
Adipose Triglyceride Lipase Deficiency Attenuates In Vitro Thrombus Formation without Affecting Platelet Activation and Bleeding In Vivo

 , , , ,
, , , ,  ,
,  , , , , ,
, , , , ,  and
and
Abstract
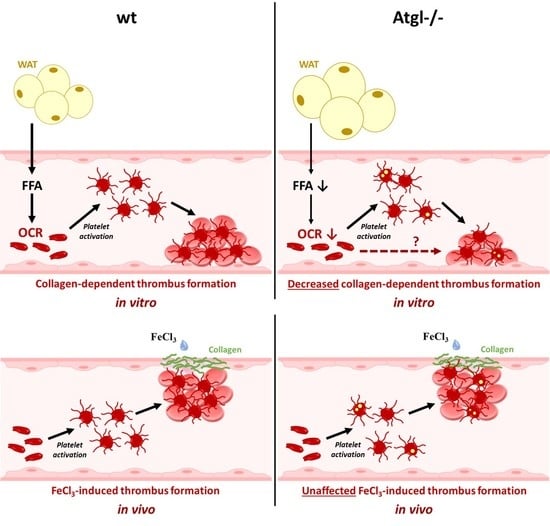
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Megakaryocyte Isolation and Differentiation
2.3. Platelet Isolation and Purification
2.4. RNA Isolation and Quantitative Real-Time PCR Analysis
2.5. Western Blotting Analysis
2.6. Electron Microscopy
2.7. Targeted Lipidomic Analysis
2.8. Flow Cytometric Analyses of P-Selectin and αIIbβ3 Expression
2.9. GPVI Staining
2.10. Platelet Aggregation Assay
2.11. Tail Bleeding and Hemoglobin Assays
2.12. Mitochondrial Respiration Measurement
2.13. In Vitro Thrombus Formation
2.14. In Vivo Thrombus Formation
2.15. Statistical Analysis
3. Results
3.1. ATGL Is Expressed in Mouse Megakaryocytes and Platelets
3.2. Little LD Formation in Atgl−/− Platelets
3.3. Platelet Mitochondrial Respiration Depends on Global Atgl Expression
3.4. Unchanged Hemostatic Function in Atgl−/− Mice
3.5. Loss of ATGL Affects Thrombus Formation In Vitro but Not In Vivo
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Machlus, K.R.; Italiano, J.E., Jr. The incredible journey: From megakaryocyte development to platelet formation. J. Cell Biol. 2013, 201, 785–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smyth, S.S.; McEver, R.P.; Weyrich, A.S.; Morrell, C.N.; Hoffman, M.R.; Arepally, G.M.; French, P.A.; Dauerman, H.L.; Becker, R.C.; Platelet Colloquium, P. Platelet functions beyond hemostasis. J. Thromb. Haemost. 2009, 7, 1759–1766. [Google Scholar] [CrossRef]
- Golebiewska, E.M.; Poole, A.W. Platelet secretion: From haemostasis to wound healing and beyond. Blood Rev. 2015, 29, 153–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Donnell, V.B.; Murphy, R.C.; Watson, S.P. Platelet lipidomics: Modern day perspective on lipid discovery and characterization in platelets. Circ. Res. 2014, 114, 1185–1203. [Google Scholar] [CrossRef] [PubMed]
- Bergmeier, W.; Hynes, R.O. Extracellular matrix proteins in hemostasis and thrombosis. Cold Spring Harb. Perspect. Biol. 2012, 4, a005132. [Google Scholar] [CrossRef]
- Kaplan, Z.S.; Jackson, S.P. The role of platelets in atherothrombosis. Hematol. Am. Soc. Hematol. Educ. Program 2011, 2011, 51–61. [Google Scholar] [CrossRef] [Green Version]
- Steinhubl, S.R.; Moliterno, D.J. The role of the platelet in the pathogenesis of atherothrombosis. Am. J. Cardiovasc. Drugs 2005, 5, 399–408. [Google Scholar] [CrossRef]
- Kaur, R.; Kaur, M.; Singh, J. Endothelial dysfunction and platelet hyperactivity in type 2 diabetes mellitus: Molecular insights and therapeutic strategies. Cardiovasc. Diabetol. 2018, 17, 121. [Google Scholar] [CrossRef]
- Wang, N.; Tall, A.R. Cholesterol in platelet biogenesis and activation. Blood 2016, 127, 1949–1953. [Google Scholar] [CrossRef] [Green Version]
- Aviram, M.; Gerald Brook, J. Platelet activation by plasma lipoproteins. Prog. Cardiovasc. Dis. 1987, 30, 61–72. [Google Scholar] [CrossRef]
- Betteridge, D.J.; Cooper, M.B.; Saggerson, E.D.; Prichard, B.N.C.; Tan, K.C.B.; Ling, E.; Barbera, G.; McCarthy, S.; Smith, C.C.T. Platelet function in patients with hypercholesterolaemia. Eur. J. Clin. Investig. 1994, 24, 30–33. [Google Scholar] [CrossRef] [PubMed]
- Milionis, H.J.; Elisaf, M.S.; Mikhailidis, D.P. Platelet function and lipid-lowering interventions. Platelets 1999, 10, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Nofer, J.-R.; Herminghaus, G.; Brodde, M.; Morgenstern, E.; Rust, S.; Engel, T.; Seedorf, U.; Assmann, G.; Bluethmann, H.; Kehrel, B.E. Impaired Platelet Activation in Familial High Density Lipoprotein Deficiency (Tangier Disease). J. Biol. Chem. 2004, 279, 34032–34037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shattil, S.J.; Anaya-Galindo, R.; Bennett, J.; Colman, R.W.; Cooper, R.A. Platelet hypersensitivity induced by cholesterol incorporation. J. Clin. Investig. 1975, 55, 636–643. [Google Scholar] [CrossRef] [Green Version]
- Zechner, R.; Madeo, F.; Kratky, D. Cytosolic lipolysis and lipophagy: Two sides of the same coin. Nat. Rev. Mol. Cell Biol. 2017, 18, 671–684. [Google Scholar] [CrossRef] [PubMed]
- Fischer, J.; Lefèvre, C.; Morava, E.; Mussini, J.-M.; Laforêt, P.; Negre-Salvayre, A.; Lathrop, M.; Salvayre, R. The gene encoding adipose triglyceride lipase (PNPLA2) is mutated in neutral lipid storage disease with myopathy. Nat. Genet. 2007, 39, 28–30. [Google Scholar] [CrossRef]
- Haemmerle, G.; Moustafa, T.; Woelkart, G.; Büttner, S.; Schmidt, A.; van de Weijer, T.; Hesselink, M.; Jaeger, D.; Kienesberger, P.C.; Zierler, K.; et al. ATGL-mediated fat catabolism regulates cardiac mitochondrial function via PPAR-α and PGC-1. Nat. Med. 2011, 17, 1076–1085. [Google Scholar] [CrossRef] [Green Version]
- Haemmerle, G.; Lass, A.; Zimmermann, R.; Gorkiewicz, G.; Meyer, C.; Rozman, J.; Heldmaier, G.; Maier, R.; Theussl, C.; Eder, S.; et al. Defective Lipolysis and Altered Energy Metabolism in Mice Lacking Adipose Triglyceride Lipase. Science 2006, 312, 734–737. [Google Scholar] [CrossRef]
- Chandak, P.G.; Radovic, B.; Aflaki, E.; Kolb, D.; Buchebner, M.; Frohlich, E.; Magnes, C.; Sinner, F.; Haemmerle, G.; Zechner, R.; et al. Efficient phagocytosis requires triacylglycerol hydrolysis by adipose triglyceride lipase. J. Biol. Chem. 2010, 285, 20192–20201. [Google Scholar] [CrossRef] [Green Version]
- Aflaki, E.; Balenga, N.A.; Luschnig-Schratl, P.; Wolinski, H.; Povoden, S.; Chandak, P.G.; Bogner-Strauss, J.G.; Eder, S.; Konya, V.; Kohlwein, S.D.; et al. Impaired Rho GTPase activation abrogates cell polarization and migration in macrophages with defective lipolysis. Cell. Mol. Life Sci. 2011, 68, 3933–3947. [Google Scholar] [CrossRef] [Green Version]
- Aflaki, E.; Doddapattar, P.; Radovic, B.; Povoden, S.; Kolb, D.; Vujic, N.; Wegscheider, M.; Koefeler, H.; Hornemann, T.; Graier, W.F.; et al. C16 ceramide is crucial for triacylglycerol-induced apoptosis in macrophages. Cell Death Dis. 2012, 3, e280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aflaki, E.; Radovic, B.; Chandak, P.G.; Kolb, D.; Eisenberg, T.; Ring, J.; Fertschai, I.; Uellen, A.; Wolinski, H.; Kohlwein, S.D.; et al. Triacylglycerol accumulation activates the mitochondrial apoptosis pathway in macrophages. J. Biol. Chem. 2011, 286, 7418–7428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lammers, B.; Chandak, P.G.; Aflaki, E.; Van Puijvelde, G.H.M.; Radovic, B.; Hildebrand, R.B.; Meurs, I.; Out, R.; Kuiper, J.; Van Berkel, T.J.C.; et al. Macrophage adipose triglyceride lipase deficiency attenuates atherosclerotic lesion development in low-density lipoprotein receptor knockout mice. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 67–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlager, S.; Goeritzer, M.; Jandl, K.; Frei, R.; Vujic, N.; Kolb, D.; Strohmaier, H.; Dorow, J.; Eichmann, T.O.; Rosenberger, A.; et al. Adipose triglyceride lipase acts on neutrophil lipid droplets to regulate substrate availability for lipid mediator synthesis. J. Leukoc. Biol. 2015, 98, 837–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salzmann, M.; Hoesel, B.; Haase, M.; Mussbacher, M.; Schrottmaier, W.C.; Kral-Pointner, J.B.; Finsterbusch, M.; Mazharian, A.; Assinger, A.; Schmid, J.A. A novel method for automated assessment of megakaryocyte differentiation and proplatelet formation. Platelets 2018, 29, 357–364. [Google Scholar] [CrossRef] [Green Version]
- Rowley, J.W.; Oler, A.J.; Tolley, N.D.; Hunter, B.N.; Low, E.N.; Nix, D.A.; Yost, C.C.; Zimmerman, G.A.; Weyrich, A.S. Genome-wide RNA-seq analysis of human and mouse platelet transcriptomes. Blood 2011, 118, e101–e111. [Google Scholar] [CrossRef] [Green Version]
- Grabner, G.F.; Eichmann, T.O.; Wagner, B.; Gao, Y.; Farzi, A.; Taschler, U.; Radner, F.P.W.; Schweiger, M.; Lass, A.; Holzer, P.; et al. Deletion of Monoglyceride Lipase in Astrocytes Attenuates Lipopolysaccharide-induced Neuroinflammation. J. Biol. Chem. 2016, 291, 913–923. [Google Scholar] [CrossRef] [Green Version]
- Matyash, V.; Liebisch, G.; Kurzchalia, T.V.; Shevchenko, A.; Schwudke, D. Lipid extraction by methyl-tert-butyl ether for high-throughput lipidomics. J. Lipid Res. 2008, 49, 1137–1146. [Google Scholar] [CrossRef] [Green Version]
- Knittelfelder, O.L.; Weberhofer, B.P.; Eichmann, T.O.; Kohlwein, S.D.; Rechberger, G.N. A versatile ultra-high performance LC-MS method for lipid profiling. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2014, 951–952, 119–128. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Jennings, N.L.; Dart, A.M.; Du, X.J. Standardizing a simpler, more sensitive and accurate tail bleeding assay in mice. World J. Exp. Med. 2012, 2, 30–36. [Google Scholar] [CrossRef]
- Eckly, A.; Hechler, B.; Freund, M.; Zerr, M.; Cazenave, J.P.; Lanza, F.; Mangin, P.H.; Gachet, C. Mechanisms underlying FeCl3-induced arterial thrombosis. J. Thromb. Haemost. 2011, 9, 779–789. [Google Scholar] [CrossRef]
- Simon, L.M.; Edelstein, L.C.; Nagalla, S.; Woodley, A.B.; Chen, E.S.; Kong, X.; Ma, L.; Fortina, P.; Kunapuli, S.; Holinstat, M.; et al. Human platelet microRNA-mRNA networks associated with age and gender revealed by integrated plateletomics. Blood 2014, 123, e37–e45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenkins, C.M.; Mancuso, D.J.; Yan, W.; Sims, H.F.; Gibson, B.; Gross, R.W. Identification, cloning, expression, and purification of three novel human calcium-independent phospholipase A2 family members possessing triacylglycerol lipase and acylglycerol transacylase activities. J. Biol. Chem. 2004, 279, 48968–48975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melchinger, H.; Jain, K.; Tyagi, T.; Hwa, J. Role of Platelet Mitochondria: Life in a Nucleus-Free Zone. Front. Cardiovasc. Med. 2019, 6, 153. [Google Scholar] [CrossRef] [PubMed]
- Knight, C.G.; Morton, L.F.; Onley, D.J.; Peachey, A.R.; Ichinohe, T.; Okuma, M.; Farndale, R.W.; Barnes, M.J. Collagen-platelet interaction: Gly-Pro-Hyp is uniquely specific for platelet Gp VI and mediates platelet activation by collagen. Cardiovasc. Res. 1999, 41, 450–457. [Google Scholar] [CrossRef] [Green Version]
- Gardiner, E.E.; Arthur, J.F.; Kahn, M.L.; Berndt, M.C.; Andrews, R.K. Regulation of platelet membrane levels of glycoprotein VI by a platelet-derived metalloproteinase. Blood 2004, 104, 3611–3617. [Google Scholar] [CrossRef] [Green Version]
- Offermanns, S. Activation of platelet function through G protein-coupled receptors. Circ. Res. 2006, 99, 1293–1304. [Google Scholar] [CrossRef] [Green Version]
- Lewis, N.; Majerus, P.W. Lipid metabolism in human platelets. II. De novo phospholipid synthesis and the effect of thrombin on the pattern of synthesis. J. Clin. Investig. 1969, 48, 2114–2123. [Google Scholar] [CrossRef]
- Majerus, P.W.; Smith, M.B.; Clamon, G.H. Lipid metabolism in human platelets. I. Evidence for a complete fatty acid synthesizing system. J. Clin. Investig. 1969, 48, 156–164. [Google Scholar] [CrossRef] [Green Version]
- Green, S.M.; Padula, M.P.; Marks, D.C.; Johnson, L. The Lipid Composition of Platelets and the Impact of Storage: An Overview. Transfus. Med. Rev. 2020, 34, 108–116. [Google Scholar] [CrossRef]
- Ruebsaamen, K.; Liebisch, G.; Boettcher, A.; Schmitz, G. Lipidomic analysis of platelet senescence. Transfusion 2010, 50, 1665–1676. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Sims, H.F.; Jenkins, C.M.; Guan, S.; Dilthey, B.G.; Gross, R.W. 12-LOX catalyzes the oxidation of 2-arachidonoyl-lysolipids in platelets generating eicosanoid-lysolipids that are attenuated by iPLA2gamma knockout. J. Biol. Chem. 2020, 295, 5307–5320. [Google Scholar] [CrossRef] [PubMed]
- Iritani, N.; Ikeda, Y.; Kajitani, H. Selectivities of 1-acylglycerophosphorylcholine acyltransferase and acyl-CoA synthetase for n-3 polyunsaturated fatty acids in platelets and liver microsomes. Biochim. Biophys. Acta 1984, 793, 416–422. [Google Scholar]
- McKean, M.L.; Smith, J.B.; Silver, M.J. Phospholipid biosynthesis in human platelets. Formation of phosphatidylcholine from 1-acyl lysophosphatidylcholine by acyl-CoA:1-acyl-sn-glycero-3-phosphocholine acyltransferase. J. Biol. Chem. 1982, 257, 11278–11283. [Google Scholar] [CrossRef]
- Koseoglu, S.; Meyer, A.F.; Kim, D.; Meyer, B.M.; Wang, Y.; Dalluge, J.J.; Haynes, C.L. Analytical characterization of the role of phospholipids in platelet adhesion and secretion. Anal. Chem. 2015, 87, 413–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossaint, J.; Kuhne, K.; Skupski, J.; Van Aken, H.; Looney, M.R.; Hidalgo, A.; Zarbock, A. Directed transport of neutrophil-derived extracellular vesicles enables platelet-mediated innate immune response. Nat. Commun. 2016, 7, 13464. [Google Scholar] [CrossRef]
- Wang, L.; Wu, Q.; Fan, Z.; Xie, R.; Wang, Z.; Lu, Y. Platelet mitochondrial dysfunction and the correlation with human diseases. Biochem. Soc. Trans. 2017, 45, 1213–1223. [Google Scholar] [CrossRef]
- Aibibula, M.; Naseem, K.M.; Sturmey, R.G. Glucose metabolism and metabolic flexibility in blood platelets. J. Thromb. Haemost. 2018, 16, 2300–2314. [Google Scholar] [CrossRef] [Green Version]
- Ferroni, P.; Basili, S.; Santilli, F.; Davi, G. Low-density lipoprotein-lowering medication and platelet function. Pathophysiol. Haemost. Thromb. 2006, 35, 346–354. [Google Scholar] [CrossRef]
- Wallace, A.; Albadawi, H.; Hoang, P.; Fleck, A.; Naidu, S.; Knuttinen, G.; Oklu, R. Statins as a preventative therapy for venous thromboembolism. Cardiovasc. Diagn. Ther. 2017, 7, S207–S218. [Google Scholar] [CrossRef]
- Le Quan Sang, K.H.; Levenson, J.; Megnien, J.L.; Simon, A.; Devynck, M.A. Platelet cytosolic Ca2+ and membrane dynamics in patients with primary hypercholesterolemia. Effects of pravastatin. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 759–764. [Google Scholar] [CrossRef] [PubMed]
- Nieswandt, B.; Watson, S.P. Platelet-collagen interaction: Is GPVI the central receptor? Blood 2003, 102, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Carestia, A.; Mena, H.A.; Olexen, C.M.; Ortiz Wilczynski, J.M.; Negrotto, S.; Errasti, A.E.; Gomez, R.M.; Jenne, C.N.; Carrera Silva, E.A.; Schattner, M. Platelets Promote Macrophage Polarization toward Pro-inflammatory Phenotype and Increase Survival of Septic Mice. Cell Rep. 2019, 28, 896–908.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owens, A.P., 3rd; Lu, Y.; Whinna, H.C.; Gachet, C.; Fay, W.P.; Mackman, N. Towards a standardization of the murine ferric chloride-induced carotid arterial thrombosis model. J. Thromb. Haemost. 2011, 9, 1862–1863. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goeritzer, M.; Schlager, S.; Kuentzel, K.B.; Vujić, N.; Korbelius, M.; Rainer, S.; Kolb, D.; Mussbacher, M.; Salzmann, M.; Schrottmaier, W.C.; et al. Adipose Triglyceride Lipase Deficiency Attenuates In Vitro Thrombus Formation without Affecting Platelet Activation and Bleeding In Vivo. Cells 2022, 11, 850. https://doi.org/10.3390/cells11050850
Goeritzer M, Schlager S, Kuentzel KB, Vujić N, Korbelius M, Rainer S, Kolb D, Mussbacher M, Salzmann M, Schrottmaier WC, et al. Adipose Triglyceride Lipase Deficiency Attenuates In Vitro Thrombus Formation without Affecting Platelet Activation and Bleeding In Vivo. Cells. 2022; 11(5):850. https://doi.org/10.3390/cells11050850
Chicago/Turabian StyleGoeritzer, Madeleine, Stefanie Schlager, Katharina B. Kuentzel, Nemanja Vujić, Melanie Korbelius, Silvia Rainer, Dagmar Kolb, Marion Mussbacher, Manuel Salzmann, Waltraud C. Schrottmaier, and et al. 2022. "Adipose Triglyceride Lipase Deficiency Attenuates In Vitro Thrombus Formation without Affecting Platelet Activation and Bleeding In Vivo" Cells 11, no. 5: 850. https://doi.org/10.3390/cells11050850