. Lentivirus Transduction of YKL-40 Reporter
2.10. Luciferase Activity
2.11. Quantitative Real-Time PCR
2.12. EV Isolation and Characterization
2.13. Quantification of Cytokine Secretion and EV Subpopulations
2.14. Transwell Migration Assay
2.15. Chip Manufacturing
2.16. Chip Operation
2.17. Cell Viability Assays
2.18. Radiation Exposure
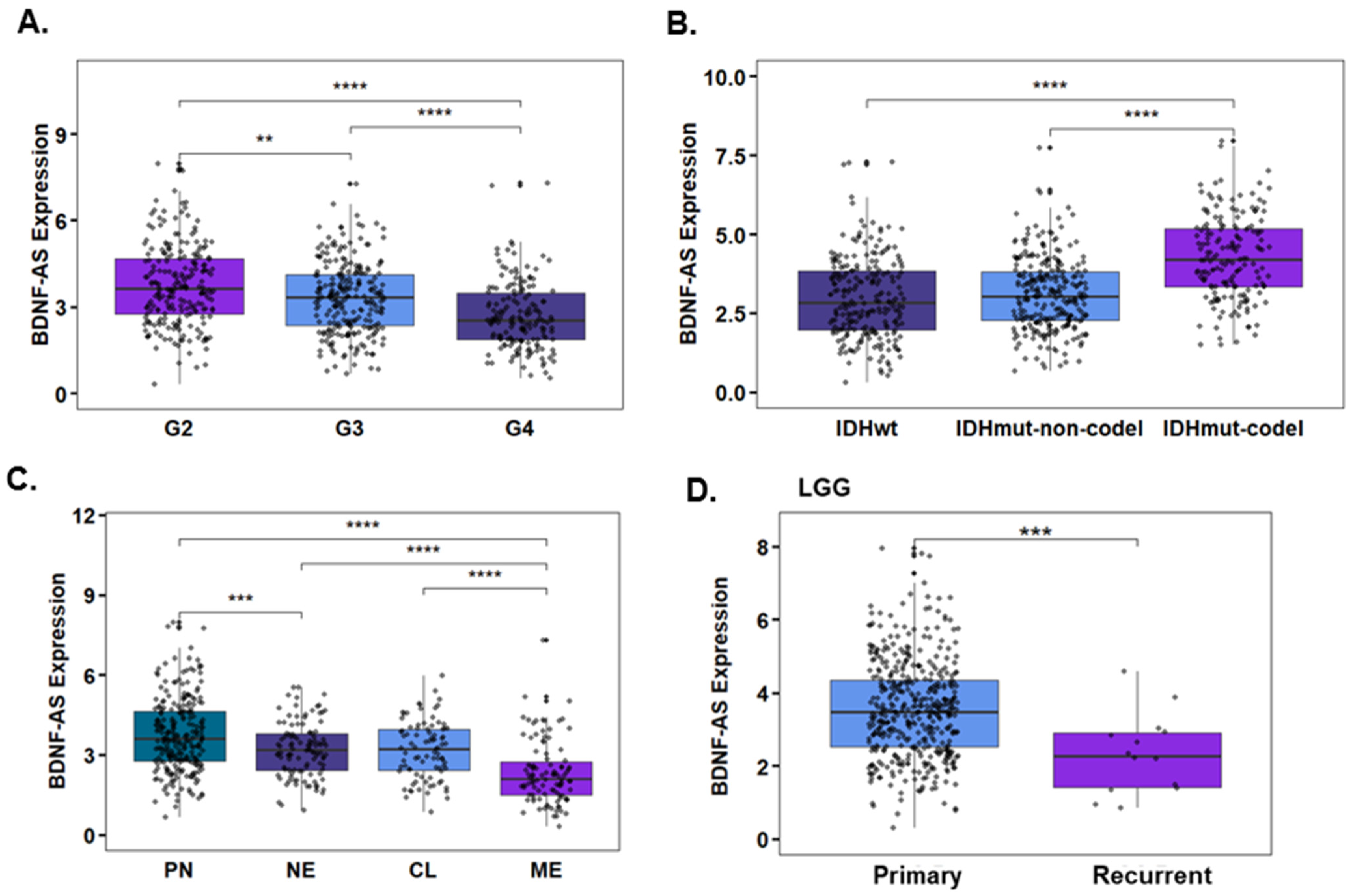
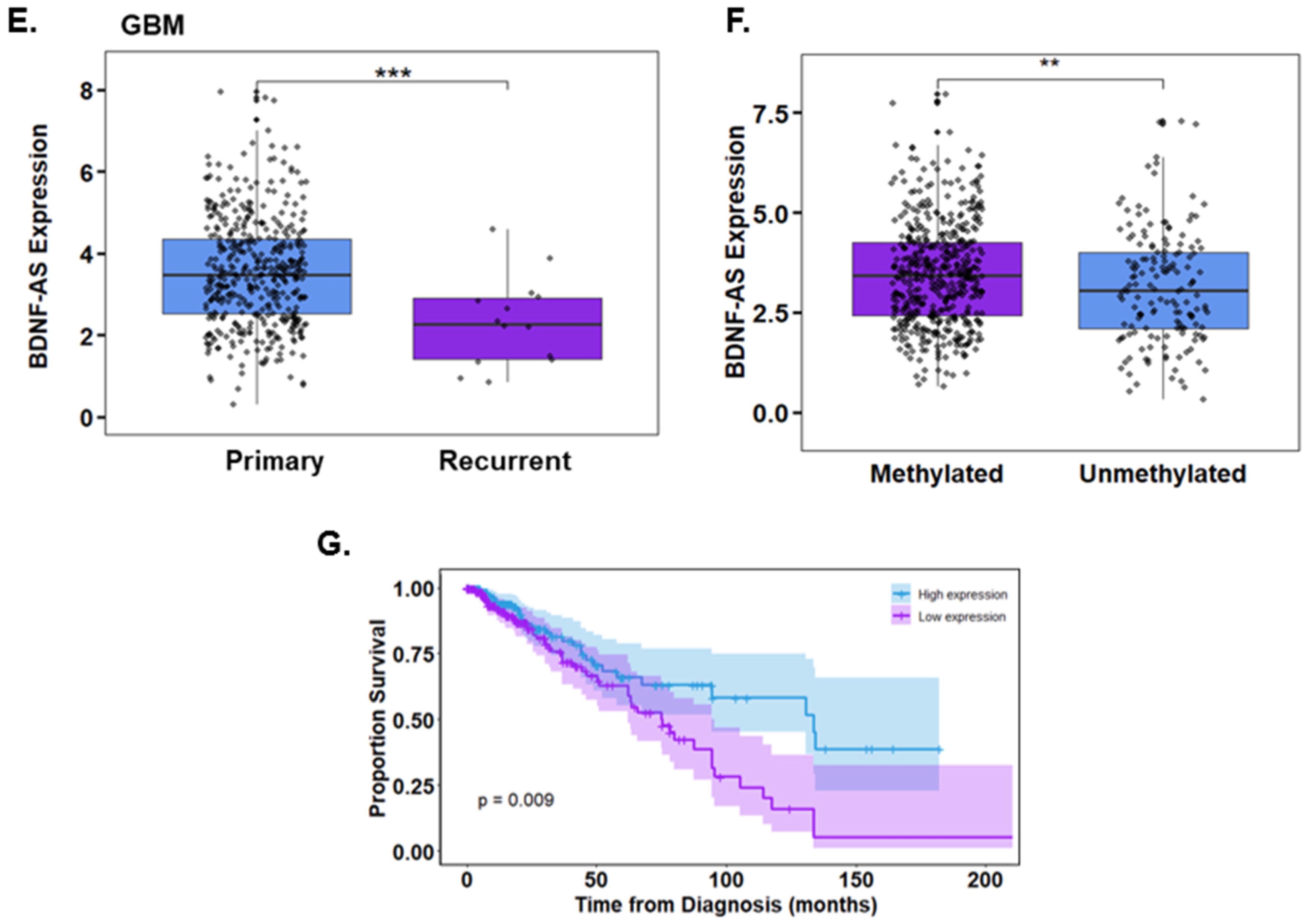
2.19. TCGA Data Analysis
2.20. Statistical Analysis
3. Results
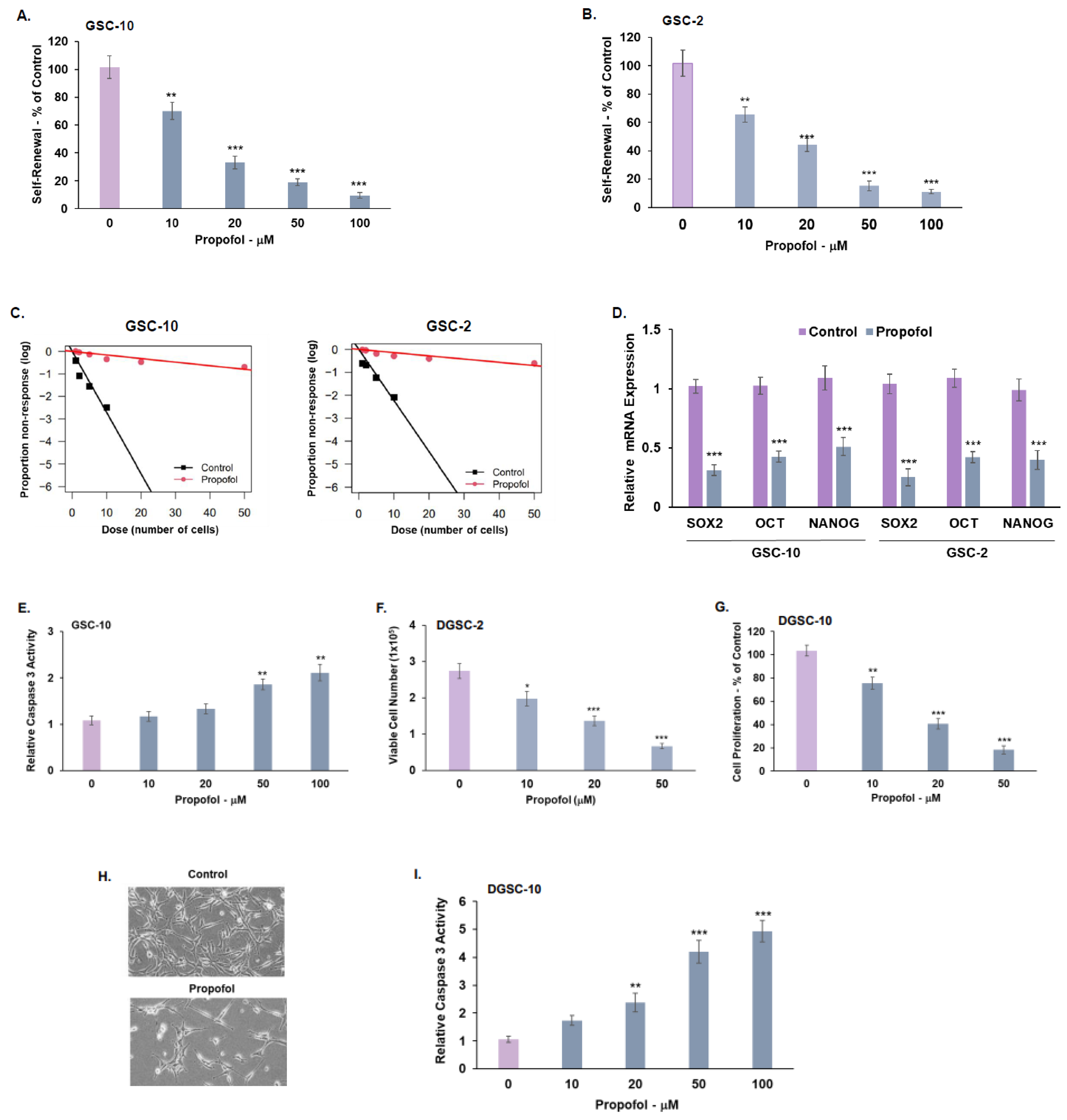
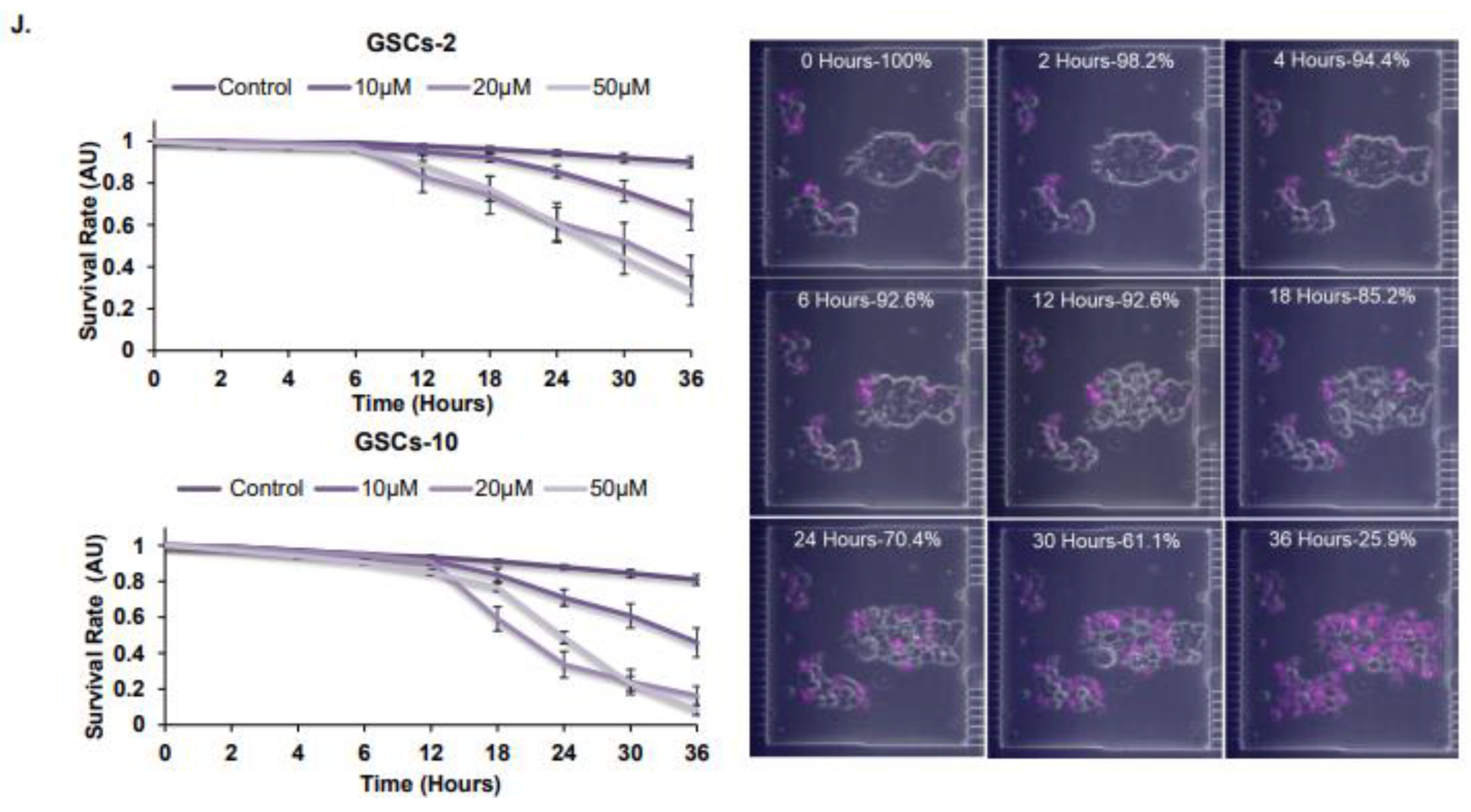
3.1. Propofol Inhibits the Self-Renewal and Stemness of GSCs and Proliferation of Differentiated Glioma Cells
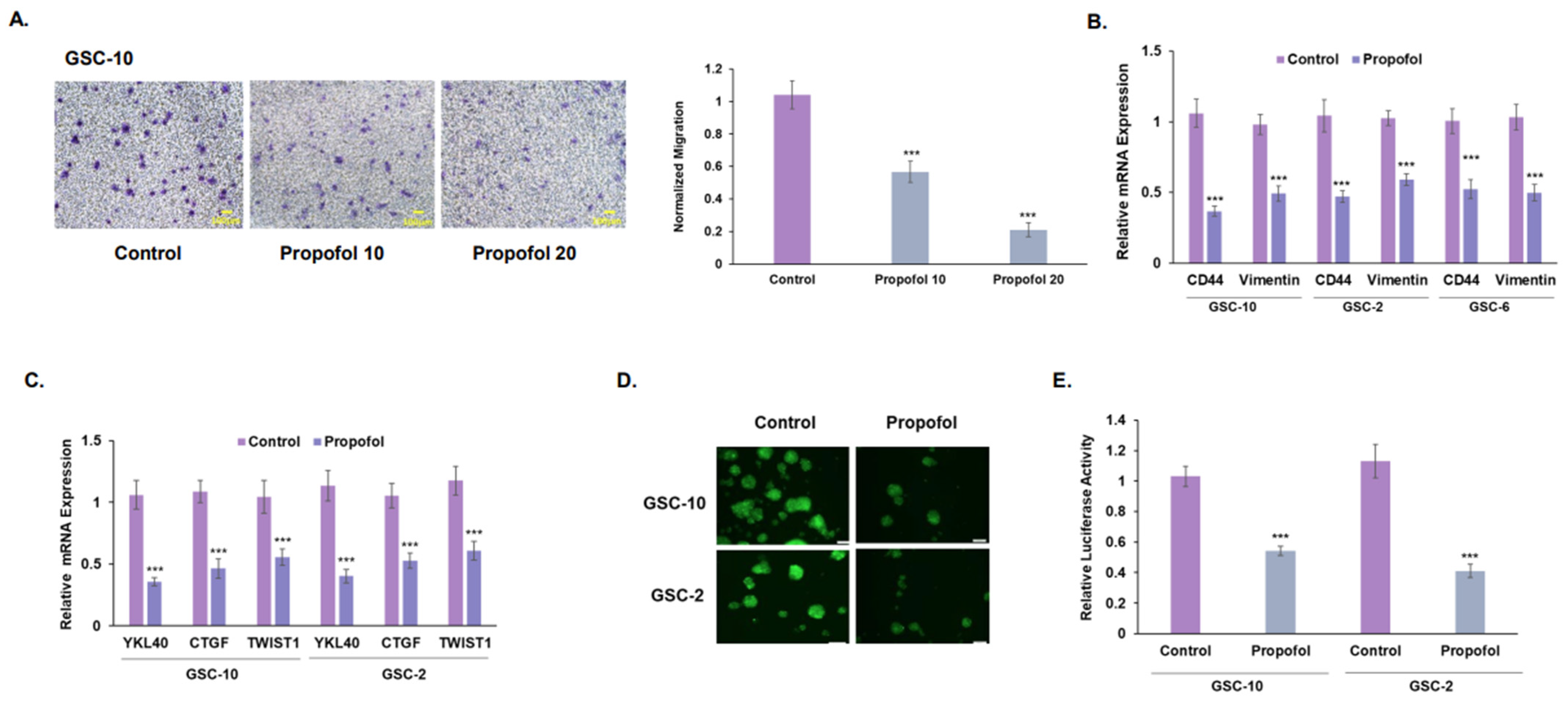
3.2. Propofol Inhibits the Migration of GSCs and Their Mesenchymal Transit
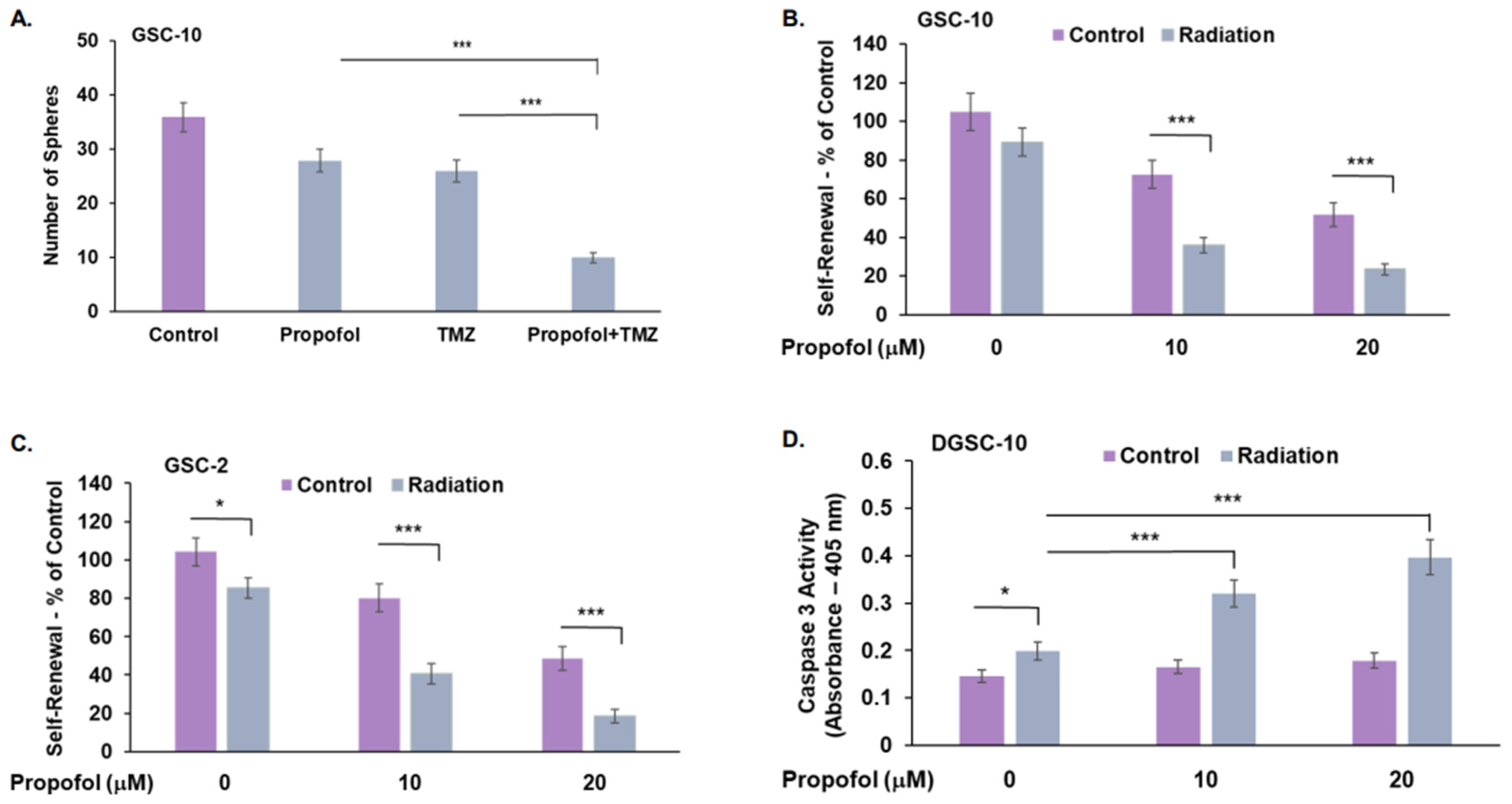
3.3. Combined Effects of Propofol, TMZ, and Radiation in GSCs and DGSCs
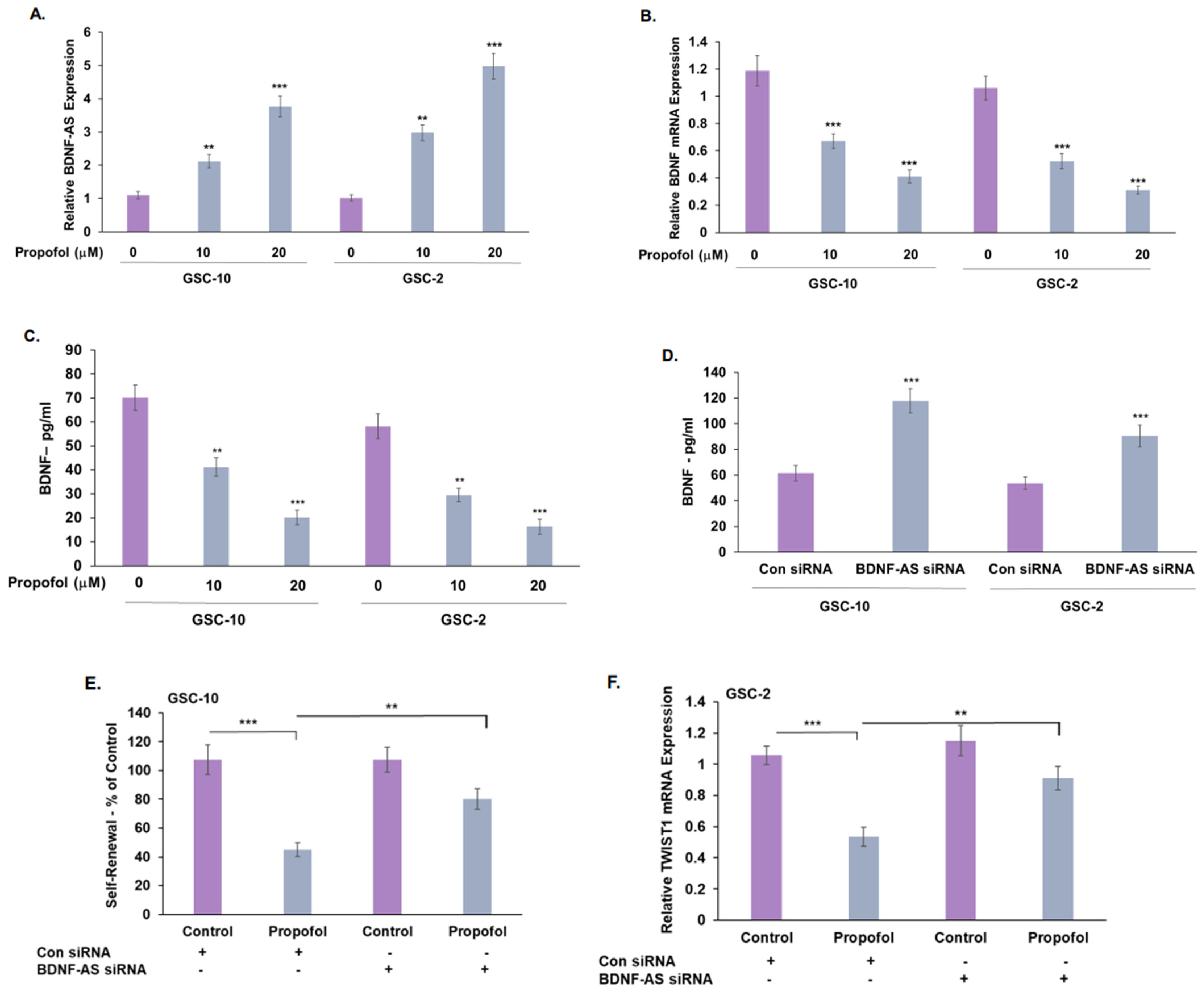
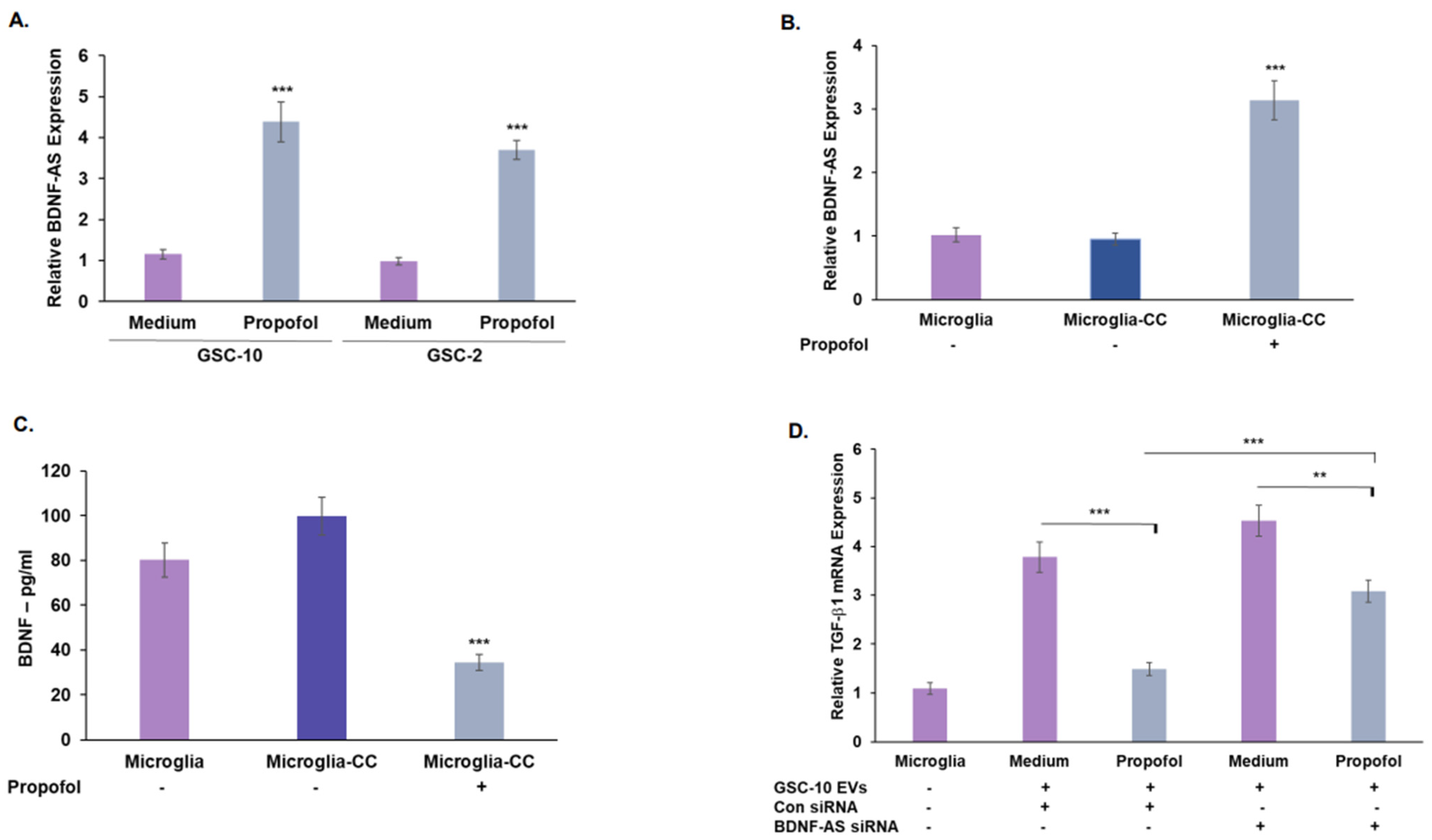
3.4. Propofol Increases the Expression of the lncRNA BDNF-AS in GSCs
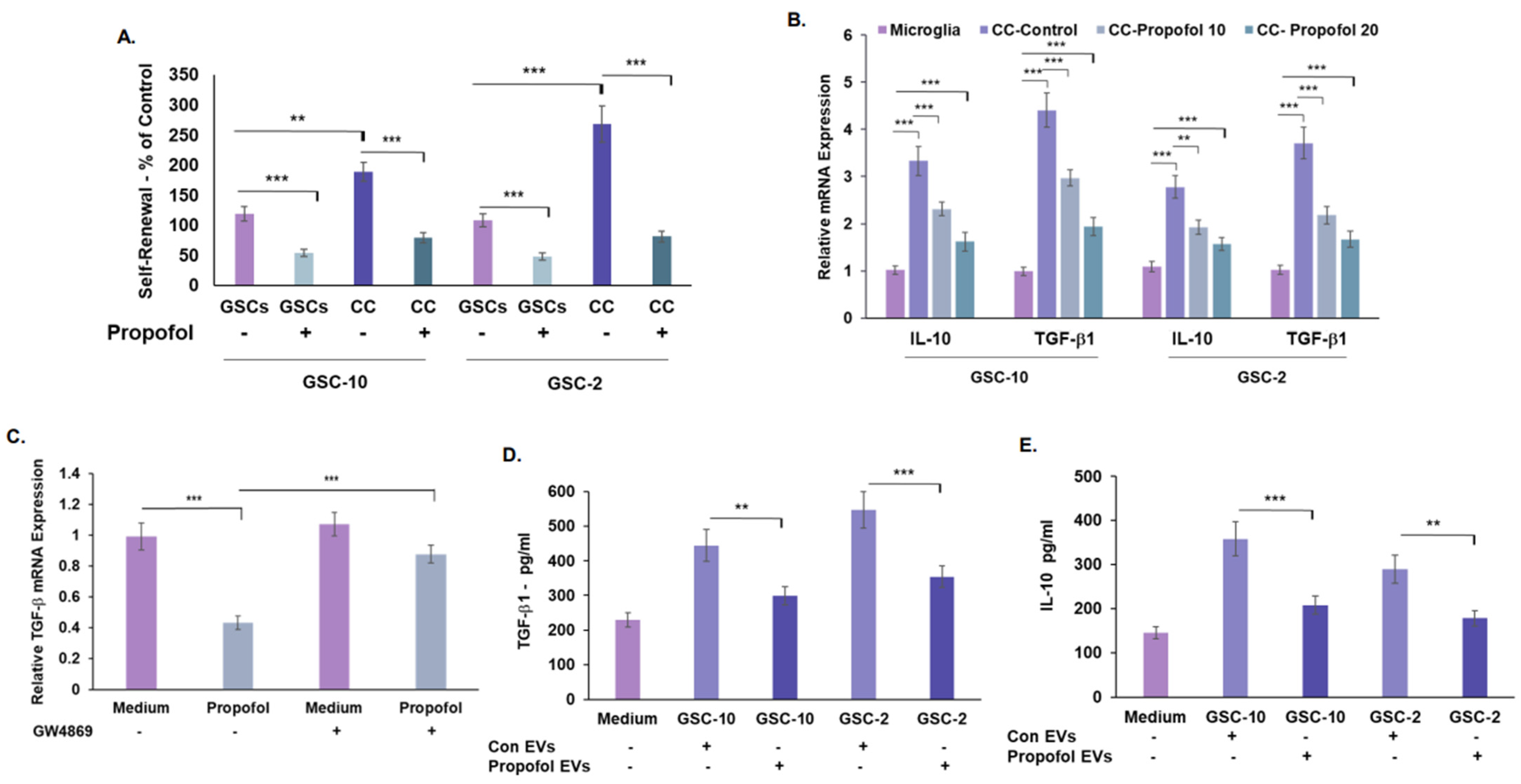
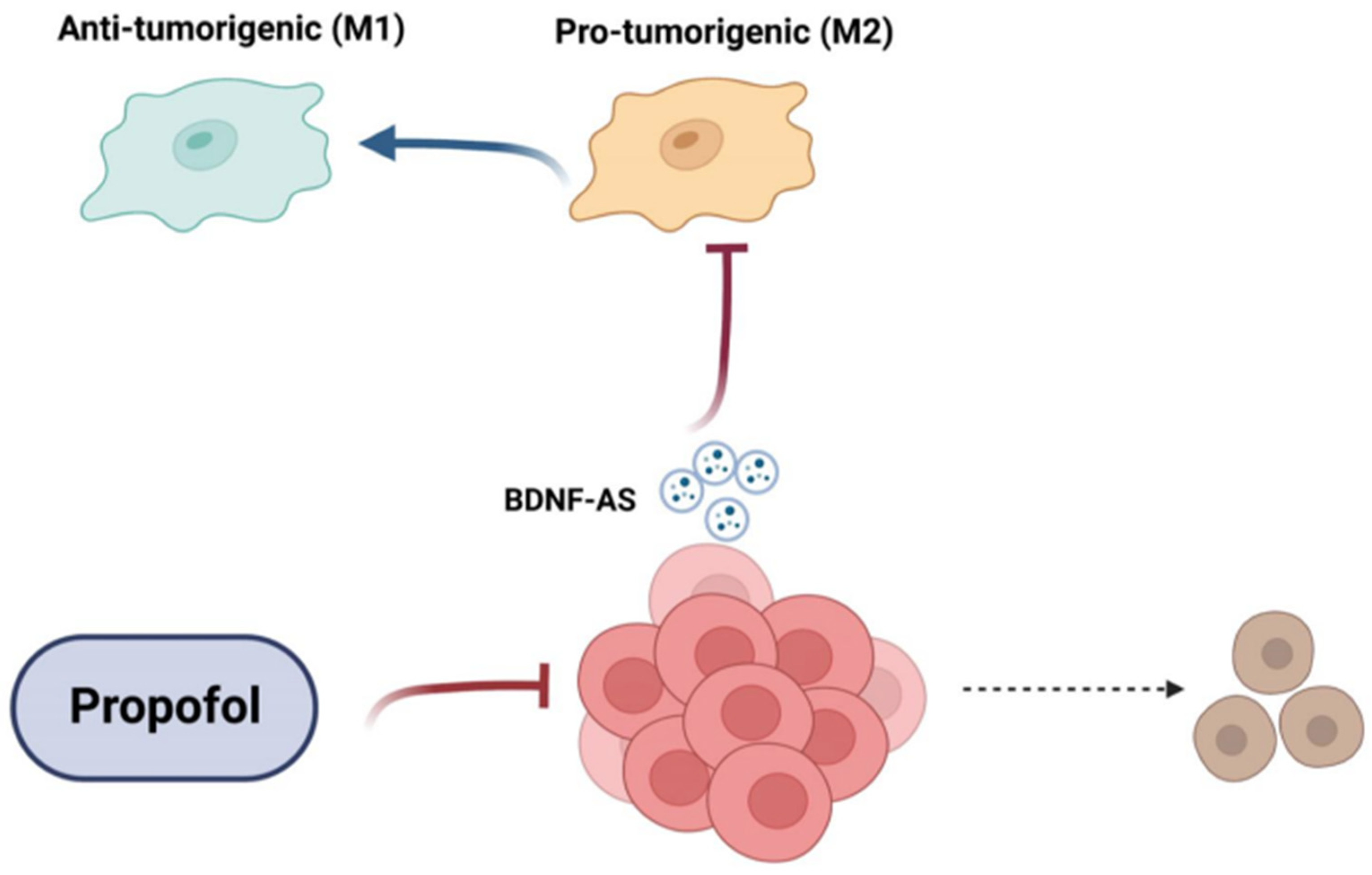
3.5. Treatment of GSCs with Propofol Decreases the Anti-Inflammatory/Pro-Tumorigenic State of Co-Cultured Microglia via the Transfer of EVs
3.6. Propofol Effects on Co-Cultured Microglia State Are Mediated by Transfer of EV-Associated BDNF-AS
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| bFGF | basic fibroblast growth factor; |
| CTGF | connective tissue growth factor; |
| EVs | extracellular vesicles; |
| EGF | epidermal growth factor; |
| FN | fibronectin; |
| GBM | glioblastoma; |
| GSC | glioma stem cell; |
| LGG | low-grade glioma; |
| miRNA | microRNA. |
References
- Furnari, F.B.; Fenton, T.; Bachoo, R.M.; Mukasa, A.; Stommel, J.M.; Stegh, A.; Hahn, W.C.; Ligon, K.L.; Louis, D.N.; Brennan, C.; et al. Malignant Astrocytic Glioma: Genetics, Biology, and Paths to Treatment. Genes Dev. 2007, 21, 2683–2710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desjardins, A.; Rich, J.N.; Quinn, J.A.; Vredenburgh, J.; Gururangan, S.; Reardon, D.A.; Friedman, A.H.; Bigner, D.D.; Friedman, H.S. Chemotherapy and Novel Therapeutic Approaches in Malignant Glioma. Front. Biosci. 2005, 10, 2645–2668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khasraw, M.; Fujita, Y.; Lee-Chang, C.; Balyasnikova, I.V.; Najem, H.; Heimberger, A.B. New Approaches to Glioblastoma. Annu. Rev. Med. 2022, 73, 279–292. [Google Scholar] [CrossRef] [PubMed]
- Gimple, R.C.; Bhargava, S.; Dixit, D.; Rich, J.N. Glioblastoma Stem Cells: Lessons from the Tumor Hierarchy in a Lethal Cancer. Genes Dev. 2019, 33, 591–609. [Google Scholar] [CrossRef] [PubMed]
- Alcantara Llaguno, S.; Parada, L.F. Cancer Stem Cells in Gliomas: Evolving Concepts and Therapeutic Implications. Curr. Opin. Neurol. 2021, 34, 868–874. [Google Scholar] [CrossRef]
- Prager, B.C.; Bhargava, S.; Mahadev, V.; Hubert, C.G.; Rich, J.N. Glioblastoma Stem Cells: Driving Resilience through Chaos. Trends Cancer 2020, 6, 223–235. [Google Scholar] [CrossRef]
- Varn, F.S.; Johnson, K.C.; Martinek, J.; Huse, J.T.; Nasrallah, M.P.; Wesseling, P.; Cooper, L.A.D.; Malta, T.M.; Wade, T.E.; Sabedot, T.S.; et al. Glioma Progression Is Shaped by Genetic Evolution and Microenvironment Interactions. Cell 2022, 185, 2184–2199.e16. [Google Scholar] [CrossRef]
- Bayik, D.; Lathia, J.D. Cancer Stem Cell–Immune Cell Crosstalk in Tumour Progression. Nat. Rev. Cancer 2021, 21, 526–536. [Google Scholar] [CrossRef]
- Dapash, M.; Hou, D.; Castro, B.; Lee-Chang, C.; Lesniak, M.S. The Interplay between Glioblastoma and Its Microenvironment. Cells 2021, 10, 2257. [Google Scholar] [CrossRef]
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The Role of Microglia and Macrophages in Glioma Maintenance and Progression. Nat. Neurosci. 2016, 19, 20–27. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Ke, S.Q.; Huang, Z.; Flavahan, W.; Fang, X.; Paul, J.; Wu, L.; Sloan, A.E.; McLendon, R.E.; Li, X.; et al. Periostin Secreted by Glioblastoma Stem Cells Recruits M2 Tumour-Associated Macrophages and Promotes Malignant Growth. Nat. Cell Biol. 2015, 17, 170–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broekman, M.L.; Maas, S.L.N.; Abels, E.R.; Mempel, T.R.; Krichevsky, A.M.; Breakefield, X.O. Multidimensional Communication in the Microenvirons of Glioblastoma. Nat. Rev. Neurol. 2018, 14, 482–495. [Google Scholar] [CrossRef] [PubMed]
- Skotland, T.; Sagini, K.; Sandvig, K.; Llorente, A. An Emerging Focus on Lipids in Extracellular Vesicles. Adv. Drug Deliv. Rev. 2020, 159, 308–321. [Google Scholar] [CrossRef] [PubMed]
- van Niel, G.; Carter, D.R.F.; Clayton, A.; Lambert, D.W.; Raposo, G.; Vader, P. Challenges and Directions in Studying Cell-Cell Communication by Extracellular Vesicles. Nat. Rev. Mol. Cell Biol. 2022, 23, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.Y.; Wu, M.Y.; Chien, Y.J.; Su, I.M.; Wang, S.C.; Kao, M.C. Anesthesia and Long-Term Oncological Outcomes: A Systematic Review and Meta-Analysis. Anesth. Analg. 2021, 132, 623–634. [Google Scholar] [CrossRef] [PubMed]
- Dubowitz, J.; Hiller, J.; Riedel, B. Anesthetic Technique and Cancer Surgery Outcomes. Curr. Opin. Anaesthesiol. 2021, 34, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Saha, P.; Das, A.; Chatterjee, N.; Chakrabarti, D.; Sinha, D. Impact of Anesthetics on Oncogenic Signaling Network: A Review on Propofol and Isoflurane. Fundam. Clin. Pharmacol. 2022, 36, 49–71. [Google Scholar] [CrossRef]
- Cata, J.P.; Hagan, K.B.; Bhavsar, S.D.O.; Arunkumar, R.; Grasu, R.; Dang, A.; Carlson, R.; Arnold, B.; Potylchansky, Y.; Lipski, I.; et al. The Use of Isoflurane and Desflurane as Inhalational Agents for Glioblastoma Surgery. A Survival Analysis. J. Clin. Neurosci. 2017, 35, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Liu, J.; Xie, H. Propofol Suppresses Osteosarcoma Cell Function by Regulating FOXO1/TUSC7. J. Pharm. Pharmacol. 2021, 73, 720–725. [Google Scholar] [CrossRef]
- Huang, Y.H.; Wu, Z.F.; Lee, M.S.; Lou, Y.S.; Wu, K.L.; Cheng, K.I.; Lai, H.C. Propofol-Based Total Intravenous Anesthesia Is Associated with Better Survival than Desflurane Anesthesia in Glioblastoma Surgery. PLoS ONE 2021, 16, e0255627. [Google Scholar] [CrossRef]
- Gao, J.; Ding, C.; Zhou, J.; Wu, G.; Han, Z.; Li, J.; Hei, F. Propofol Suppresses Lung Cancer Tumorigenesis by Modulating the Circ-ERBB2/MiR-7-5p/FOXM1 Axis. Thorac. Cancer 2021, 12, 824–834. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Liu, H.; Dilger, J.P.; Lin, J. Effect of Propofol on Breast Cancer Cell, the Immune System, and Patient Outcome. BMC Anesthesiol. 2018, 18, 77. [Google Scholar] [CrossRef] [Green Version]
- Iwakiri, H.; Nishihara, N.; Nagata, O.; Matsukawa, T.; Ozaki, M.; Sessler, D.I. Individual Effect-Site Concentrations of Propofol Are Similar at Loss of Consciousness and at Awakening. Anesth. Analg. 2005, 100, 107–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller-Wirtz, L.M.; Maurer, F.; Brausch, T.; Kiefer, D.; Floss, M.; Doneit, J.; Volk, T.; Sessler, D.I.; Fink, T.; Lehr, T.; et al. Exhaled Propofol Concentrations Correlate with Plasma and Brain Tissue Concentrations in Rats. Anesth. Analg. 2021, 132, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Hasselbach, L.A.; Irtenkauf, S.M.; Lemke, N.W.; Nelson, K.K.; Berezovsky, A.D.; Carlton, E.T.; Transou, A.D.; Mikkelsen, T.; deCarvalho, A.C. Optimization of High Grade Glioma Cell Culture from Surgical Specimens for Use in Clinically Relevant Animal Models and 3D Immunochemistry. J. Vis. Exp. 2014, e51088. [Google Scholar] [CrossRef] [Green Version]
- Bier, A.; Hong, X.; Cazacu, S.; Goldstein, H.; Rand, D.; Xiang, C.; Jiang, W.; Ben-Asher, H.W.; Attia, M.; Brodie, A.; et al. MiR-504 Modulates the Stemness and Mesenchymal Transition of Glioma Stem Cells and Their Interaction with Microglia via Delivery by Extracellular Vesicles. Cell Death Dis. 2020, 11, 899. [Google Scholar] [CrossRef] [PubMed]
- Giladi, N.D.; Ziv-Av, A.; Lee, H.K.; Finniss, S.; Cazacu, S.; Xiang, C.; Ben-Asher, H.W.; de Carvalho, A.; Mikkelsen, T.; Poisson, L.; et al. RTVP-1 Promotes Mesenchymal Transformation of Glioma via a STAT-3/IL-6-Dependent Positive Feedback Loop. Oncotarget 2015, 6, 22680–22697. [Google Scholar] [CrossRef] [PubMed]
- Penning, D.H.; Cazacu, S.; Brodie, A.; Jevtovic-Todorovic, V.; Kalkanis, S.N.; Lewis, M.; Brodie, C. Neuron-Glia Crosstalk Plays a Major Role in the Neurotoxic Effects of Ketamine via Extracellular Vesicles. Front. Cell Dev. Biol. 2021, 9, 691648. [Google Scholar] [CrossRef]
- Unterman, I.; Bloch, I.; Cazacu, S.; Kazimirsky, G.; Ben-Zeev, B.; Berman, B.P.; Brodie, C.; Tabach, Y. Expanding the Mecp2 Network Using Comparative Genomics Reveals Potential Therapeutic Targets for Rett Syndrome. Elife 2021, 10. [Google Scholar] [CrossRef]
- Jiang, W.; Finniss, S.; Cazacu, S.; Xiang, C.; Brodie, Z.; Mikkelsen, T.; Poisson, L.; Shackelford, D.B.; Brodie, C.; Jiang, W.; et al. Repurposing Phenformin for the Targeting of Glioma Stem Cells and the Treatment of Glioblastoma. Oncotarget 2016, 7, 56456–56470. [Google Scholar] [CrossRef] [Green Version]
- Bier, A.; Giladi, N.; Kronfeld, N.; Lee, H.K.; Cazacu, S.; Finniss, S.; Xiang, C.; Poisson, L.; deCarvalho, A.C.; Slavin, S.; et al. MicroRNA-137 Is Downregulated in Glioblastoma and Inhibits the Stemness of Glioma Stem Cells by Targeting RTVP-1. Oncotarget 2013, 4, 665–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.K.; Bier, A.; Cazacu, S.; Finniss, S.; Xiang, C.; Twito, H.; Poisson, L.M.; Mikkelsen, T.; Slavin, S.; Jacoby, E.; et al. MicroRNA-145 Is Downregulated in Glial Tumors and Regulates Glioma Cell Migration by Targeting Connective Tissue Growth Factor. PLoS ONE 2013, 8, e54652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziv-Av, A.; Giladi, N.D.; Lee, H.K.; Cazacu, S.; Finniss, S.; Xiang, C.; Pauker, M.H.; Barda-Saad, M.; Poisson, L.; Brodie, C. RTVP-1 Regulates Glioma Cell Migration and Invasion via Interaction with N-WASP and HnRNPK. Oncotarget 2015, 6, 19826–19840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glick, Y.; Avrahami, D.; Michaely, E.; Gerber, D. High-Throughput Protein Expression Generator Using a Microfluidic Platform. J. Vis. Exp. 2012, e3849. [Google Scholar] [CrossRef] [Green Version]
- Desyatnik, I.; Krasner, M.; Frolov, L.; Ronen, M.; Guy, O.; Wasserman, D.; Tzur, A.; Avrahami, D.; Barbiro-Michaely, E.; Gerber, D. An Integrated Microfluidics Approach for Personalized Cancer Drug Sensitivity and Resistance Assay. Adv. Biosyst. 2019, 3, 1900001. [Google Scholar] [CrossRef] [PubMed]
- Ceccarelli, M.; Barthel, F.P.; Malta, T.M.; Noushmehr, H.; Iavarone, A.; Verhaak, R.G.W. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell 2016, 164, 550–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Yang, T.; Liu, J.; Liu, Y.; Xing, N.; He, J.; Yang, J.; Ai, Y. Propofol Inhibits the Migration and Invasion of Glioma Cells by Blocking the PI3K/AKT Pathway Through MiR-206/ROCK1 Axis. Onco. Targets. Ther. 2020, 13, 361–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.; Zheng, L.; Yang, C.; Zhang, W.; Wang, H. Propofol Inhibits Proliferation and Migration of Glioma Cells by Up-Regulating LncRNA GAS5. Toxicol. Vitr. 2022, 80, 105321. [Google Scholar] [CrossRef]
- Wang, X.; Prager, B.C.; Wu, Q.; Kim, L.J.Y.; Gimple, R.C.; Shi, Y.; Yang, K.; Morton, A.R.; Zhou, W.; Zhu, Z.; et al. Reciprocal Signaling between Glioblastoma Stem Cells and Differentiated Tumor Cells Promotes Malignant Progression. Cell Stem Cell 2018, 22, 514–528.e5. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Zhao, H.; Weng, H.; Ma, D. Lasting Effects of General Anesthetics on the Brain in the Young and Elderly: “Mixed Picture” of Neurotoxicity, Neuroprotection and Cognitive Impairment. J. Anesth. 2019, 33, 321–335. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Varn, F.S.; Park, S.H.; Yoon, B.W.; Park, H.R.; Lee, C.; Verhaak, R.G.W.; Paek, S.H. Perspective of Mesenchymal Transformation in Glioblastoma. Acta Neuropathol. Commun. 2021, 9, 1–20. [Google Scholar] [CrossRef]
- Davis, M.E. Glioblastoma: Overview of Disease and Treatment. CJON 2016, 20, S2–S8. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Pan, S.; Jiang, W.; Xue, F.; Zhu, X. Effects of Propofol on the Development of Cancer in Humans. Cell Prolif. 2020, 53, e12867. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, C.; Yan, T.; Wang, J.; Liang, W. Long Noncoding RNA BDNF-AS Is Downregulated in Cervical Cancer and Has Anti-Cancer Functions by Negatively Associating with BDNF. Arch. Biochem. Biophys. 2018, 646, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Pruunsild, P.; Kazantseval, A.; Aid, T.; Palm, K.; Timmusk, T. Dissecting the Human BDNF Locus: Bidirectional Transcription, Complex Splicing, and Multiple Promoters. Genomics 2007, 90, 397–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rui, S.; Jun, M.; Xiaobai, Z.J.L.; Yunhui, L.; Xuelei, R.; Shuyuan, S.; Chunqing, Y.; Di, W.; Heng, C.; Zhen, L.; et al. PABPC1-Induced Stabilization of BDNF-AS Inhibits Malignant Progression of Glioblastoma Cells through STAU1-Mediated Decay. Cell Death Dis. 2020, 11, 81. [Google Scholar]
- Villeneuve, J.; Tremblay, P.; Vallières, L. Tumor Necrosis Factor Reduces Brain Tumor Growth by Enhancing Macrophage Recruitment and Microcyst Formation. Cancer Res. 2005, 65, 3928–3936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trajkovic, K.; Hsu, C.; Chiantia, S.; Rajendran, L.; Wenzel, D.; Wieland, F.; Schwille, P.; Brügger, B.; Simons, M. Ceramide Triggers Budding of Exosome Vesicles into Multivesicular Endosomes. Science 2008, 319, 1244–1247. [Google Scholar] [CrossRef]
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a Cancer Stem Cell in Human Brain Tumors. Cancer Res. 2003, 63, 5821–5828. [Google Scholar] [PubMed]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.L.; Rich, J.N. Cancer Stem Cells in Glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef] [Green Version]
- Fedele, M.; Cerchia, L.; Pegoraro, S.; Sgarra, R.; Manfioletti, G. Proneural-Mesenchymal Transition: Phenotypic Plasticity to Acquire Multitherapy Resistance in Glioblastoma. Int. J. Mol. Sci. 2019, 20, 2746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; Li, Y.; Liu, X.; Wang, Z.; Zhang, C.; Wu, F.; Jiang, H.; Zhang, W.; Bao, Z.; Wang, Y.; et al. ALDH1A3 Induces Mesenchymal Differentiation and Serves as a Predictor for Survival in Glioblastoma. Cell Death Dis. 2018, 9, 1190–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Gao, Y.; Zhang, F. Propofol Inhibits Pancreatic Cancer Proliferation and Metastasis by Up-Regulating MiR-328 and down-Regulating ADAM8. Basic Clin. Pharmacol. Toxicol. 2019, 125, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.-S.; Jan, C.-R.; Liang, W.-Z. Evaluation of Cytotoxicity of Propofol and Its Related Mechanism in Glioblastoma Cells and Astrocytes. Environ. Toxicol. 2017, 32, 2440–2454. [Google Scholar] [CrossRef] [PubMed]
- Edgunlu, T.G.; Avci, C.B.; Ozates, N.P.; Bagca, B.G.; Celik, S.K.; Boluk, A.; Ugur, B. In Vitro Effects of Propofol on Cytotoxic, Apoptotic and PI3K-Akt Signaling Pathway Genes on Brain Cancer Cells. Anticancer Agents Med. Chem. 2021, 22, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Chen, H.; Tian, C.; Zheng, D. Propofol Represses Cell Growth and Metastasis by Modulating the Circular RNA Non-SMC Condensin I Complex Subunit G/MicroRNA-200a-3p/RAB5A Axis in Glioma. World Neurosurg. 2021, 153, e46–e58. [Google Scholar] [CrossRef]
- Fan, X.; Gong, M.; Yu, H.; Yang, H.; Wang, S.; Wang, R. Propofol Enhances Stem-like Properties of Glioma via GABAAR-dependent Src Modulation of ZDHHC5-EZH2 Palmitoylation Mechanism. Stem Cell Res. Ther. 2022, 13, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ghafouri-Fard, S.; Khoshbakht, T.; Taheri, M.; Ghanbari, M. A Concise Review on the Role of BDNF-AS in Human Disorders. Biomed. Pharmacother. 2021, 142, 112051. [Google Scholar] [CrossRef]
- Chen, J.-W.E.; Lumibao, J.; Leary, S.; Sarkaria, J.N.; Steelman, A.J.; Gaskins, H.R.; Harley, B.A.C. Crosstalk between Microglia and Patient-Derived Glioblastoma Cells Inhibit Invasion in a Three-Dimensional Gelatin Hydrogel Model. J. Neuroinflamm. 2020, 17, 346. [Google Scholar] [CrossRef]
- Zhi, H.; Lian, J. LncRNA BDNF-AS Suppresses Colorectal Cancer Cell Proliferation and Migration by Epigenetically Repressing GSK-3β Expression. Cell Biochem. Funct. 2019, 37, 340–347. [Google Scholar] [CrossRef]
- Meng, L.; Liu, B.; Ji, R.U.I.; Jiang, X.I.N.; Yan, X.; Xin, Y. Targeting the BDNF/TrkB Pathway for the Treatment of Tumors. Oncol. Lett. 2019, 17, 2031. [Google Scholar] [CrossRef] [Green Version]
- Colucci-D’amato, L.; Speranza, L.; Volpicelli, F. Molecular Sciences Neurotrophic Factor BDNF, Physiological Functions and Therapeutic Potential in Depression, Neurodegeneration and Brain Cancer. Int. J. Mol. Sci. 2020, 21, 7777. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Diao, C.; Wang, X.; Xie, Y.; Liu, Y.; Gao, X.; Han, J.; Li, S. LncRNA BDNF-AS Inhibits Proliferation, Migration, Invasion and EMT in Oesophageal Cancer Cells by Targeting MiR-214. J. Cell. Mol. Med. 2018, 22, 3729–3739. [Google Scholar] [CrossRef] [PubMed]
- Lv, X.; Gu, C.; Guo, S. Activation of BDNF-AS/ADAR/P53 Positive Feedback Loop Inhibits Glioblastoma Cell Proliferation. Neurochem. Res. 2020, 45, 508–518. [Google Scholar] [CrossRef] [PubMed]
- Ikushima, H.; Todo, T.; Ino, Y.; Takahashi, M.; Miyazawa, K.; Miyazono, K. Autocrine TGF-β Signaling Maintains Tumorigenicity of Glioma-Initiating Cells through Sry-Related HMG-Box Factors. Cell Stem Cell 2009, 5, 504–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nizar, R.; Cazacu, S.; Xiang, C.; Krasner, M.; Barbiro-Michaely, E.; Gerber, D.; Schwartz, J.; Fried, I.; Yuval, S.; Brodie, A.; et al. Propofol Inhibits Glioma Stem Cell Growth and Migration and Their Interaction with Microglia via BDNF-AS and Extracellular Vesicles. Cells 2023, 12, 1921. https://doi.org/10.3390/cells12151921
Nizar R, Cazacu S, Xiang C, Krasner M, Barbiro-Michaely E, Gerber D, Schwartz J, Fried I, Yuval S, Brodie A, et al. Propofol Inhibits Glioma Stem Cell Growth and Migration and Their Interaction with Microglia via BDNF-AS and Extracellular Vesicles. Cells. 2023; 12(15):1921. https://doi.org/10.3390/cells12151921
Chicago/Turabian StyleNizar, Rephael, Simona Cazacu, Cunli Xiang, Matan Krasner, Efrat Barbiro-Michaely, Doron Gerber, Jonathan Schwartz, Iris Fried, Shira Yuval, Aharon Brodie, and et al. 2023. "Propofol Inhibits Glioma Stem Cell Growth and Migration and Their Interaction with Microglia via BDNF-AS and Extracellular Vesicles" Cells 12, no. 15: 1921. https://doi.org/10.3390/cells12151921