
Conoidin A, a Covalent Inhibitor of Peroxiredoxin 2, Reduces Growth of Glioblastoma Cells by Triggering ROS Production

Abstract
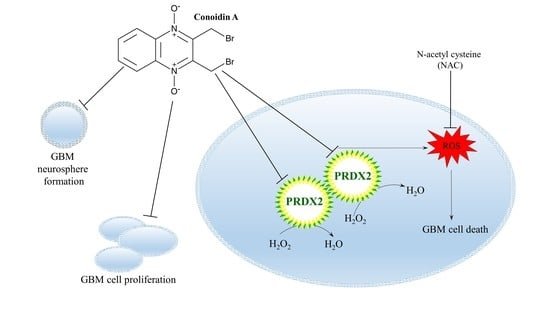
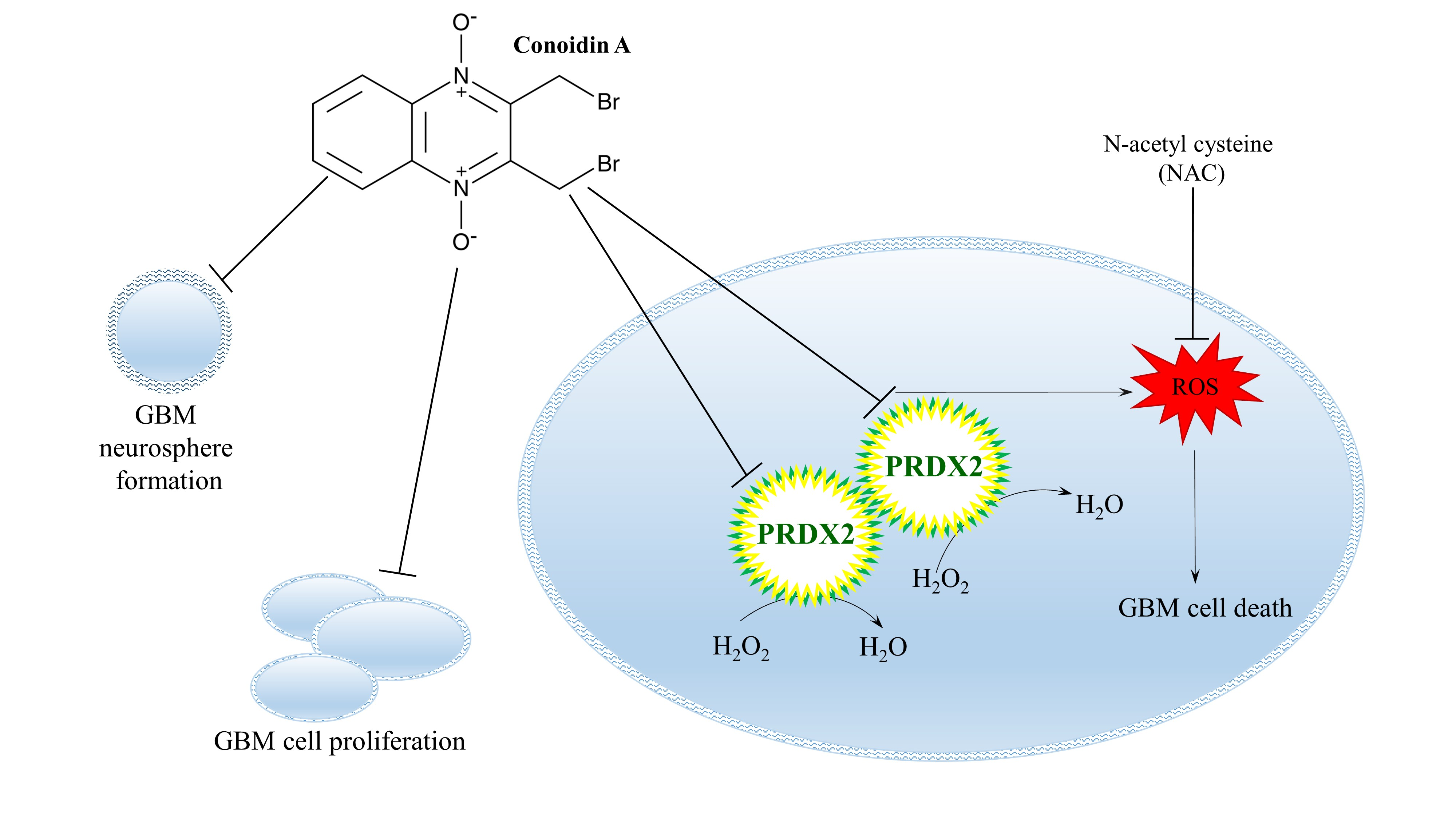
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Cell Culture and Handling
2.3. Chemicals
2.4. Gene Expression Analysis
2.5. Immunoblotting
2.6. MTT Cell Viability Assay
2.7. Colony Formation Assay
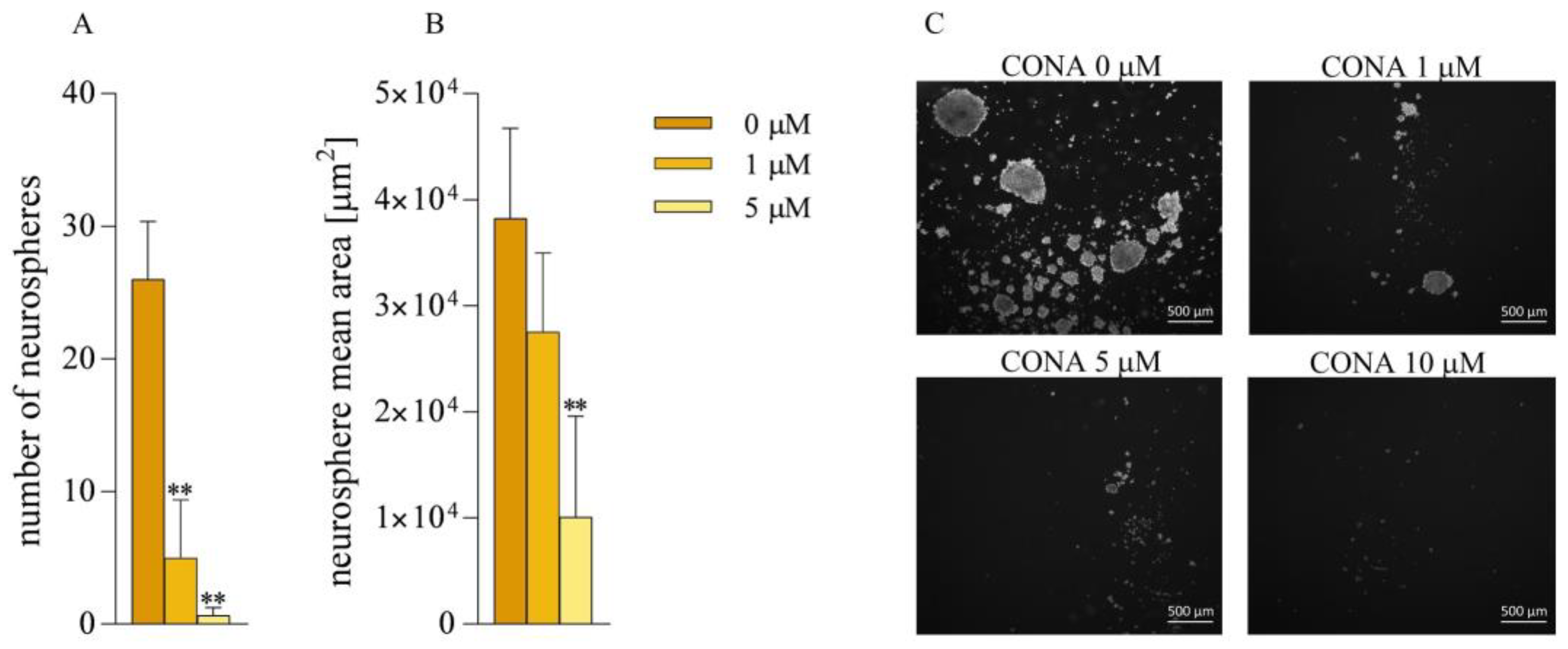
2.8. Formation of Neurospheres
2.9. Measurement of Intracellular ROS
2.10. Statistical Analysis
3. Results
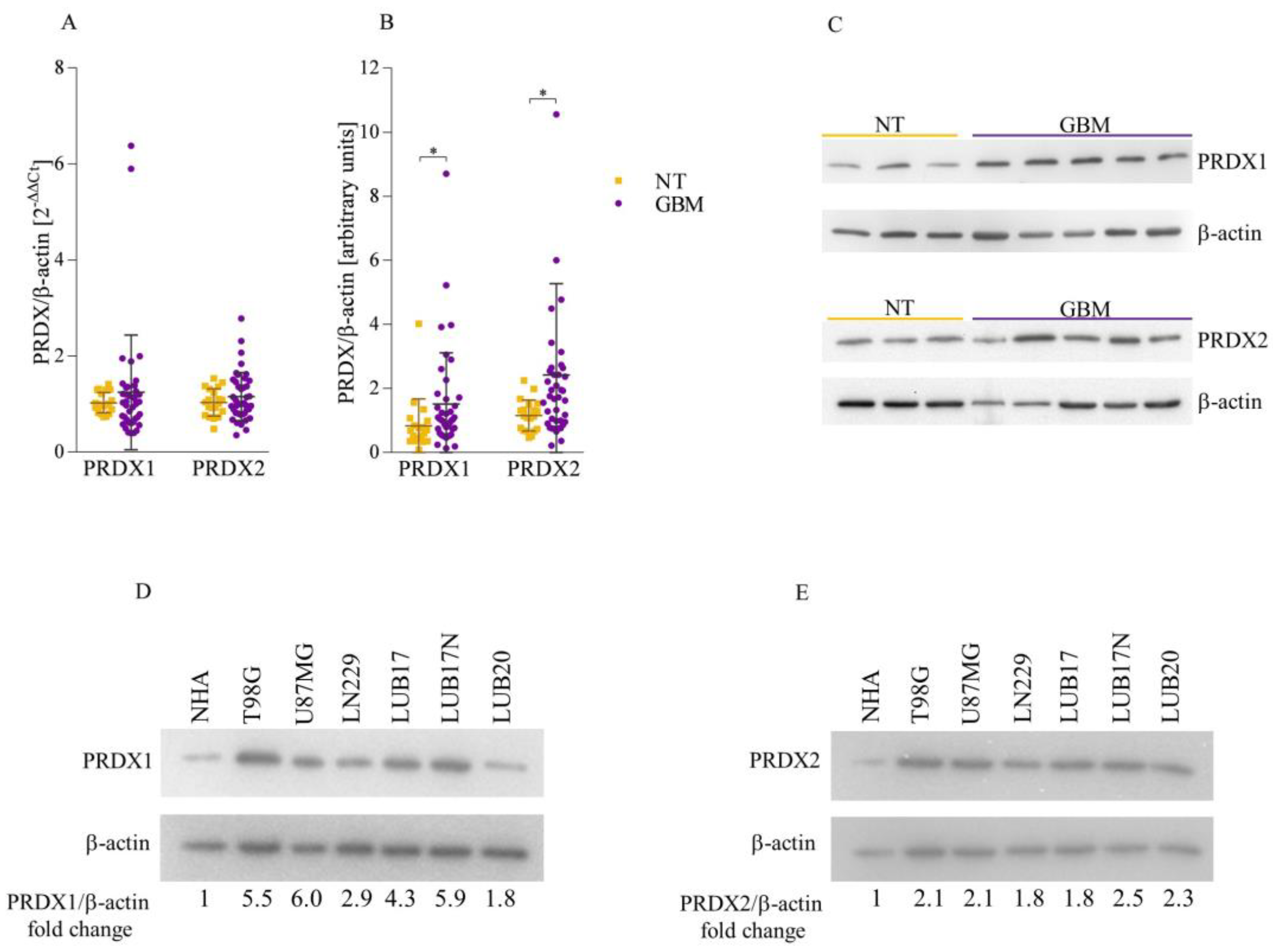
3.1. Both PRDX1 and PRDX2 Are Upregulated in GBM Tissues and Cells
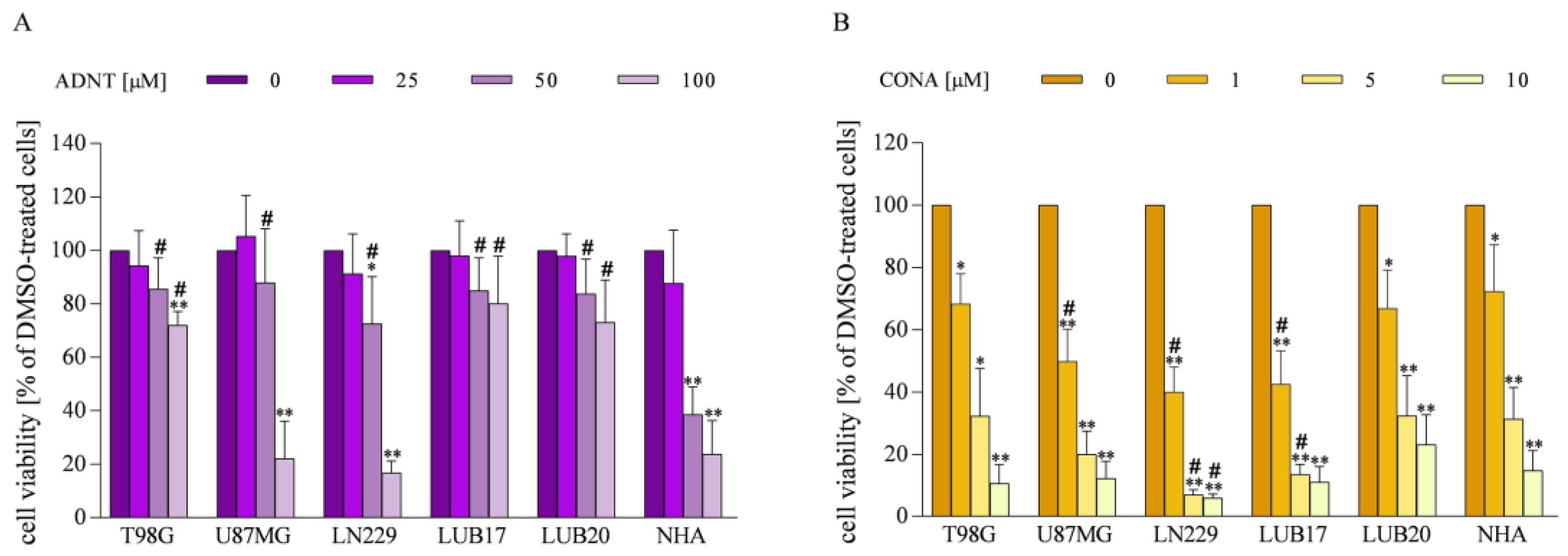
3.2. Impact of ADNT and CONA on Viability of GBM Cells and NHAs
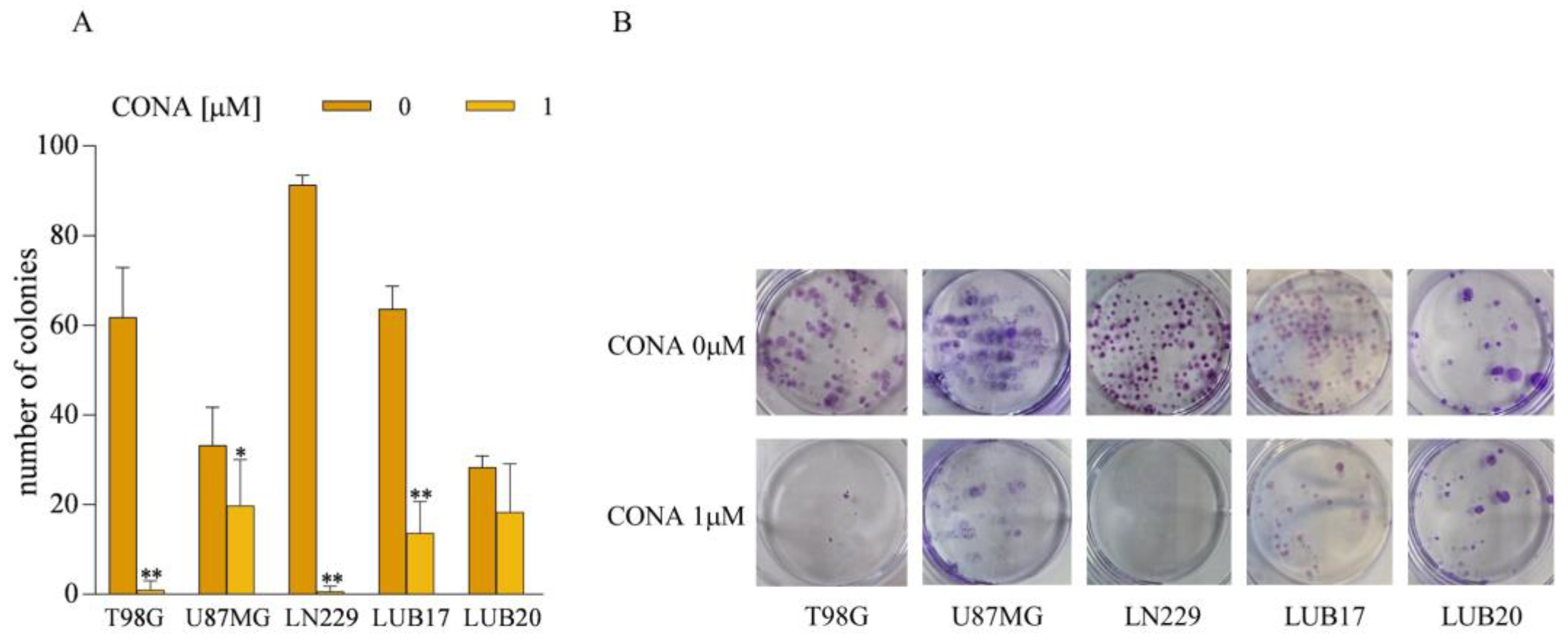
3.3. CONA Reduces Clonogenicity of GBM Cells
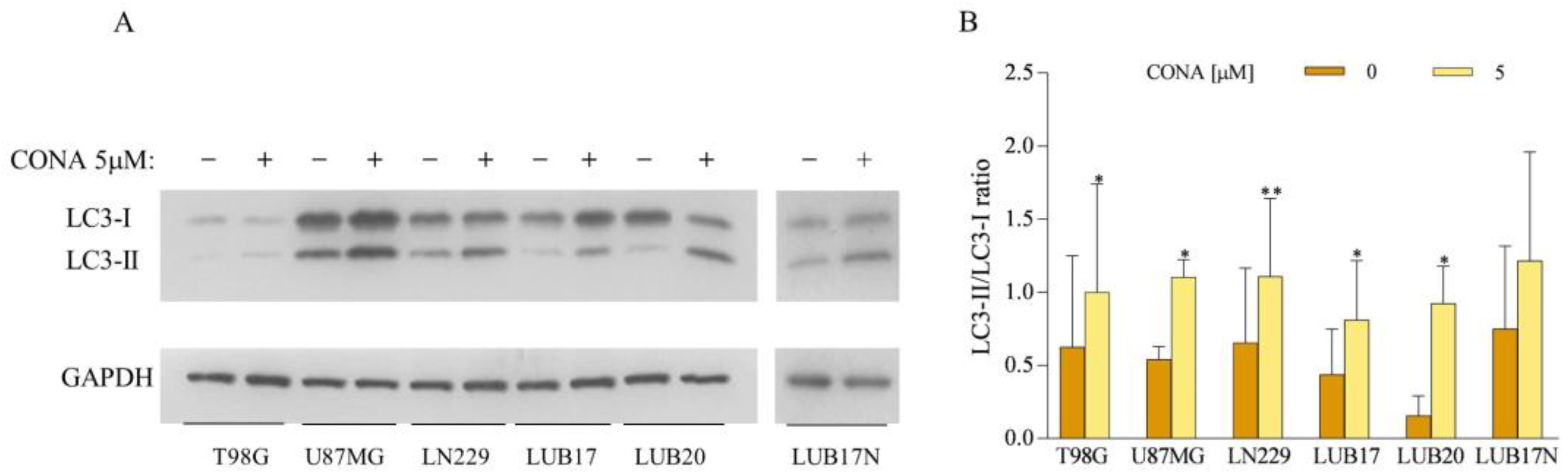
3.4. CONA Induces Autophagosome Accumulation in GBM Cells
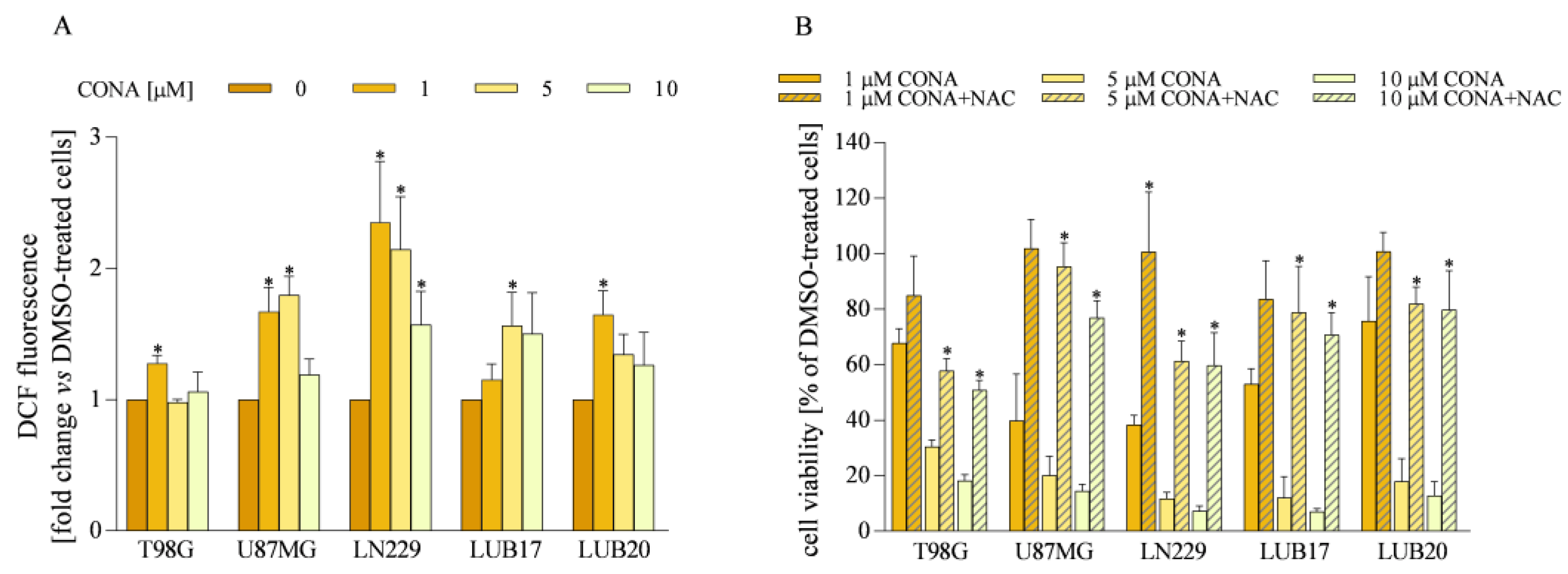
3.5. ROS Generation Induced by CONA Contributes to GBM Cell Death
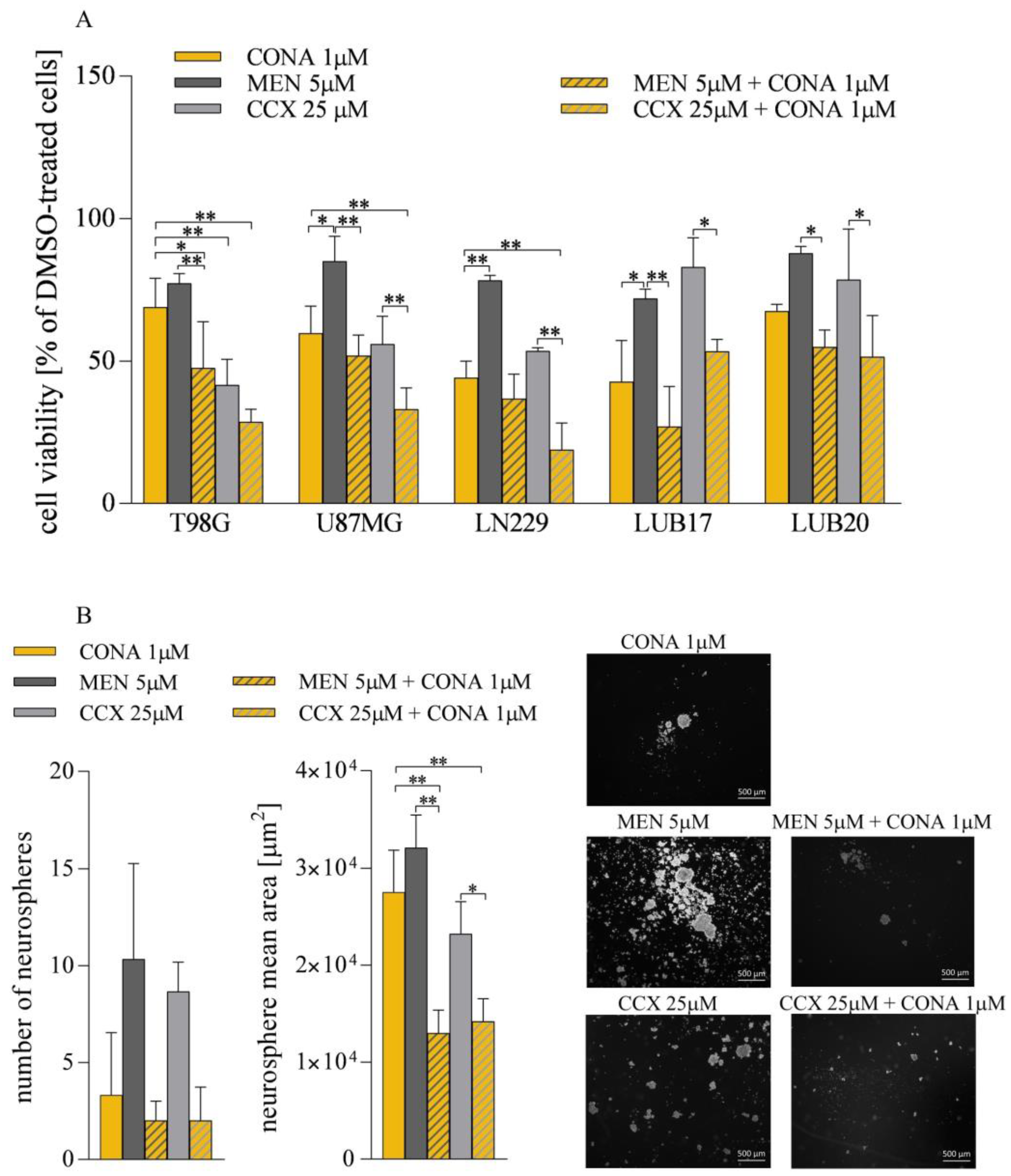
3.6. ROS Generators Potentiate the Anticancer Activity of CONA
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ostrom, Q.T.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2014–2018. Neuro-Oncology 2021, 23 (Suppl. S2), iii1–iii105. [Google Scholar] [CrossRef]
- Ghosh, D.; Nandi, S.; Bhattacharjee, S. Combination therapy to checkmate Glioblastoma: Clinical challenges and advances. Clin. Transl. Med. 2018, 7, 33. [Google Scholar] [CrossRef] [Green Version]
- Salazar-Ramiro, A.; Ramírez-Ortega, D.; de la Cruz, V.P.; Hérnandez-Pedro, N.Y.; González-Esquivel, D.F.; Sotelo, J.; Pineda, B. Role of Redox Status in Development of Glioblastoma. Front. Immunol. 2016, 7, 156. [Google Scholar] [CrossRef] [Green Version]
- Olivier, C.; Oliver, L.; Lalier, L.; Vallette, F.M. Drug Resistance in Glioblastoma: The Two Faces of Oxidative Stress. Front. Mol. Biosci. 2021, 7, 620677. [Google Scholar] [CrossRef]
- Ria, F.; Landriscina, M.; Remiddi, F.; Rosselli, R.; Iacoangeli, M.; Scerrati, M.; Pani, G.; Borrello, S.; Galeotti, T. The level of manganese superoxide dismutase content is an independent prognostic factor for glioblastoma. Biological mechanisms and clinical implications. Br. J. Cancer 2001, 84, 529–534. [Google Scholar] [CrossRef] [Green Version]
- Haapasalo, H.; Kyläniemi, M.; Paunul, N.; Kinnula, V.L.; Soini, Y. Expression of antioxidant enzymes in astrocytic brain tumors. Brain Pathol. 2003, 13, 155–164. [Google Scholar] [CrossRef]
- Deighton, R.F.; Le Bihan, T.; Martin, S.F.; Gerth, A.M.; McCulloch, M.; Edgar, J.M.; Kerr, L.E.; Whittle, I.R.; McCulloch, J. Interactions among mitochondrial proteins altered in glioblastoma. J. Neurooncol. 2014, 118, 247–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Lee, S.; Lee, S.; Kang, S.W. 2-Cys Peroxiredoxins: Emerging hubs determining redox dependency of mammalian signaling networks. Int. J. Cell Biol. 2014, 2014, 715867. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.H.; Kim, S.U.; Lee, B.K.; Kim, H.S.; Song, I.S.; Shin, H.J.; Han, Y.H.; Chang, K.T.; Kim, J.M.; Lee, D.S.; et al. Prx I suppresses K-ras-driven lung tumorigenesis by opposing redox-sensitive ERK/cyclin D1 pathway. Antioxid. Redox Signal. 2013, 19, 482–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Leary, P.C.; Terrile, M.; Bajor, M.; Gaj, P.; Hennessy, B.T.; Mills, G.B.; Zagozdzon, A.; O’Connor, D.P.; Brennan, D.J.; Connor, K.; et al. Peroxiredoxin-1 protects estrogen receptor α from oxidative stress-induced suppression and is a protein biomarker of favorable prognosis in breast cancer. Breast Cancer Res. 2014, 16, R79. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Bogner, P.N.; Ramnath, N.; Park, Y.; Yu, J.; Park, Y.M. Elevated peroxiredoxin 1, but not NF-E2-related factor 2, is an independent prognostic factor for disease recurrence and reduced survival in stage I non-small cell lung cancer. Clin. Cancer Res. 2007, 13, 3875–3882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trzeciecka, A.; Klossowski, S.; Bajor, M.; Zagozdzon, R.; Gaj, P.; Muchowicz, A.; Malinowska, A.; Czerwoniec, A.; Barankiewicz, J.; Domagala, A.; et al. Dimeric peroxiredoxins are druggable targets in human Burkitt lymphoma. Oncotarget 2016, 7, 1717–1731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, L.; Xiong, Y.; Wang, R.; Xiang, L.; Zhou, H.; Fu, Z. The critical role of peroxiredoxin-2 in colon cancer stem cells. Aging 2021, 13, 11170–11187. [Google Scholar] [CrossRef] [PubMed]
- Whitaker, H.C.; Patel, D.; Howat, W.J.; Warren, A.Y.; Kay, J.D.; Sangan, T.; Marioni, J.C.; Mitchell, J.; Aldridge, S.; Luxton, H.J.; et al. Peroxiredoxin-3 is overexpressed in prostate cancer and promotes cancer cell survival by protecting cells from oxidative stress. Br. J. Cancer 2013, 109, 983–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, N.; Xiao, M.B.; Ni, W.K.; Jiang, F.; Lu, C.H.; Ni, R.Z. High expression of peroxiredoxin 4 affects the survival time of colorectal cancer patients, but is not an independent unfavorable prognostic factor. Mol. Clin. Oncol. 2014, 2, 767–772. [Google Scholar] [CrossRef] [Green Version]
- Ahn, H.M.; Yoo, J.W.; Lee, S.; Lee, H.J.; Lee, H.S.; Lee, D.S. Peroxiredoxin 5 promotes the epithelial-mesenchymal transition in colon cancer. Biochem. Biophys. Res. Commun. 2017, 487, 580–586. [Google Scholar] [CrossRef]
- Hu, X.; Lu, E.; Pan, C.; Xu, Y.; Zhu, X. Overexpression and biological function of PRDX6 in human cervical cancer. J. Cancer 2020, 11, 2390–2400. [Google Scholar] [CrossRef]
- Liu, C.X.; Yin, Q.Q.; Zhou, H.C.; Wu, Y.L.; Pu, J.X.; Xia, L.; Liu, W.; Huang, X.; Jiang, T.; Wu, M.X.; et al. Adenanthin targets peroxiredoxin I and II to induce differentiation of leukemic cells. Nat. Chem. Biol. 2012, 8, 486–493. [Google Scholar] [CrossRef]
- Hou, J.K.; Huang, Y.; He, W.; Yan, Z.W.; Fan, L.; Liu, M.H.; Xiao, W.L.; Sun, H.D.; Chen, G.Q. Adenanthin targets peroxiredoxin I/II to kill hepatocellular carcinoma cells. Cell Death Dis. 2014, 5, e1400. [Google Scholar] [CrossRef] [Green Version]
- Haraldsen, J.D.; Liu, G.; Botting, C.H.; Walton, J.G.; Storm, J.; Phalen, T.J.; Kwok, L.Y.; Soldati-Favre, D.; Heintz, N.H.; Müller, S.; et al. Identification of conoidin A as a covalent inhibitor of peroxiredoxin II. Org. Biomol. Chem. 2009, 7, 3040–3048. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.H.; Jin, J.O.; Yu, K.J.; Kim, H.S.; Lee, P.C. Inhibition of peroxiredoxin 2 suppresses Wnt/β-catenin signaling in gastric cancer. Biochem. Biophys. Res. Commun. 2019, 512, 250–255. [Google Scholar] [CrossRef] [PubMed]
- Järvelä, S.; Rantala, I.; Rodriguez, A.; Kallio, H.; Parkkila, S.; Kinnula, V.L.; Soini, Y.; Haapasalo, H. Specific expression profile and prognostic significance of peroxiredoxins in grade II-IV astrocytic brain tumors. BMC Cancer 2010, 10, 104. [Google Scholar] [CrossRef] [Green Version]
- Odreman, F.; Vindigni, M.; Gonzales, M.L.; Niccolini, B.; Candiano, G.; Zanotti, B.; Skrap, M.; Pizzolitto, S.; Stanta, G.; Vindigni, A. Proteomic studies on low- and high-grade human brain astrocytomas. J. Proteome Res. 2005, 4, 698–708. [Google Scholar] [CrossRef] [PubMed]
- Dittmann, L.M.; Danner, A.; Gronych, J.; Wolter, M.; Stühler, K.; Grzendowski, M.; Becker, N.; Bageritz, J.; Goidts, V.; Toedt, G.; et al. Downregulation of PRDX1 by promoter hypermethylation is frequent in 1p/19q-deleted oligodendroglial tumours and increases radio- and chemosensitivity of Hs683 glioma cells in vitro. Oncogene 2012, 31, 3409–3418. [Google Scholar] [CrossRef] [Green Version]
- Wirthschaft, P.; Bode, J.; Simon, A.E.M.; Hoffmann, E.; van Laack, R.; Krüwel, T.; Dietrich, F.; Bucher, D.; Hahn, A.; Sahm, F.; et al. A PRDX1-p38α heterodimer amplifies MET-driven invasion of IDH-wildtype and IDH-mutant gliomas. Int. J. Cancer 2018, 143, 1176–1187. [Google Scholar] [CrossRef] [Green Version]
- Smith-Pearson, P.S.; Kooshki, M.; Spitz, D.R.; Poole, L.B.; Zhao, W.; Robbins, M.E. Decreasing peroxiredoxin II expression decreases glutathione, alters cell cycle distribution, and sensitizes glioma cells to ionizing radiation and H2O2. Free Radic. Biol. Med. 2008, 45, 1178–1189. [Google Scholar] [CrossRef] [Green Version]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Despotović, A.; Mirčić, A.; Misirlić-Denčić, S.; Harhaji-Trajković, L.; Trajković, V.; Zogović, N.; Tovilović-Kovačević, G. Combination of Ascorbic Acid and Menadione Induces Cytotoxic Autophagy in Human Glioblastoma Cells. Oxidative Med. Cell. Longev. 2022, 2022, 2998132. [Google Scholar] [CrossRef]
- Zhu, J.; May, S.; Ulrich, C.; Stockfleth, E.; Eberle, J. High ROS Production by Celecoxib and Enhanced Sensitivity for Death Ligand-Induced Apoptosis in Cutaneous SCC Cell Lines. Int. J. Mol. Sci. 2021, 22, 3622. [Google Scholar] [CrossRef]
- Szeliga, M. Comprehensive analysis of the expression levels and prognostic values of PRDX family genes in glioma. Neurochem. Int. 2022, 153, 105256. [Google Scholar] [CrossRef] [PubMed]
- Dahou, H.; Minati, M.A.; Jacquemin, P.; Assi, M. Genetic Inactivation of Peroxiredoxin-I Impairs the Growth of Human Pancreatic Cancer Cells. Antioxidants 2021, 10, 570. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wei, J.; Zhang, H.; Zheng, X.; Zhou, H.; Luo, Y.; Yang, J.; Deng, Q.; Huang, S.; Fu, Z. PRDX2 promotes the proliferation of colorectal cancer cells by increasing the ubiquitinated degradation of p53. Cell Death Dis. 2021, 12, 605. [Google Scholar] [CrossRef] [PubMed]
- Nakaso, K.; Kitayama, M.; Mizuta, E.; Fukuda, H.; Ishii, T.; Nakashima, K.; Yamada, K. Co-induction of heme oxygenase-1 and peroxiredoxin I in astrocytes and microglia around hemorrhagic region in the rat brain. Neurosci. Lett. 2000, 293, 49–52. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Liu, X.; Zhang, J.; Bao, G.; Xu, G.; Sun, Y.; Shen, Q.; Lian, M.; Huang, Y.; Cui, Z. Expression of Peroxiredoxin 1 After Traumatic Spinal Cord Injury in Rats. Cell. Mol. Neurobiol. 2015, 35, 1217–1226. [Google Scholar] [CrossRef]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010, 17, 98–110. [Google Scholar] [CrossRef] [Green Version]
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.M.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavaré, S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014. [Google Scholar] [CrossRef]
- Skaga, E.; Kulesskiy, E.; Fayzullin, A.; Sandberg, C.J.; Potdar, S.; Kyttälä, A.; Langmoen, I.A.; Laakso, A.; Gaál-Paavola, E.; Perola, M.; et al. Intertumoral heterogeneity in patient-specific drug sensitivities in treatment-naïve glioblastoma. BMC Cancer 2019, 19, 628. [Google Scholar] [CrossRef] [Green Version]
- Cifarelli, C.P.; Jacques, A.; Bobko, A. Heterogeneity of radiation response in mesenchymal subtype glioblastoma: Molecular profiling and reactive oxygen species generation. J. Neurooncol. 2021, 152, 245–255. [Google Scholar] [CrossRef]
- Caverzán, M.D.; Beaugé, L.; Chesta, C.A.; Palacios, R.E.; Ibarra, L.E. Photodynamic therapy of Glioblastoma cells using doped conjugated polymer nanoparticles: An in vitro comparative study based on redox status. J. Photochem. Photobiol. B 2020, 212, 112045. [Google Scholar] [CrossRef]
- Murnan, K.M.; Horbinski, C.; Stegh, A.H. Redox Homeostasis and Beyond: The Role of Wild-Type Isocitrate Dehydrogenases for the Pathogenesis of Glioblastoma. Antioxid. Redox Signal. 2023. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Fu, X.; Liu, Y.; Yu, D.; Cai, S.J.; Yang, C. Blockade of Glutathione Metabolism in IDH1-Mutated Glioma. Mol. Cancer Ther. 2020, 19, 221–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.; Gao, F.; Zheng, S.; Zhang, C.; Martinez-Ledesma, E.; Ezhilarasan, R.; Ding, J.; Li, X.; Feng, N.; Multani, A.; et al. EGFR Amplification Induces Increased DNA Damage Response and Renders Selective Sensitivity to Talazoparib (PARP Inhibitor) in Glioblastoma. Clin. Cancer Res. 2020, 26, 1395–1407. [Google Scholar] [CrossRef] [PubMed]
- Franken, N.A.; Rodermond, H.M.; Stap, J.; Haveman, J.; van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef]
- Khaitan, D.; Chandna, S.; Arya, M.B.; Dwarakanath, B.S. Establishment and characterization of multicellular spheroids from human glioma cell line; Implications for tumor therapy. J. Transl. Med. 2006, 4, 12. [Google Scholar] [CrossRef] [Green Version]
- Van Loenhout, J.; Freire Boullosa, L.; Quatannens, D.; De Waele, J.; Merlin, C.; Lambrechts, H.; Lau, H.W.; Hermans, C.; Lin, A.; Lardon, F.; et al. Auranofin and Cold Atmospheric Plasma Synergize to Trigger Distinct Cell Death Mechanisms and Immunogenic Responses in Glioblastoma. Cells 2021, 10, 2936. [Google Scholar] [CrossRef]
- Szeliga, M.; Rola, R. Menadione Potentiates Auranofin-Induced Glioblastoma Cell Death. Int. J. Mol. Sci. 2022, 23, 15712. [Google Scholar] [CrossRef]
- Feng, T.; Zhao, R.; Sun, F.; Lu, Q.; Wang, X.; Hu, J.; Wang, S.; Gao, L.; Zhou, Q.; Xiong, X.; et al. TXNDC9 regulates oxidative stress-induced androgen receptor signaling to promote prostate cancer progression. Oncogene 2020, 39, 356–367. [Google Scholar] [CrossRef]
- Tan, X.; Chen, J.; Keep, R.F.; Xi, G.; Hua, Y. Prx2 (Peroxiredoxin 2) as a Cause of Hydrocephalus After Intraventricular Hemorrhage. Stroke 2020, 51, 1578–1586. [Google Scholar] [CrossRef]
- Bian, L.; Zhang, J.; Wang, M.; Keep, R.F.; Xi, G.; Hua, Y. Intracerebral Hemorrhage-Induced Brain Injury in Rats: The Role of Extracellular Peroxiredoxin 2. Transl. Stroke Res. 2020, 11, 288–295. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szeliga, M.; Rola, R. Conoidin A, a Covalent Inhibitor of Peroxiredoxin 2, Reduces Growth of Glioblastoma Cells by Triggering ROS Production. Cells 2023, 12, 1934. https://doi.org/10.3390/cells12151934
Szeliga M, Rola R. Conoidin A, a Covalent Inhibitor of Peroxiredoxin 2, Reduces Growth of Glioblastoma Cells by Triggering ROS Production. Cells. 2023; 12(15):1934. https://doi.org/10.3390/cells12151934
Chicago/Turabian StyleSzeliga, Monika, and Radosław Rola. 2023. "Conoidin A, a Covalent Inhibitor of Peroxiredoxin 2, Reduces Growth of Glioblastoma Cells by Triggering ROS Production" Cells 12, no. 15: 1934. https://doi.org/10.3390/cells12151934