
Ubiquitination-Proteasome System (UPS) and Autophagy Two Main Protein Degradation Machineries in Response to Cell Stress

Abstract
:1. Introduction
2. Ubiquitin Pool
3. UPS
3.1. Mechanism of UPS
3.2. Molecular Chaperones and Ubiquitin E3 Ligases Involved in Cell
4. Autophagy
4.1. Mechanisms of Autophagy
4.2. Cargo Receptors of Autophagy
4.3. UPR and Autophagy
4.4. Chaperone-Mediated Autophagy
5. Crosstalk between UPS and Autophagy
5.1. UPR Mediates Crosstalk between UPS and Autophagy
5.2. Proteasome Inhibitors Regulates Proteins Involved in Autophagy
5.3. Inhibition of Autophagy Affects UPS
6. UPS and Autophagy in Disease
6.1. Neurodegenerative Disease
6.2. Cancers
6.2.1. UPR and Cancers
6.2.2. UPS and Cancers
6.2.3. Autophagy and Cancer
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Chen, X.; Yin, X.M. Coordination of Autophagy and the Proteasome in Resolving Endoplasmic Reticulum Stress. Vet. Pathol. 2011, 48, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Qiu, X. Ubiquitin at the crossroad of cell death and survival. Chin. J. Cancer 2013, 32, 640–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, J.; Qi, L. Quality Control in the Endoplasmic Reticulum: Crosstalk between ERAD and UPR pathways. Trends Biochem. Sci. 2018, 43, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Tan, J.; Miao, Y.; Zhang, Q. Crosstalk of ER stress-mediated autophagy and ER-phagy: Involvement of UPR and the core autophagy machinery. J. Cell. Physiol. 2018, 233, 3867–3874. [Google Scholar] [CrossRef]
- Qu, J.; Zou, T.; Lin, Z. The Roles of the Ubiquitin–Proteasome System in the Endoplasmic Reticulum Stress Pathway. Int. J. Mol. Sci. 2021, 22, 1526. [Google Scholar] [CrossRef]
- Lindholm, D.; Korhonen, L.; Eriksson, O.; Kõks, S. Recent Insights into the Role of Unfolded Protein Response in ER Stress in Health and Disease. Front. Cell Dev. Biol. 2017, 5, 48–63. [Google Scholar] [CrossRef] [Green Version]
- B’chir, W.; Maurin, A.-C.; Carraro, V.; Averous, J.; Jousse, C.; Muranishi, Y.; Parry, L.; Stepien, G.; Fafournoux, P.; Bruhat, A. The eIF2α/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 2013, 41, 7683–7699. [Google Scholar] [CrossRef] [Green Version]
- Reiss, Y.; Gur, E.; Ravid, T. Releasing the Lockdown: An Emerging Role for the Ubiquitin-Proteasome System in the Breakdown of Transient Protein Inclusions. Biomolecules 2020, 10, 1168. [Google Scholar] [CrossRef]
- Ding, W.; Yin, X. Sorting, recognition and activation of the misfolded protein degradation pathways through macroautophagy and the proteasome. Autophagy 2008, 4, 141–150. [Google Scholar] [CrossRef] [Green Version]
- Ji, C.H.; Kwon, Y.T. Crosstalk and Interplay between the Ubiquitin-Proteasome System and Autophagy. Mol. Cells 2017, 40, 441–449. [Google Scholar]
- Akutsu, M.; Dikic, I.; Bremm, A. Ubiquitin chain diversity at a glance. Cell Sci. Glance 2016, 129, 875–880. [Google Scholar] [CrossRef] [Green Version]
- Dantuma, N.P.; Lindsten, K. Stressing the ubiquitin-proteasome system. Cardiovasc. Res. 2009, 85, 263–271. [Google Scholar] [CrossRef] [Green Version]
- Kubota, H. Quality Control against Misfolded Proteins in the Cytosol: A Network for Cell Survival. J. Biochem. 2009, 146, 609–616. [Google Scholar] [CrossRef]
- Ryu, H.; Ryu, K. Quantification of oxidative stress in live mouse embryonic fibroblasts by monitoring the responses of polyubiquitin genes. Biochem. Biophys. Res. Commun. 2011, 404, 470–475. [Google Scholar] [CrossRef]
- Marinovic, A.C.; Zheng, B.; Mitch, W.E.; Price, S.R. Ubiquitin (UbC) Expression in Muscle Cells Is Increased by Glucocorticoids through a Mechanism Involving Sp1 and MEK1. J. Biol. Chem. 2002, 277, 16673–16681. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.; Ryu, K.-Y. Effect of cellular ubiquitin levels on the regulation of oxidative stress response and proteasome function via Nrf1. Biochem. Biophys. Res. Commun. 2017, 2, 234–240. [Google Scholar] [CrossRef]
- Kim, M.-N.; Choi, J.; Ryu, H.-W.; Ryu, K.-Y. Disruption of polyubiquitin gene UbC leads to attenuated resistance against arsenite-induced toxicity in mouse embryonic fibroblasts. Biochim. Biophys. Acta 2015, 1853, 996–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianchi, M.; Crinelli, R.; Arbore, V.; Magnani, M. Induction of ubiquitin C (UbC) gene transcription is mediated by HSF1: Role of proteotoxic and oxidative stress. FEBS Open Bio 2018, 8, 1471–1485. [Google Scholar] [CrossRef] [Green Version]
- Ryu, K.-Y.; Maehr, R.; Gilchrist, C.A.; Long, M.A.; Bouley, D.M.; Mueller, B.; Ploegh, H.L.; Kopito, R.R. The mouse polyubiquitin gene UbC is essential for fetal liver development, cell-cycle progression and stress tolerance. EMBO J. 2007, 26, 2693–2706. [Google Scholar] [CrossRef]
- Höhfeld, J.; Cyr, D.M.; Patterson, C. From the cradle to the grave: Molecular chaperones that may choose between folding and degradation. EMBO Rep. 2001, 2, 885–890. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.A.; Jeon, Y.J. How Is the Fidelity of Proteins Ensured in Terms of Both Quality and Quantity at the Endoplasmic Reticulum Mechanistic Insights into E3 Ubiquitin Ligases. Int. J. Mol. Sci. 2021, 22, 2078. [Google Scholar] [CrossRef] [PubMed]
- Fang, S.; Ferrone, M.; Yang, C.; Jensen, J.P.; Tiwari, S.; Weissman, A.M. The Tumor Autocrine Motility Factor Receptor, gp78, is a Ubiquitin Protein Ligase Implicated in Degradation from the Endoplasmic Reticulum. Proc. Natl. Acad. Sci. USA 2001, 98, 14422–14427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballar, P.; Ors, A.U.; Yang, H.; Fang, S. Differential regulation of CFTRDeltaF508 degradation by ubiquitin ligases gp78 and Hrd1. Int. J. Biochem. Cell. Biol. 2010, 1, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.-F.; Tang, J.-J.; Li, P.-S.; Shen, Y.; Li, J.-G.; Miao, H.-H.; Li, B.-L.; Song, B.-L. Ablation of gp78 in liver improves hyperlipidemia and insulin resistance by inhibiting SREBP to decrease lipid biosynthesis. Cell Metab. 2012, 2, 213–225. [Google Scholar] [CrossRef] [Green Version]
- Omura, T.; Kaneko, M.; Okuma, Y.; Orba, Y.; Nagashima, K.; Takahashi, R.; Fujitani, N.; Matsumura, S.; Hata, A.; Kubota, K.; et al. A ubiquitin ligase HRD1 promotes the degradation of Pael receptor, a substrate of Parkin. J. Neurochem. 2006, 99, 1456–1469. [Google Scholar] [CrossRef] [Green Version]
- Ron, I.; Rapaport, D.; Horowitz, M. Interaction between parkin and mutant glucocerebrosidase variants: A possible link between Parkinson disease and Gaucher disease. Hum. Mol. Genet. 2010, 19, 3771–3781. [Google Scholar] [CrossRef] [Green Version]
- Lips, C.; Ritterhoff, T.; Weber, A.; Janowska, M.K.; Mustroph, M.; Sommer, T.; Klevit, R.E. Who with whom: Functional coordination of E2 enzymes by RING E3 ligases during poly-ubiquitylation. EMBO J. 2020, 39, e104863. [Google Scholar] [CrossRef]
- Gardner, R.G.; Shearer, A.G.; Hampton, R.Y. In Vivo Action of the HRD Ubiquitin Ligase Complex: Mechanisms of Endoplasmic Reticulum Quality Control and Sterol Regulation. Mol. Cell. Biol. 2001, 21, 4276–4291. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Qiu, Q.; Gao, B.; Kong, S.; Lin, Z.; Fang, D. Hrd1-mediated BLIMP-1 ubiquitination promotes dendritic cell MHCII expression for CD4 T cell priming during inflammation. J. Exp. Med. 2014, 211, 2467–2479. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Zhang, B.; Pan, X.; Huang, H.; Xie, Z.; Ma, Y.; Hu, B.; Wang, J.; Chen, Z.; Shi, P. Octyl itaconate inhibits osteoclastogenesis by suppressing Hrd1 and activating Nrf2 signaling. FASEB J. 2019, 33, 12929–12940. [Google Scholar] [CrossRef] [Green Version]
- Fujita, H.; Yagishita, N.; Aratani, S.; Saito-Fujita, T.; Morota, S.; Yamano, Y.; Hansson, M.J.; Inazu, M.; Kokuba, H.; Sudo, K.; et al. The E3 ligase synoviolin controls body weight and mitochondrial biogenesis through negative regulation of PGC-1β. EMBO J. 2015, 34, 1042–1055. [Google Scholar] [CrossRef] [Green Version]
- Shrestha, N.; Liu, T.; Ji, Y.; Reinert, R.B.; Torres, M.; Li, X.; Zhang, M.; Tang, C.-H.A.; Hu, C.-C.A.; Liu, C.; et al. Sel1L-Hrd1 ER-associated degradation maintains β cell identity via TGF-β signaling. J. Clin. Investig. 2020, 130, 3499–3510. [Google Scholar] [CrossRef]
- Kikkert, M.; Doolman, R.; Dai, M.; Avner, R.; Hassink, G.; van Voorden, S.; Thanedar, S.; Roitelman, J.; Chau, V.; Wiertz, E. Human HRD1 Is an E3 Ubiquitin Ligase Involved in Degradation of Proteins from the Endoplasmic Reticulum. J. Biol. Chem. 2004, 279, 3525–3534. [Google Scholar] [CrossRef] [Green Version]
- Christianson, J.C.; Shaler, T.A.; Tyler, R.E.; Kopito, R.R. OS-9 and GRP94 deliver mutant α1-antitrypsin to the Hrd1–SEL1L ubiquitin ligase complex for ERAD. Nat. Cell Biol. 2008, 10, 272–282. [Google Scholar] [CrossRef] [Green Version]
- Yamasaki, S.; Yagishita, N.; Sasaki, T.; Nakazawa, M.; Kato, Y.; Yamadera, T.; Bae, E.; Toriyama, S.; Ikeda, R.; Zhang, L.; et al. Cytoplasmic destruction of p53 by the endoplasmic reticulum-resident ubiquitin ligase ‘Synoviolin’. EMBO J. 2007, 26, 113–122. [Google Scholar] [CrossRef]
- Kaneko, M.; Ishiguro, M.; Niinuma, Y.; Uesugi, M.; Nomura, Y. Human HRD1 protects against ER stress-induced apoptosis through ER-associated degradation1. FEBS Lett. 2002, 532, 147–152. [Google Scholar] [CrossRef] [Green Version]
- Moon, H.W.; Han, H.G.; Jeon, Y.J. Protein Quality Control in the Endoplasmic Reticulum and Cancer. Int. J. Mol. Sci. 2018, 19, 3020. [Google Scholar] [CrossRef] [Green Version]
- Scott, N.A.; Sharpe, L.J.; Brown, A.J. The E3 ubiquitin ligase MARCHF6 as a metabolic integrator in cholesterol synthesis and beyond. Biochim. et Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2020, 1866, 158837. [Google Scholar] [CrossRef]
- Hassink, G.; Kikkert, M.; Van Voorden, S.; Lee, S.-J.; Spaapen, R.; van Laar, T.; Coleman, C.S.; Bartee, E.; Früh, K.; Chau, V.; et al. TEB4 is a C4HC3 RING finger-containing ubiquitin ligase of the endoplasmic reticulum. Biochem. J. 2005, 388, 647–655. [Google Scholar] [CrossRef] [Green Version]
- Tan, J.M.; Van Der Stoel, M.M.; Berg, M.V.D.; Van Loon, N.M.; Moeton, M.; Scholl, E.; Van Der Wel, N.N.; Kovačević, I.; Hordijk, P.L.; Loregger, A.; et al. The MARCH6-SQLE Axis Controls Endothelial Cholesterol Homeostasis and Angiogenic Sprouting. Cell Rep. 2020, 32, 107944. [Google Scholar] [CrossRef]
- Gao, Y.; Xuan, C.; Jin, M.; An, Q.; Zhuo, B.; Chen, X.; Wang, L.; Wang, Y.; Sun, Q.; Shi, Y. Ubiquitin ligase RNF5 serves an important role in the development of human glioma. Oncol. Lett. 2019, 18, 4659–4666. [Google Scholar] [CrossRef] [Green Version]
- Roy, S.J.; Glazkova, I.; Fréchette, L.; Iorio-Morin, C.; Binda, C.; Pétrin, D.; Trieu, P.; Robitaille, M.; Angers, S.; Hébert, T.E.; et al. Novel, Gel-free Proteomics Approach Identifies RNF5 and JAMP as Modulators of GPCR Stability. Mol. Endocrinol. 2013, 27, 1245–1266. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.-C.; Fu, S.-J.; Hsu, P.-H.; Chang, P.-T.; Huang, J.-J.; Chiu, Y.-C.; Liao, Y.-F.; Jow, G.-M.; Tang, C.-Y.; Jeng, C.-J. Identification of MKRN1 as a second E3 ligase for Eag1 potassium channels reveals regulation via differential degradation. J. Biol. Chem. 2021, 296, 100484. [Google Scholar] [CrossRef]
- Gong, B.; Chen, F.; Pan, Y.; Arrieta-Cruz, I.; Yoshida, Y.; Haroutunian, V.; Pasinetti, G.M. SCFFbx2-E3-ligase-mediated degradation of BACE1 attenuates Alzheimer’s disease amyloidosis and improves synaptic function. Aging Cell 2010, 9, 1018–1031. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, Y.; Chiba, T.; Tokunaga, F.; Kawasaki, H.; Iwai, K.; Suzuki, T.; Ito, Y.; Matsuoka, K.; Yoshida, M.; Tanaka, K.; et al. E3 ubiquitin ligase that recognizes sugar chains. Nature 2002, 418, 438–442. [Google Scholar] [CrossRef]
- Zhang, Y.-W.; Brognard, J.; Coughlin, C.; You, Z.; Dolled-Filhart, M.; Aslanian, A.; Manning, G.; Abraham, R.T.; Hunter, T.; Zhang, Y.-W.; et al. The F Box Protein Fbx6 Regulates Chk1 Stability and Cellular Sensitivity to Replication Stress. Mol. Cell 2009, 35, 442–453. [Google Scholar] [CrossRef] [Green Version]
- Hill, M.S.; Ruiz, A.; Schmitt, K.; Stephens, E.B. Identification of amino acids within the second alpha helical domain of the human immunodeficiency virus type 1 Vpu that are critical for preventing CD4 cell surface expression. Virology 2009, 397, 104–112. [Google Scholar] [CrossRef] [Green Version]
- Tervo, H.M.; Homann, S.; Ambiel, I.; Fritz, J.V.; Fackler, O.T.; Keppler, O.T. β-TrCP is dispensable for Vpu’s ability to overcome the CD317/Tetherin-imposed restriction to HIV-1 release. Retrovirology 2011, 8, 9. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Zhang, J.; Sun, H.; Jiang, C.; Dong, Y.; Shan, Q.; Su, S.; Xie, Y.; Xu, N.; Lou, X.; et al. Ubiquitination of Inositol-requiring Enzyme 1 (IRE1) by the E3 Ligase CHIP Mediates the IRE1/TRAF2/JNK Pathway. J. Biol. Chem. 2014, 289, 30567–30577. [Google Scholar] [CrossRef] [Green Version]
- Seo, H.-R.; Jeong, D.; Lee, S.; Lee, H.-S.; Lee, S.-A.; Kang, S.W.; Kwon, A.J. CHIP and BAP1 Act in Concert to Regulate INO80 Ubiquitination and Stability for DNA Replication. Mol. Cells 2021, 44, 101–115. [Google Scholar] [CrossRef]
- Dickey, C.A.; Koren, J.; Zhang, Y.-J.; Xu, Y.-F.; Jinwal, U.K.; Birnbaum, M.; Monks, B.; Sun, M.; Cheng, J.Q.; Patterson, C.; et al. Akt and CHIP coregulate tau degradation through coordinated interactions. Proc. Natl. Acad. Sci. USA 2008, 105, 3622–3627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Shen, S.; Song, S.; He, S.; Cui, Y.; Xing, G.; Wang, J.; Yin, Y.; Fan, L.; He, F.; et al. The E3 Ligase Smurf1 Regulates Wolfram Syndrome Protein Stability at the Endoplasmic Reticulum. J. Biol. Chem. 2011, 286, 18037–18047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, Q.; Zhang, H.; Zhang, P.; Li, Y.; Xu, M.; Li, X.; Li, X.; Dong, L. Oncogenic Smurf1 promotes PTEN wild-type glioblastoma growth by mediating PTEN ubiquitylation. Oncogene 2020, 36, 5902–5915. [Google Scholar] [CrossRef] [PubMed]
- Yen, L.; Cao, Z.; Wu, X.; Ingalla, E.R.Q.; Baron, C.; Young, L.J.T.; Gregg, J.P.; Cardiff, R.D.; Borowsky, A.D.; Sweeney, C.; et al. Loss of Nrdp1 enhances ErbB2/ErbB3-dependent breast tumor cell growth. Cancer Res. 2006, 23, 11279–11286. [Google Scholar] [CrossRef] [Green Version]
- Puissant, A.; Fenouille, N.; Auberger, P. When autophagy meets cancer through p62/SQSTM1. Am. J. Cancer Res. 2012, 2, 397–413. [Google Scholar]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef]
- Rashid, H.-O.; Yadav, R.K.; Kim, H.-R.; Chae, H.-J. ER stress: Autophagy induction, inhibition and selection. Autophagy 2015, 11, 1956–1977. [Google Scholar] [CrossRef]
- Zhao, J.; Zhai, B.; Gygi, S.P.; Goldberg, A.L. mTOR inhibition activates overall protein degradation by the ubiquitin proteasome system as well as by autophagy. Proc. Natl. Acad. Sci. USA 2015, 112, 15790–15797. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; He, S.; Ma, B. Autophagy and autophagy-related proteins in cancer. Mol. Cancer 2020, 19, 1–16. [Google Scholar] [CrossRef]
- Shang, L.; Chen, S.; Du, F.; Li, S.; Zhao, L.; Wang, X. Nutrient starvation elicits an acute autophagic response mediated by Ulk1 dephosphorylation and its subsequent dissociation from AMPK. Proc. Natl. Acad. Sci. USA 2011, 108, 4788–4793. [Google Scholar] [CrossRef] [Green Version]
- Turco, E.; Savova, A.; Gere, F.; Ferrari, L.; Romanov, J.; Schuschnig, M.; Martens, S. Reconstitution defines the roles of p62, NBR1 and TAX1BP1 in ubiquitin condensate formation and autophagy initiation. Nat. Commun. 2021, 12, 1–16. [Google Scholar] [CrossRef]
- Kocaturk, N.M.; Gozuacik, D. Crosstalk between Mammalian Autophagy and the Ubiquitin-Proteasome System. Front. Cell Dev. Biol. 2018, 6, 128–154. [Google Scholar] [CrossRef]
- Duran, A.; Amanchy, R.; Linares, J.F.; Joshi, J.; Abu-Baker, S.; Porollo, A.; Hansen, M.; Moscat, J.; Diaz-Meco, M.T. p62 Is a Key Regulator of Nutrient Sensing in the mTORC1 Pathway. Mol. Cell 2011, 44, 134–146. [Google Scholar] [CrossRef] [Green Version]
- Linares, J.F.; Duran, A.; Yajima, T.; Pasparakis, M.; Moscat, J.; Diaz-Meco, M.T. K63 Polyubiquitination and Activation of mTOR by the p62-TRAF6 Complex in Nutrient-Activated Cells. Mol. Cell 2013, 51, 283–296. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Qi, Q.; Zhou, W.; Feng, Z.; Huang, B.; Chen, A.; Zhang, D.; Li, W.; Zhang, Q.; Jiang, Z.; et al. Inhibition of glioma growth by flavokawain B is mediated through endoplasmic reticulum stress induced autophagy. Autophagy 2018, 14, 2007–2022. [Google Scholar] [CrossRef] [Green Version]
- García-Navas, R.; Munder, M.; Mollinedo, F. Depletion ofL-arginine induces autophagy as a cytoprotective response to endoplasmic reticulum stress in human T lymphocytes. Autophagy 2014, 8, 1557–1576. [Google Scholar] [CrossRef] [Green Version]
- He, H.; Du, Z.; Lin, J.; Wu, W.; Yu, Y. DUSP4 inhibits autophagic cell death in PTC by inhibiting JNK-BCL2-Beclin1 signaling. Biochem. Cell. Biol. 2021, 5, 554–561. [Google Scholar] [CrossRef]
- Liu, F.; Chang, L.; Hu, J. Activating transcription factor 6 regulated cell growth, migration and inhibiteds cell apoptosis and autophagy via MAPK pathway in cervical cancer. J. Reprod. Immunol. 2020, 139, 103120. [Google Scholar] [CrossRef]
- Rouschop, K.M.; Beucken, T.V.D.; Dubois, L.; Niessen, H.; Bussink, J.; Savelkouls, K.; Keulers, T.; Mujcic, H.; Landuyt, W.; Voncken, J.W.; et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J. Clin. Investig. 2010, 120, 120–127. [Google Scholar] [CrossRef]
- Margariti, A.; Li, H.; Chen, T.; Martin, D.; Vizcay-Barrena, G.; Alam, S.; Karamariti, E.; Xiao, Q.; Zampetaki, A.; Zhang, Z.; et al. XBP1 mRNA Splicing Triggers an Autophagic Response in Endothelial Cells through BECLIN-1 Transcriptional Activation. J. Biol. Chem. 2013, 288, 859–872. [Google Scholar] [CrossRef] [Green Version]
- Nie, T.; Tao, K.; Zhu, L.; Huang, L.; Hu, S.; Yang, R.; Xu, P.; Mao, Z.; Yang, Q. Chaperone-mediated autophagy controls the turnover of E3 ubiquitin ligase MARCHF5 and regulates mitochondrial dynamics. Autophagy 2021, 17, 2923–2938. [Google Scholar] [CrossRef]
- Dong, S.; Wang, Q.; Kao, Y.-R.; Diaz, A.; Tasset, I.; Kaushik, S.; Thiruthuvanathan, V.; Zintiridou, A.; Nieves, E.; Dzieciatkowska, M.; et al. Chaperone-mediated autophagy sustains haematopoietic stem-cell function. Nature 2021, 591, 117–123. [Google Scholar] [CrossRef]
- Wang, E.; Yu, J.; Wong, S.H.; Cheng, A.; Chan, F.K.; Ng, S.S.M.; Cho, C.H.; Sung, J.J.Y.; Wu, W.K. A novel crosstalk between two major protein degradation systems. Autophagy 2013, 10, 1500–1508. [Google Scholar] [CrossRef]
- Wilde, I.B.; Brack, M.; Winget, J.M.; Mayor, T. Proteomic Characterization of Aggregating Proteins after the Inhibition of the Ubiquitin Proteasome System. J. Proteome Res. 2011, 10, 1062–1072. [Google Scholar] [CrossRef]
- Wojcik, S. Crosstalk between autophagy and proteasome protein degradation systems: Possible implications for cancer therapy. Folia Histochem. Et Cytobiol. 2014, 51, 249–264. [Google Scholar] [CrossRef] [Green Version]
- Ding, W.-X.; Ni, H.-M.; Gao, W.; Yoshimori, T.; Stolz, D.B.; Ron, D.; Yin, X.-M. Linking of Autophagy to Ubiquitin-Proteasome System Is Important for the Regulation of Endoplasmic Reticulum Stress and Cell Viability. Am. J. Pathol. 2007, 171, 513–524. [Google Scholar] [CrossRef] [Green Version]
- Luan, Q.; Jin, L.; Jiang, C.C.; Tay, K.H.; Lai, F.; Liu, X.Y.; Liu, Y.L.; Guo, S.T.; Li, C.Y.; Yan, X.G.; et al. RIPK1 regulates survival of human melanoma cells upon endoplasmic reticulum stress through autophagy. Autophagy 2015, 11, 975–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardner, B.M.; Walter, P. Unfolded proteins are Ire1-activating ligands that directly induce the unfolded protein response. Science 2011, 6051, 1891–1894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ron, D.; Hubbard, S.R. How IRE1 reacts to ER stress. Cell 2008, 1, 24–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gade, P.; Manjegowda, S.B.; Nallar, S.C.; Maachani, U.B.; Cross, A.S.; Kalvakolanu, D.V. Regulation of the Death-Associated Protein Kinase 1 Expression and Autophagy via ATF6 Requires Apoptosis Signal-Regulating Kinase 1. Mol. Cell. Biol. 2014, 34, 4033–4048. [Google Scholar] [CrossRef] [Green Version]
- Milani, M.; Rzymski, T.; Mellor, H.R.; Pike, L.; Bottini, A.; Generali, D.; Harris, A. The Role of ATF4 Stabilization and Autophagy in Resistance of Breast Cancer Cells Treated with Bortezomib. Cancer Res. 2009, 69, 4415–4423. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.K.K.; Wu, Y.C.; Yu, L.; Li, Z.J.; Sung, J.J.Y.; Cho, C.H. Induction of autophagy by proteasome inhibitor is associated with proliferative arrest in colon cancer cells. Biochem. Biophys. Res. Commun. 2008, 2, 258–263. [Google Scholar] [CrossRef]
- Wu, W.K.K.; Volta, V.; Cho, C.H.; Wu, Y.C.; Li, H.T.; Yu, L.; Li, Z.J.; Sung, J.J.Y. Repression of protein translation and mTOR signaling by proteasome inhibitor in colon cancer cells. Biochem. Biophys. Res. Commun. 2009, 4, 598–601. [Google Scholar] [CrossRef]
- Wu, W.K.K.; Cho, C.H.; Lee, C.W.; Wu, K.; Fan, D.; Yu, J.; Sung, J.J.Y. Proteasome inhibition: A new therapeutic strategy to cancer treatment. Cancer Lett. 2010, 1, 15–22. [Google Scholar] [CrossRef]
- Gao, Z.; Gammoh, N.; Wong, P.-M.; Erdjument-Bromage, H.; Tempst, P.; Jiang, X. Processing of autophagic protein LC3 by the 20S proteasome. Autophagy 2010, 1, 126–137. [Google Scholar] [CrossRef] [Green Version]
- Choe, J.-Y.; Jung, H.-Y.; Park, K.-Y.; Kim, S.-K. Enhanced p62 expression through impaired proteasomal degradation is involved in caspase-1 activation in monosodium urate crystal-induced interleukin-1 expression. Rheumatology 2014, 53, 1043–1053. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.J.; Ye, L.; Huang, W.F.; Guo, L.J.; Xu, Z.G.; Wu, H.L.; Yang, C.; Liu, H.F. p62 links the autophagy pathway and the ubiqutin–proteasome system upon ubiquitinated protein degradation. Cell. Mol. Biol. Lett. 2016, 21, 29. [Google Scholar] [CrossRef] [Green Version]
- Fusco, C.; Micale, L.; Egorov, M.; Monti, M.; D’Addetta, E.V.; Augello, B.; Cozzolino, F.; Calcagnì, A.; Fontana, A.; Polishchuk, R.S.; et al. The E3-ubiquitin ligase TRIM50 interacts with HDAC6 and p62, and promotes the sequestration and clearance of ubiquitinated proteins into the aggresome. PLoS ONE 2012, 7, e40440. [Google Scholar] [CrossRef] [Green Version]
- Jain, A.; Lamark, T.; Sjøttem, E.; Bowitz Larsen, K.; Atesoh Awuh, J.; Øvervatn, A.; McMahon, M.; Hayes, J.D.; Johansen, T. p62/SQSTM1 Is a Target Gene for Transcription Factor NRF2 and Creates a Positive Feedback Loop by Inducing Antioxidant Response Element-driven Gene Transcription. J. Biol. Chem. 2010, 285, 22576–22591. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.; Lachenmayer, M.L.; Wu, S.; Liu, W.; Kundu, M.; Wang, R.; Komatsu, M.; Oh, Y.J.; Zhao, Y.; Yue, Z. Proteotoxic Stress Induces Phosphorylation of p62/SQSTM1 by ULK1 to Regulate Selective Autophagic Clearance of Protein Aggregates. PLoS Genet. 2015, 11, e1004987. [Google Scholar] [CrossRef] [PubMed]
- Kulka, L.A.M.; Fangmann, P.-V.; Panfilova, D.; Olzscha, H. Impact of HDAC Inhibitors on Protein Quality Control Systems: Consequences for Precision Medicine in Malignant Disease. Front. Cell Dev. Biol. 2020, 8, 425. [Google Scholar] [CrossRef]
- Wu, W.K.; Sakamoto, K.M.; Milani, M.; Aldana-Masankgay, G.; Fan, D.; Wu, K.; Lee, C.W.; Cho, C.H.; Yu, J.; Sung, J.J. Macroautophagy modulates cellular response to proteasome inhibitors in cancer therapy. Drug Resist. Updates 2010, 13, 87–92. [Google Scholar] [CrossRef]
- Kawaguchi, Y.; Kovacs, J.J.; McLaurin, A.; Vance, J.M.; Ito, A.; Yao, T.-P. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 2003, 115, 727–738. [Google Scholar] [CrossRef] [Green Version]
- Behl, C. BAG3 and friends: Co-chaperones in selective autophagy during aging and disease. Autophagy 2011, 7, 795–798. [Google Scholar] [CrossRef]
- Gamerdinger, M.; Hajieva, P.; Kaya, A.M.; Wolfrum, U.; Hartl, F.U.; Behl, C. Protein quality control during aging involves recruitment of the macroautophagy pathway by BAG3. EMBO J. 2009, 28, 889–901. [Google Scholar] [CrossRef] [Green Version]
- Cohen-Kaplan, V.; Livneh, I.; Avni, N.; Cohen-Rosenzweig, C.; Ciechanover, A. The ubiquitin-proteasome system and autophagy: Coordinated and independent activities. Int. J. Biochem. Cell Biol. 2016, 79, 403–418. [Google Scholar] [CrossRef]
- Korolchuk, V.; Mansilla, A.; Menzies, F.M.; Rubinsztein, D.C. Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol. Cell 2009, 4, 517–527. [Google Scholar] [CrossRef]
- Takahashi, M.; Kitaura, H.; Kakita, A.; Kakihana, T.; Katsuragi, Y.; Nameta, M.; Zhang, L.; Iwakura, Y.; Nawa, H.; Higuchi, M.; et al. USP10 Is a Driver of Ubiquitinated Protein Aggregation and Aggresome Formation to Inhibit Apoptosis. iScience 2018, 9, 433–450. [Google Scholar] [CrossRef] [Green Version]
- Alves da Costa, C.; El Manaa, W.; Duplan, E.; Checler, F. The Endoplasmic Reticulum Stress/Unfolded Protein Response and Their Contributions to Parkinson’s Disease Physiopathology. Cells 2020, 9, 2495. [Google Scholar] [CrossRef]
- Du, X.-Y.; Xie, X.-X.; Liu, R.-T. The Role of α-Synuclein Oligomers in Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 8645. [Google Scholar] [CrossRef] [PubMed]
- Colla, E. Linking the Endoplasmic Reticulum to Parkinson’s Disease and Alpha-Synucleinopathy. Front. Neurosci. 2019, 13, 560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, S.E.; Hallett, P.J.; Moens, T.; Smith, G.; Mangano, E.; Kim, H.T.; Goldberg, A.L.; Liu, J.-L.; Isacson, O.; Tofaris, G.K. Enhanced ubiquitin-dependent degradation by Nedd4 protects against α-synuclein accumulation and toxicity in animal models of Parkinson’s disease. Neurobiol. Dis. 2014, 100, 79–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wasner, K.; Grünewald, A.; Klein, C. Parkin-linked Parkinson’s disease: From clinical insights to pathogenic mechanisms and novel therapeutic approaches. Neurosci. Res. 2020, 159, 34–39. [Google Scholar] [CrossRef]
- Brahmachari, S.; Lee, S.; Kim, S.; Yuan, C.; Karuppagounder, S.S.; Ge, P.; Shi, R.; Kim, E.J.; Liu, A.; Kim, D.; et al. Parkin interacting substrate zinc finger protein 746 is a pathological mediator in Parkinson’s disease. Brian 2019, 8, 2380–2401. [Google Scholar] [CrossRef]
- Ding, D.; Ao, X.; Liu, Y.; Wang, Y.-Y.; Fa, H.-G.; Wang, M.-Y.; He, Y.-Q.; Wang, J.-X. Post-translational modification of Parkin and its research progress in cancer. Cancer Commun. 2019, 1, 77. [Google Scholar] [CrossRef] [Green Version]
- Ashkenazi, A.; Bento, C.F.; Ricketts, T.; Vicinanza, M.; Siddiqi, F.; Pavel, M.; Squitieri, F.; Hardenberg, M.; Imarisio, S.; Menzies, F.M.; et al. Polyglutamine tracts regulate beclin 1-dependent autophagy. Nature 2017, 7652, 108–111. [Google Scholar] [CrossRef] [Green Version]
- Jin, S.; Tian, S.; Chen, Y.; Zhang, C.; Xie, W.; Xia, X.; Cui, J.; Wang, R. USP19 modulates autophagy and antiviral immune responses by deubiquitinating Beclin-1. EMBO J. 2016, 8, 866–880. [Google Scholar] [CrossRef] [Green Version]
- Budzik, J.M.; Swaney, D.L.; Jimenez-Morales, D.; Johnson, J.R.; Garelis, N.E.; Repasy, T.; Roberts, A.W.; Popov, L.M.; Parry, T.J.; Pratt, D.; et al. Dynamic post-translational modification profiling of Mycobacterium tuberculosisinfected primary macrophages. eLife 2020, 9, e51461. [Google Scholar] [CrossRef]
- Wang, M.; Kaufman, R.J. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 2016, 529, 326–335. [Google Scholar] [CrossRef]
- Lin, Y.; Jiang, M.; Chen, W.; Zhao, T.; Wei, Y. Cancer and ER stress: Mutual crosstalk between autophagy, oxidative stress and inflammatory response. Biomed. Pharmacother. 2019, 118, 109249–109258. [Google Scholar] [CrossRef]
- Wang, P.; Han, L.; Yu, M.; Cao, Z.; Li, X.; Shao, Y.; Zhu, G. The Prognostic Value of PERK in Cancer and Its Relationship with Immune Cell Infiltration. Front. Mol. Biosci. 2021, 8, 648752–648765. [Google Scholar] [CrossRef]
- Iwawaki, T.; Akai, R.; Yamanaka, S.; Kohno, K. Function of IRE1 Alpha in the Placenta Is Essential for Placental Development and Embryonic Viability. Proc. Natl. Acad. Sci. USA 2009, 106, 16657–16662. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Alam, G.N.; Ning, Y.; Visioli, F.; Dong, Z.; Nör, J.E.; Polverini, P.J. The Unfolded Protein Response Induces the Angiogenic Switch in Human Tumor Cells through the PERK/ATF4 Pathway. Cancer Res. 2012, 72, 5396–5406. [Google Scholar] [CrossRef] [Green Version]
- Coker-Gurkan, A.; Ayhan-Sahin, B.; Keceloglu, G.; Obakan-Yerlikaya, P.; Arisan, E.-D.; Palavan-Unsal, N. Atiprimod induce apoptosis in pituitary adenoma: Endoplasmic reticulum stress and autophagy pathways. J. Cell. Biochem. 2019, 120, 19749–19763. [Google Scholar] [CrossRef]
- Chen, X.; Iliopoulos, D.; Zhang, Q.; Tang, Q.; Greenblatt, M.B.; Hatziapostolou, M.; Lim, E.; Tam, W.L.; Ni, M.; Chen, Y.; et al. XBP1 promotes triple-negative breast cancer by controlling the HIF1α pathway. Nature 2014, 508, 103–107. [Google Scholar] [CrossRef]
- Cubillos-Ruiz, J.R.; Silberman, P.C.; Rutkowski, M.R.; Chopra, S.; Perales-Puchalt, A.; Song, M.; Zhang, S.; Bettigole, S.E.; Gupta, D.; Holcomb, K.; et al. ER Stress Sensor XBP1 Controls Anti-tumor Immunity by Disrupting Dendritic Cell Homeostasis. Cell 2015, 7, 1527–1538. [Google Scholar] [CrossRef] [Green Version]
- Luo, Q.; Shi, W.; Dou, B.; Wang, J.; Peng, W.; Liu, X.; Zhao, D.; Tang, F.; Wu, Y.; Li, X.; et al. XBP1-IGFBP3 Signaling Pathway Promotes NSCLC Invasion and Metastasis. Front. Oncol. 2021, 11, 654995. [Google Scholar] [CrossRef]
- Meng, J.; Liu, K.; Shao, Y.; Feng, X.; Ji, Z.; Chang, B.; Wang, Y.; Xu, L.; Yang, G. ID1 confers cancer cell chemoresistance through STAT3/ATF6-mediated induction of autophagy. Cell Death Dis. 2020, 11, 137–152. [Google Scholar] [CrossRef] [Green Version]
- Spaan, C.N.; Smit, W.L.; Jeude, J.F.V.L.D.; Meijer, B.J.; Muncan, V.; Brink, G.R.V.D.; Heijmans, J. Expression of UPR effector proteins ATF6 and XBP1 reduce colorectal cancer cell proliferation and stemness by activating PERK signaling. Cell Death Dis. 2019, 10, 490. [Google Scholar] [CrossRef]
- Jia, Z.; Wang, M.; Li, S.; Li, X.; Bai, X.-Y.; Xu, Z.; Yang, Y.; Li, B.; Li, Y.; Wu, H. 2U-box ubiquitin ligase PPIL2 suppresses breast cancer invasion and metastasis by altering cell morphology and promoting SNAI1 ubiquitination and degradation. Cell Death Dis. 2018, 9, 63. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Ben-Neriah, Y. Phosphorylation meets ubiquitination: The control of NF-[kappa]B activity. Annu. Rev. Immunol. 2000, 18, 621–663. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Huo, R.; Yan, X.; Xu, T. IRF3 Negatively Regulates Toll-Like Receptor-Mediated NF-κB Signaling by Targeting TRIF for Degradation in Teleost Fish. Front. Immunol. 2018, 9, 867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakamoto, H.; Egashira, S.; Saito, N.; Kirisako, T.; Miller, S.; Sasaki, Y.; Matsumoto, T.; Shimonishi, M.; Komatsu, T.; Terai, T.; et al. Gliotoxin suppresses NF-κB activation by selectively inhibiting linear ubiquitin chain assembly complex (LUBAC). ACS Chem. Biol. 2015, 3, 675–681. [Google Scholar] [CrossRef]
- Wu, L.; He, Y.; Hu, Y.; Lu, H.; Cao, Z.; Yi, X.; Wang, J. Real-time surface plasmon resonance monitoring of site-specific phosphorylation of p53 protein and its interaction with MDM2 protein. Analyst 2019, 144, 6033–6040. [Google Scholar] [CrossRef]
- Tu, Y.; Chen, C.; Pan, J.; Xu, J.; Zhou, Z.-G.; Wang, C.-Y. The Ubiquitin Proteasome Pathway (UPP) in the regulation of cell cycle control and DNA damage repair and its implication in tumorigenesis. Int. J. Clin. Exp. Pathol. 2012, 5, 726–738. [Google Scholar]
- Grim, J.E.; Knoblaugh, S.E.; Guthrie, K.A.; Hagar, A.; Swanger, J.; Hespelt, J.; Delrow, J.J.; Small, T.; Grady, W.M.; Nakayama, K.I. Fbw7 and p53 cooperatively suppress advanced and chromosomally unstable intestinal cancer. Mol. Cell. Biol. 2012, 11, 2160–2167. [Google Scholar] [CrossRef] [Green Version]
- Fischer, E.S.; Böhm, K.; Lydeard, J.R.; Yang, H.; Stadler, M.B.; Cavadini, S.; Nagel, J.; Serluca, F.; Acker, V.; Lingaraju, G.M. Structure of the DDB1-CRBN E3 ubiquitin ligase in complex with thalidomide. Nature 2014, 7512, 49–53. [Google Scholar] [CrossRef] [Green Version]
- Senft, D.; Qi, J.; Ronai, Z. Ubiquitin ligases in oncogenic transformation and cancer therapy. Nat. Rev. Cancer 2018, 18, 69–88. [Google Scholar] [CrossRef]
- Hirata, Y.; Katagiri, K.; Nagaoka, K.; Morishita, T.; Kudoh, Y.; Hatta, T.; Naguro, I.; Kano, K.; Udagawa, T.; Natsume, T.; et al. TRIM48 Promotes ASK1 Activation and Cell Death through Ubiquitination-Dependent Degradation of the ASK1-Negative Regulator PRMT1. Cell Rep. 2017, 9, 2447–2457. [Google Scholar] [CrossRef] [Green Version]
- Zeng, T.; Wang, Q.; Fu, J.; Lin, Q.; Bi, J.; Ding, W.; Qiao, Y.; Zhang, S.; Zhao, W.; Lin, H.; et al. Impeded Nedd4-1-mediated Ras degradation underlies Ras-driven tumorigenesis. Cell Rep. 2014, 7, 871–882. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.-W.; Jin, D.-H.; Shin, J.-S.; Moon, J.-H.; Na, Y.-S.; Jung, K.-A.; Kim, S.-M.; Kim, J.C.; Kim, K.-P.; Hong, Y.S.; et al. Ring finger protein 149 is an E3 ubiquitin ligase active on wild-type v-Raf murine sarcoma viral oncogene homolog B1 (BRAF). J. Biol. Chem. 2012, 28, 24017–24025. [Google Scholar] [CrossRef] [Green Version]
- Brahimi-Horn, C.; Pouysségur, J. When hypoxia signalling meets the ubiquitin-proteasomal pathway, new targets for cancer therapy. Crit. Rev. Oncol. Hematol. 2005, 53, 115–123. [Google Scholar] [CrossRef]
- Rashid, M.; Zadeh, L.R.; Baradaran, B.; Molavi, O.; Ghesmati, Z.; Sabzichi, M.; Ramezani, F. Up-down regulation of HIF-1α in cancer progression. Gene 2021, 798, 145796. [Google Scholar] [CrossRef]
- Buratti, J.; Ji, L.; Keren, B.; Lee, Y.; Booke, S.; Erdin, S.; Kim, S.Y.; Palculict, T.B.; Meiner, V.; Chae, J.H.; et al. De novo variants inSIAH1, encoding an E3 ubiquitin ligase, are associated with developmental delay, hypotonia and dysmorphic features. J. Med. Genet. 2021, 58, 205–212. [Google Scholar] [CrossRef]
- Ji, L.; Jiang, B.; Jiang, X.; Charlat, O.; Chen, A.; Mickanin, C.; Bauer, A.; Xu, W.; Yan, X.; Cong, F. The SIAH E3 ubiquitin ligases promote Wnt/β-catenin signaling through mediating Wnt-induced Axin degradation. Genes Dev. 2017, 31, 904–915. [Google Scholar] [CrossRef] [Green Version]
- Datta, K.; Suman, S.; Kumar, S.; Fornace, A.J., Jr. Colorectal Carcinogenesis, Radiation Quality, and the Ubiquitin-Proteasome Pathway. J. Cancer 2016, 7, 174–183. [Google Scholar] [CrossRef] [Green Version]
- Kuchay, S.; Duan, S.; Schenkein, E.; Peschiaroli, A.; Saraf, A.; Florens, L.; Washburn, M.; Pagano, M. FBXL2- and PTPL1-mediated degradation of p110-free p85β regulatory subunit controls the PI(3)K signalling cascade. Nat. Cell. Biol. 2013, 5, 472–480. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.; Anjomani-Virmouni, S.; Koundouros, N.; Dimitriadi, M.; Choo-Wing, R.; Valle, A.; Zheng, Y.; Chiu, Y.-H.; Agnihotri, S.; Zadeh, G.; et al. PARK2 depletion connects energy and oxidative stress to PI3K/Akt activation via PTEN S-nitrosylation. Mol. Cell 2017, 65, 999–1013.e7. [Google Scholar] [CrossRef] [Green Version]
- Chan, C.-H.; Li, C.-F.; Yang, W.-L.; Gao, Y.; Lee, S.-W.; Feng, Z.; Huang, H.-Y.; Tsai, K.K.C.; Flores, L.G.; Shao, Y.; et al. The Skp2-SCF E3 ligase regulates Akt ubiquitination, glycolysis, herceptin sensitivity, and tumorigenesis. Cell 2012, 149, 1098–1111. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.-L.; Wang, J.; Chan, C.-H.; Lee, S.-W.; Campos, A.D.; Lamothe, B.; Hur, L.; Grabiner, B.C.; Lin, X.; Darnay, B.G.; et al. The E3 ligase TRAF6 regulates Akt ubiquitination and activation. Science 2009, 5499, 1134–1138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, S.; Skaar, J.R.; Kuchay, S.; Toschi, A.; Kanarek, N.; Ben-Neriah, Y.; Pagano, M. mTOR generates an auto-amplification loop by triggering the βTrCP- and CK1αdependent degradation of DEPTOR. Mol. Cell. 2011, 2, 317–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eskelinen, E.-L. The dual role of autophagy in cancer. Curr. Opin. Pharm. 2011, 4, 294–300. [Google Scholar] [CrossRef]
- Jin, S.; Wei, J.; You, L.; Liu, H.; Qian, W. Autophagy regulation and its dual role in blood cancers: A novel target for therapeutic development (Review). Oncol. Rep. 2018, 6, 2473–2481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, S.; Tortola, L.; Perlot, T.; Wirnsberger, G.; Novatchkova, M.; Nitsch, R.; Sykacek, P.; Frank, L.; Schramek, D.; Komnenovic, V.; et al. A dual role for autophagy in a murine model of lung cancer. Nat. Commun. 2014, 5, 3056–3070. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Wu, Y.; Lian, X. Targeted inhibition of GRP78 by HA15 promotes apoptosis of lung cancer cells accompanied by ER stress and autophagy. Biol. Open 2020, 11, bio053298. [Google Scholar] [CrossRef] [PubMed]
- Zong, H.; Yin, B.; Zhou, H.; Cai, D.; Ma, B.; Xiang, Y. Inhibition of mTOR pathway attenuates migration and invasion of gallbladder cancer via EMT inhibition. Mol. Biol. Rep. 2014, 7, 4507–4512. [Google Scholar] [CrossRef]
- Hanzhi, C.; Cui, H.; Kang, L.; Zhang, X.; Jin, Z.; Lu, L.; Fan, Z. Metformin inhibits thyroid cancer cell growth, migration, and EMT through the mTOR pathway. Tumour Biol. 2015, 8, 6295–6304. [Google Scholar]
- Guo, S.; Liang, X.; Guo, M.; Zhang, X.; Li, Z. Migration inhibition of water stress proteins from Nostoc commune Vauch via activation of autophagy in DLD-1 cells. Int. J. Biol. Macromol. 2018, 119, 669–676. [Google Scholar] [CrossRef]
- Huang, H.; Song, J.; Liu, Z.; Pan, L.; Xu, G. Autophagy activation promotes bevacizumab resistance in glioblastoma by suppressing Akt/mTOR signaling pathway. Oncol. Lett. 2016, 2, 1487–1494. [Google Scholar] [CrossRef]
- Cufí, S.; Vazquez-Martin, A.; Oliveras-Ferraros, C.; Corominas-Faja, B.; Cuyàs, E.; López-Bonet, E.; Martin-Castillo, B.; Joven, J.; Menendez, J.A. The anti-malarial chloroquine overcomes primary resistance and restores sensitivity to trastuzumab in HER2-positive breast cancer. Sci. Rep. 2013, 3, 2469. [Google Scholar] [CrossRef] [Green Version]
- Cufí, S.; Vazquez-Martin, A.; Oliveras-Ferraros, C.; Corominas-Faja, B.; Urruticoechea, A.; Martin-Castillo, B.; Menendez, J.A. Autophagy-related gene 12 (ATG12) is a novel determinant of primary resistance to HER2-targeted therapies: Utility of transcriptome analysis of the autophagy interactome to guide breast cancer treatment. Oncotarget 2012, 12, 1600–1614. [Google Scholar] [CrossRef] [Green Version]


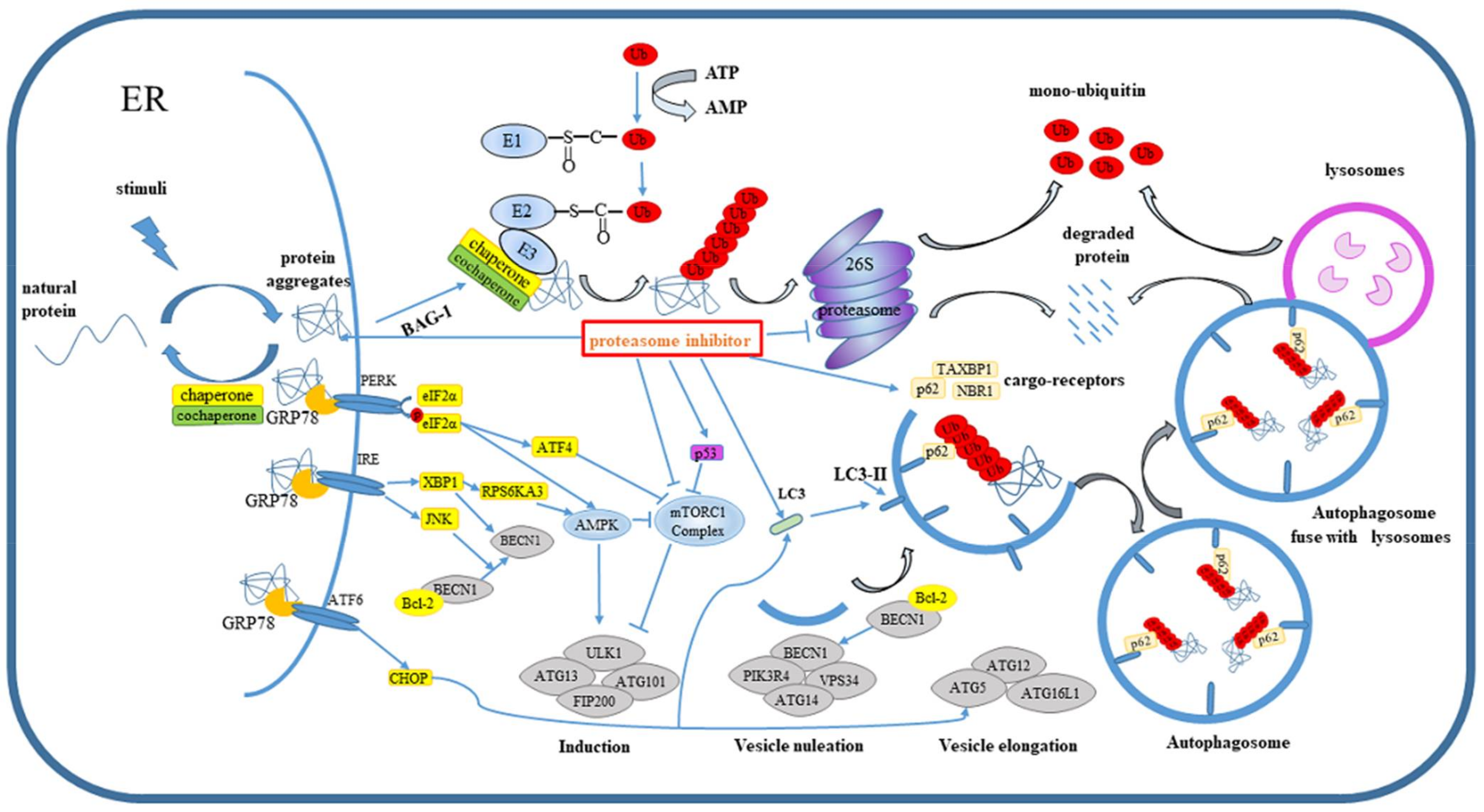{kind=link}
| Ubiquitin E3 Ligase | Substrates |
|---|---|
| GP78 | CD3—δ [22], CFTR [23], HMGCR [24]. |
| Parkin | α-synuclien, synphilin-1, Pae1 [25], glucocerebrosidase (GCase) [26]. |
| E3 ubiquitin–protein ligase synoviolin (HRD1/3) [21,27] | Hmg1p [28], pre-B cell receptor (pre-BCR), pro-arginine vasopressin (AVP) B-lymphocyte-induced maturation protein 1 (BLIMP1) [29], NRF2 [30], peroxisome proliferator activated receptor γ coactivator-1 β (PGC1β) [31], transforming growth factor beta (TGF-β) [32], T-cell receptor alpha (TCR-α), [33] alpha1-antitrypsin [34], p53 [35], amyloid precursor protein (APP) [36], et al. |
| membrane-associated RING C3HC4 finger 6 (MARCHF6) [37,38,39] | squalene epoxidase (SQLE) [40], HMGCR [38], perilipin-2 (PLIN2) [38], et al. |
| RNF5 [41] | CFTR [41], JNK-associated membrane protein (JAMP) [42], et al. |
| Makorin ring-finger protein 1 (MKRN1) | Potassium voltage-gated channel subfamily H member 1 (KCNH1), Eag1 [43] |
| SCF Fbx2 [37] | β-secretase (BACE1) [44], CFTR [45], et al. |
| SCF Fbx6 [37] | Chk [46] |
| SCFβ-TrCP1/2 | CD4 [47], Tetherin [48] |
| CHIP [24] | (CFTR) [19,20,21], GR [22], Pael [23], ErbB2 [23], IRE [49], INO80 [50], tau [51], et al. |
| SMAD ubiquitination regulatory factor 1 (Smurf1) | Wolframin (WFS1) [52], phosphatase and tensin homolog (PTEN) [53], et al. |
| Neuregulin receptor degradation pathway protein 1 (Nrdp1) [37] | Erb-B2 receptor tyrosine kinase 3 (ErbB3) [54], et al. |
| Signal Pathway | Cell Stimuli | E3 Enzyme |
|---|---|---|
| NF-κB pathway [111,112] | Cytokines, UV, virus, oxide, et al. | β-TrCP [122], IRF3 [123], Linear Ubiquitin Chain Assembly Complex (LUBAC) [124] |
| p53 pathway | DNA damage | MDM2 [125,126], FBXW7 [127] |
| DDB1/DDB2-XPC/HR23B pathway | DNA damage | DDB1-CRBN ubiquitin E3 ligase [128] |
| MAPK pathway [129] | UV, Osmotic pressure change, heat shock, et al. | TRIM48 [130], NEDD4 [123], RNF [131,132], et al. |
| HIFα pathway [133] | hypoxia | pVHL E3 ligase complex [134]. |
| wnt/β-catein pathway | DNA damage, hypoxia, virus, et al. | SIAH1 [135,136,137]. |
| PI3K/AKT/mTOR pathway | Hypoxia, high glucose, cytokines, et al. | FBXL12 [138], parkin [139,140], TRAF6 [141,142], β-TRCP [143], et al. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Li, S.; Wu, H. Ubiquitination-Proteasome System (UPS) and Autophagy Two Main Protein Degradation Machineries in Response to Cell Stress. Cells 2022, 11, 851. https://doi.org/10.3390/cells11050851
Li Y, Li S, Wu H. Ubiquitination-Proteasome System (UPS) and Autophagy Two Main Protein Degradation Machineries in Response to Cell Stress. Cells. 2022; 11(5):851. https://doi.org/10.3390/cells11050851
Chicago/Turabian StyleLi, Yanan, Shujing Li, and Huijian Wu. 2022. "Ubiquitination-Proteasome System (UPS) and Autophagy Two Main Protein Degradation Machineries in Response to Cell Stress" Cells 11, no. 5: 851. https://doi.org/10.3390/cells11050851