
Intersections of Ubiquitin-Proteosome System and Autophagy in Promoting Growth of Glioblastoma Multiforme: Challenges and Opportunities


Abstract
:
1. Introduction
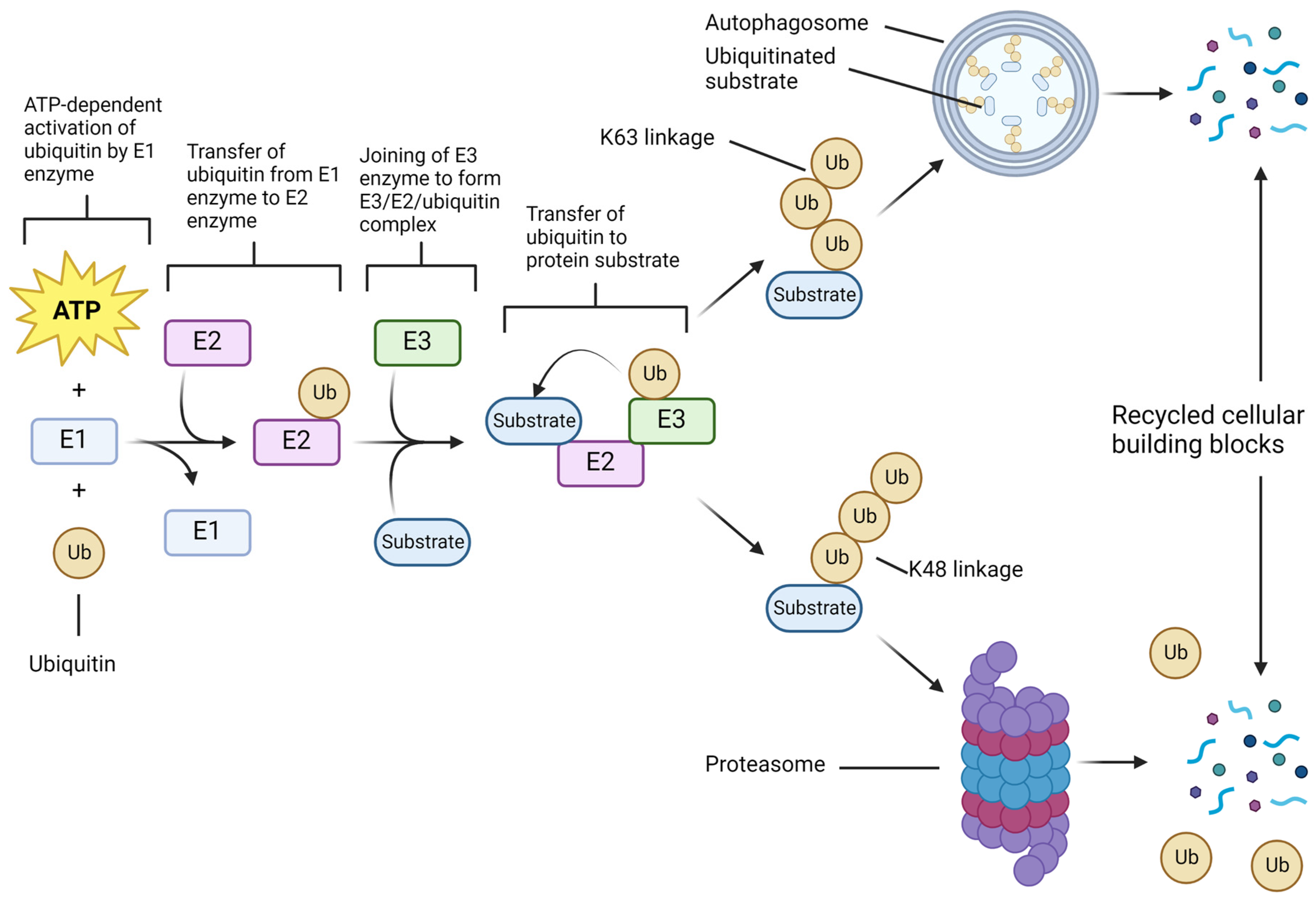
2. UPS Molecular Components and Functions in Cell Survival Show Therapeutic Opportunities in GBM
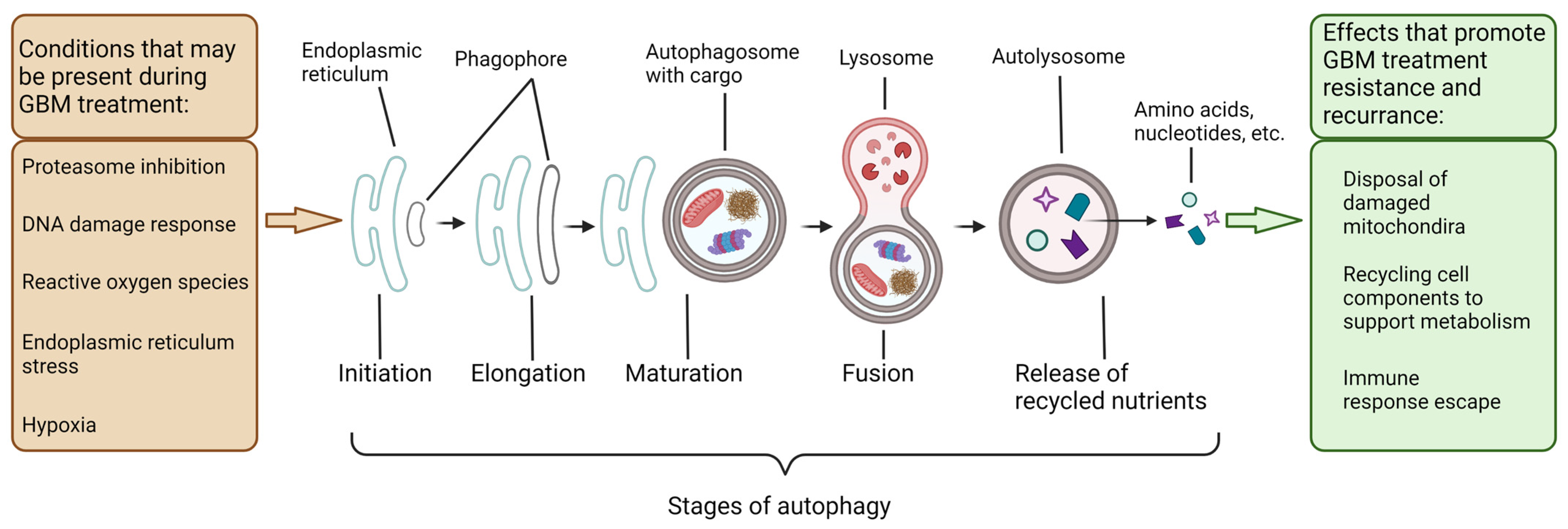
3. Autophagy in the Degradation and Salvaging of Cellular Components to Support GBM Growth Indicating Potential Therapeutic Targets
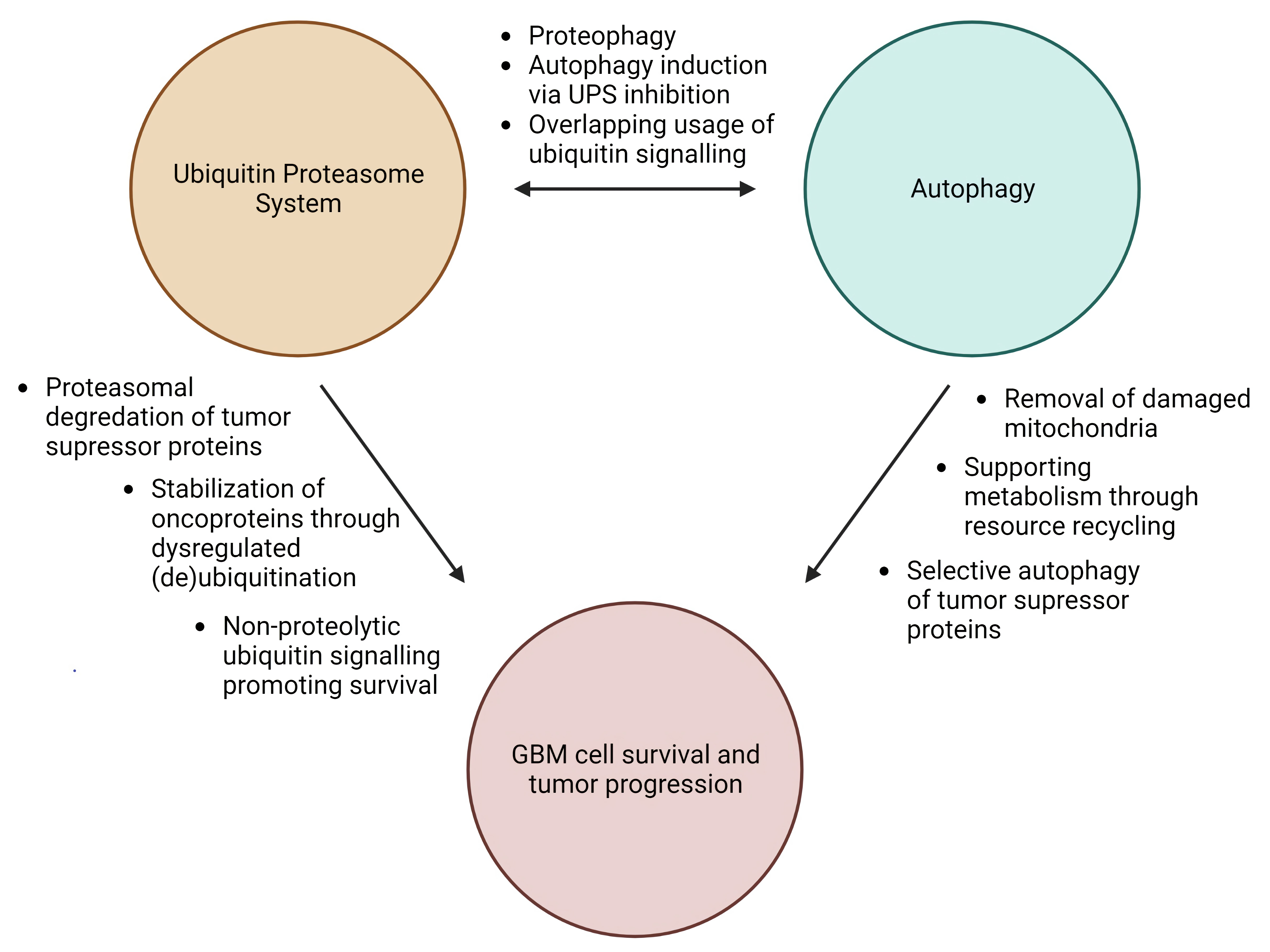
4. Molecular Components in the Intersections of UPS and Autophagy Pose Challenges and Provide Therapeutic Opportunities in GBM
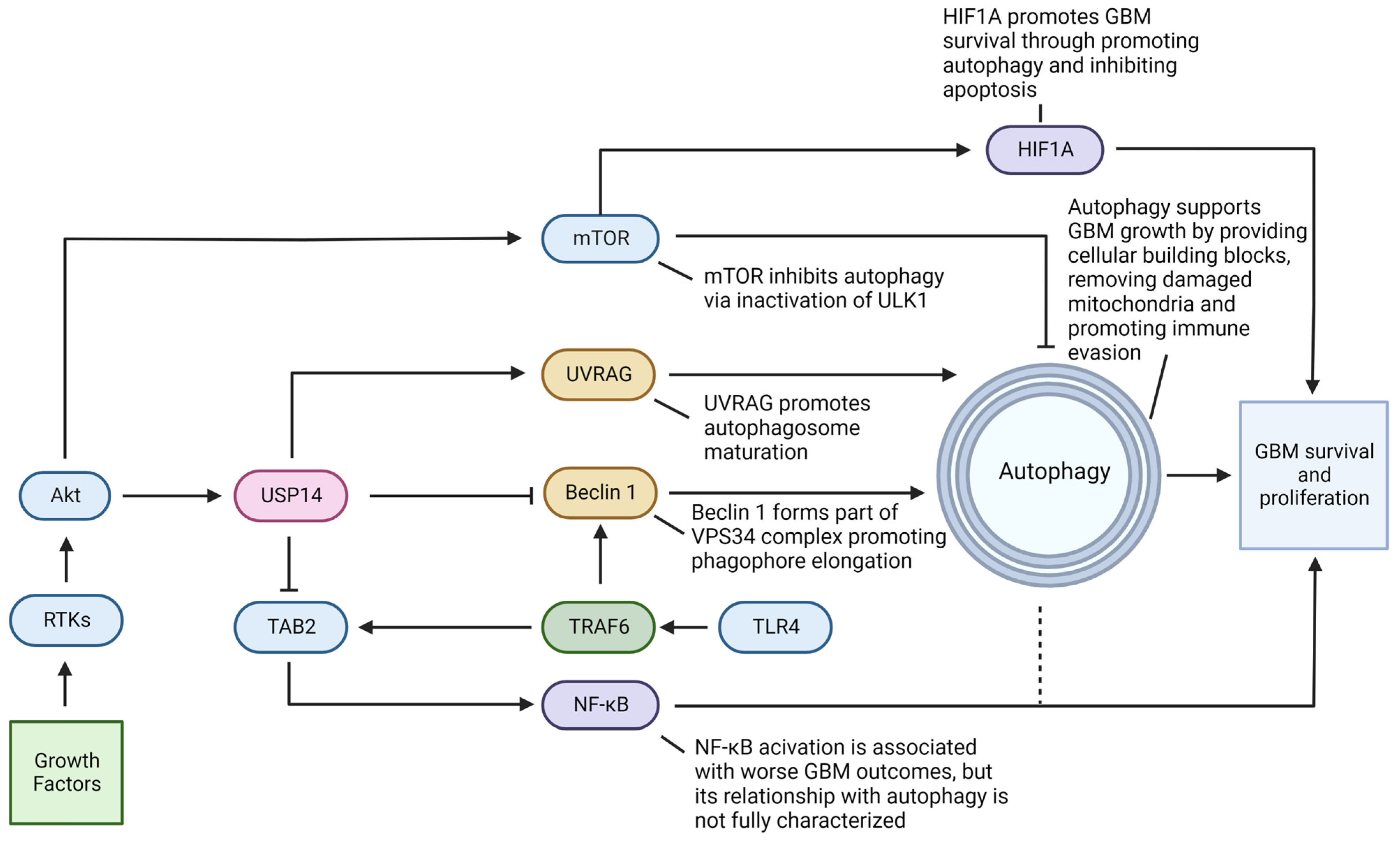
4.1. Ubiquitin-Specific Protease 14 (USP14) Acts in Promoting GBM Survival and Progression
4.2. Etoposide Induced 2.4 Transcript (EI24) Acts as a Promoter of GBM Growth
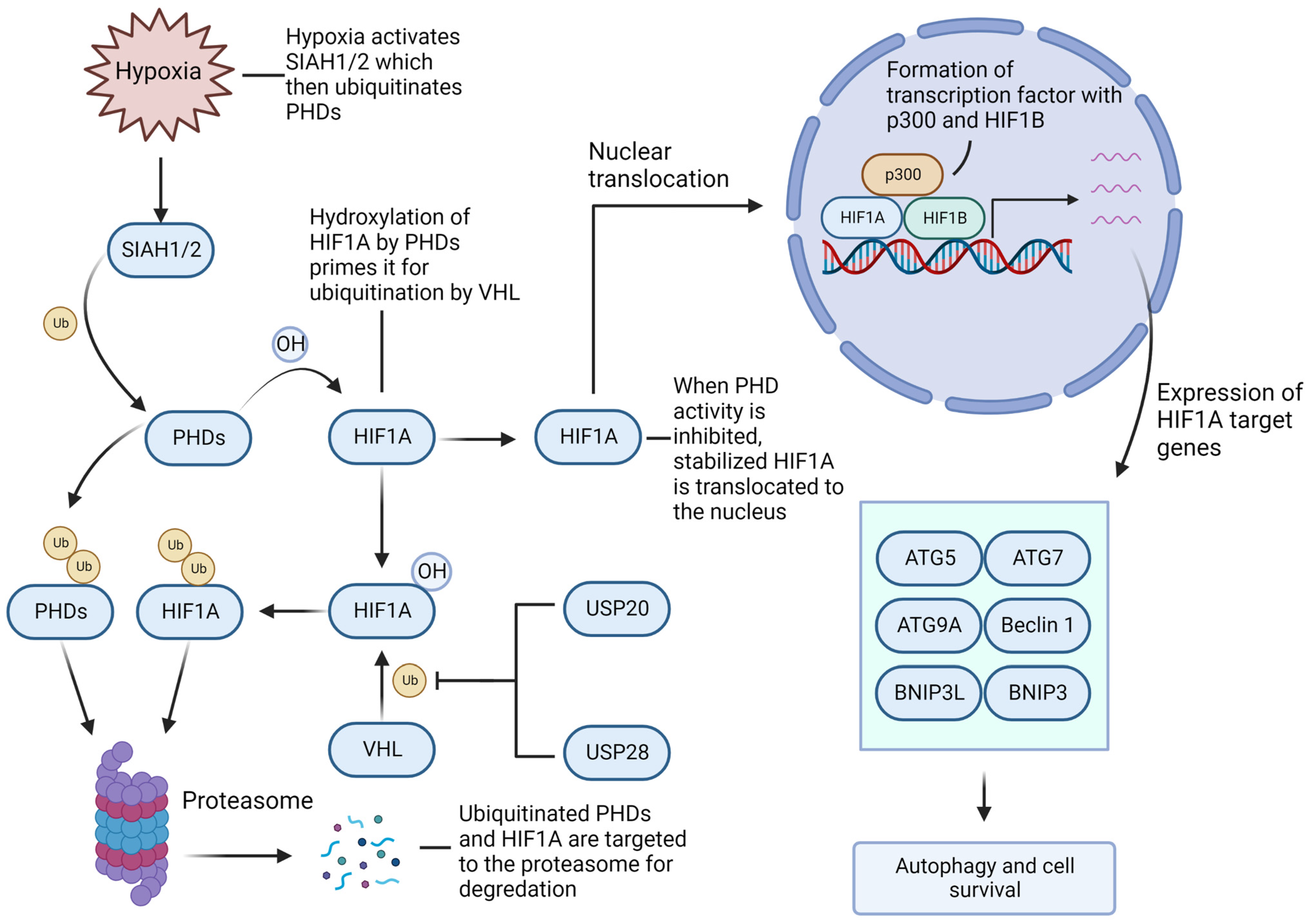
4.3. Hypoxia-Inducible Factor-1A/2A (HIF1A/2A) Facilitates GBM Growth and Recurrence
4.4. Histone Deacetylase 6 (HDAC6) Lends a Hand in Rapid GBM Progression
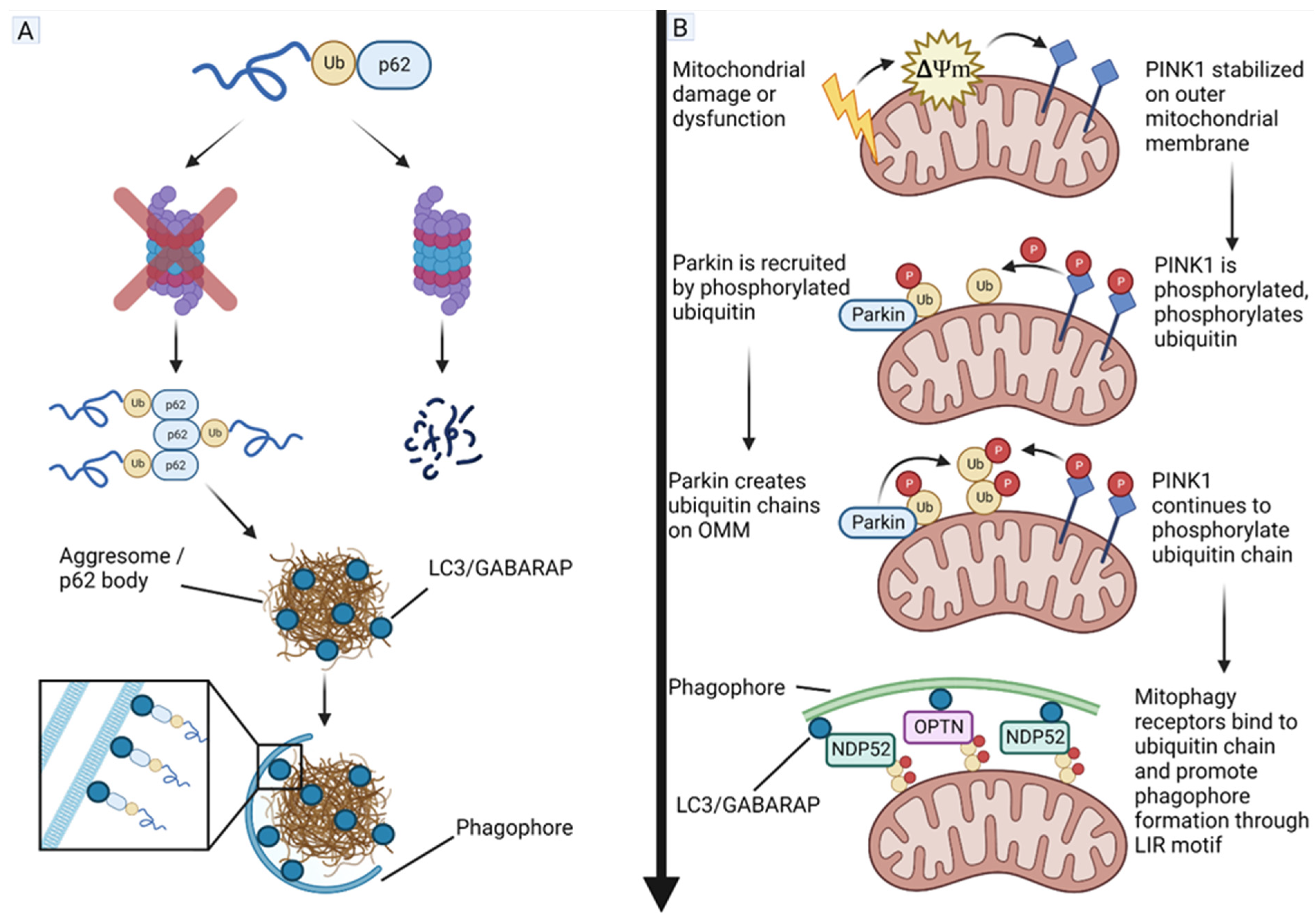
4.5. Phosphatase and Tensin Homolog (PTEN)-Induced Kinase 1 (PINK1) and Parkin Act as Tumor Suppressors in GBM
4.6. microRNAs (miRNAs) and Their Roles in Promoting or Inhibiting GBM Growth
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Grochans, S.; Cybulska, A.M.; Simińska, D.; Korbecki, J.; Kojder, K.; Chlubek, D.; Baranowska-Bosiacka, I. Epidemiology of Glioblastoma Multiforme–Literature Review. Cancers 2022, 14, 2412. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kim, D.-Y. Present and Future of Anti-Glioblastoma Therapies: A Deep Look into Molecular Dependencies/Features. Molecules 2020, 25, 4641. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colwell, N.; Larion, M.; Giles, A.J.; Seldomridge, A.N.; Sizdahkhani, S.; Gilbert, M.R.; Park, D.M. Hypoxia in the glioblastoma microenvi-ronment: Shaping the phenotype of cancer stem-like cells. Neuro Oncol. 2017, 19, 887–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpson, J.E.; Gammoh, N. The impact of autophagy during the development and survival of glioblastoma. Open Biol. 2020, 10, 200184. [Google Scholar] [CrossRef] [PubMed]
- Maksoud, S. The Role of the Ubiquitin Proteasome System in Glioma: Analysis Emphasizing the Main Molecular Players and Therapeutic Strategies Identified in Glioblastoma Multiforme. Mol. Neurobiol. 2021, 58, 3252–3269. [Google Scholar] [CrossRef]
- Rousseau, A.; Bertolotti, A. Regulation of proteasome assembly and activity in health and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 697–712. [Google Scholar] [CrossRef] [Green Version]
- Kirkin, V.; Rogov, V.V. A Diversity of Selective Autophagy Receptors Determines the Specificity of the Autophagy Pathway. Mol. Cells 2019, 76, 268–285. [Google Scholar] [CrossRef]
- Nam, T.; Han, J.H.; Devkota, S.; Lee, H.-W. Emerging Paradigm of Crosstalk between Autophagy and the Ubiquitin-Proteasome System. Mol. Cells 2017, 40, 897–905. [Google Scholar] [CrossRef]
- Grumati, P.; Dikic, I. Ubiquitin signaling and autophagy. J. Biol. Chem. 2018, 293, 5404–5413. [Google Scholar] [CrossRef]
- Kocaturk, N.M.; Gozuacik, D. Crosstalk between Mammalian Autophagy and the Ubiquitin-Proteasome System. Front. Cell Dev. Biol. 2018, 6, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun-Wang, J.L.; Ivanova, S.; Zorzano, A. The dialogue between the ubiquitin-proteasome system and autophagy: Implications in ageing. Ageing Res. Rev. 2020, 64, 101203. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, Y.; Xuan, C.; Liu, Y.; Shi, H.; Gao, Y. Identification of ubiquitination-related genes in human glioma as indicators of patient prognosis. PLoS ONE 2021, 16, e0250239. [Google Scholar] [CrossRef] [PubMed]
- Vriend, J.; Klonisch, T. Genes of the Ubiquitin Proteasome System Qualify as Differential Markers in Malignant Glioma of Astrocytic and Oligodendroglial Origin. Cell. Mol. Neurobiol. 2022. ahead of print. [Google Scholar] [CrossRef]
- Schulman, B.A.; Harper, J.W. Ubiquitin-like protein activation by E1 enzymes: The apex for downstream signalling pathways. Nat. Rev. Mol. Cell Biol. 2009, 10, 319–331. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Zhao, B. UBA6 and Its Bispecific Pathways for Ubiquitin and FAT10. Int. J. Mol. Sci. 2019, 20, 2250. [Google Scholar] [CrossRef] [Green Version]
- Barghout, S.H.; Schimmer, A.D. E1 Enzymes as Therapeutic Targets in Cancer. Pharmacol. Rev. 2020, 73, 1–58. [Google Scholar] [CrossRef]
- Metzger, M.B.; Pruneda, J.N.; Klevit, R.E.; Weissman, A.M. RING-type E3 ligases: Master manipulators of E2 ubiquitin-conjugating enzymes and ubiquitination. Biochim. Biophys. Acta 2014, 1843, 47–60. [Google Scholar] [CrossRef] [Green Version]
- Scheffner, M.; Kumar, S. Mammalian HECT ubiquitin-protein ligases: Biological and pathophysiological aspects. Biochim. Biophys. Acta 2014, 1843, 61–74. [Google Scholar] [CrossRef]
- Chen, Y.; Jin, J. The application of ubiquitin ligases in the PROTAC drug design. Acta Biochim. et Biophys. Sin. 2020, 52, 776–790. [Google Scholar] [CrossRef]
- Cotton, T.; Lechtenberg, B.C. Chain reactions: Molecular mechanisms of RBR ubiquitin ligases. Biochem. Soc. Trans. 2020, 48, 1737–1750. [Google Scholar] [CrossRef]
- Mabbitt, P.D.; Loreto, A.; Déry, M.-A.; Fletcher, A.J.; Stanley, M.; Pao, K.-C.; Wood, N.T.; Coleman, M.P.; Virdee, S. Structural basis for RING-Cys-Relay E3 ligase activity and its role in axon integrity. Nat. Chem. Biol. 2020, 16, 1227–1236. [Google Scholar] [CrossRef] [PubMed]
- Clague, M.J.; Heride, C.; Urbé, S. The demographics of the ubiquitin system. Trends Cell Biol. 2015, 25, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Grou, C.P.; Pinto, M.P.; Mendes, A.V.; Domingues, P.; Azevedo, J.E. The de novo synthesis of ubiquitin: Identification of deubiqui-tinases acting on ubiquitin precursors. Sci. Rep. 2015, 5, 12836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Bie, P.; Ciechanover, A. Faculty Opinions recommendation of Ubiquitination of E3 ligases: Self-regulation of the ubiquitin system via proteolytic and non-proteolytic mechanisms. Cell Death Differ. 2011, 18, 1393–1402. [Google Scholar] [CrossRef] [Green Version]
- Sahtoe, D.D.; Sixma, T.K. Layers of DUB regulation. Trends Biochem. Sci. 2015, 40, 456–467. [Google Scholar] [CrossRef]
- Liess, A.K.; Kucerova, A.; Schweimer, K.; Yu, L.U.; Roumeliotis, T.I.; Diebold, M.; Lorenz, S. Autoinhibition Mechanism of the Ubiqui-tin-Conjugating Enzyme UBE2S by Autoubiquitination. Structure 2019, 27, 1195–1210.e7. [Google Scholar] [CrossRef]
- Vlachostergios, P.J.; Voutsadakis, I.A.; Papandreou, C.N. The role of ubiquitin-proteasome system in glioma survival and growth. Growth Factors 2013, 31, 106–113. [Google Scholar] [CrossRef]
- Scholz, N.; Kurian, K.M.; Siebzehnrubl, F.A.; Licchesi, J.D.F. Targeting the Ubiquitin System in Glioblastoma. Front. Oncol. 2020, 10, 574011. [Google Scholar] [CrossRef]
- Akutsu, M.; Dikic, I.; Bremm, A. Ubiquitin chain diversity at a glance. J. Cell Sci. 2016, 129, 875–880. [Google Scholar] [CrossRef]
- Huang, Q.; Zhang, X. Emerging Roles and Research Tools of Atypical Ubiquitination. Proteomics 2020, 20, e1900100. [Google Scholar] [CrossRef]
- Magits, W.; Sablina, A.A. The regulation of the protein interaction network by monoubiquitination. Curr. Opin. Struct. Biol. 2022, 73, 102333. [Google Scholar] [CrossRef] [PubMed]
- Ohtake, F. Branched ubiquitin code: From basic biology to targeted protein degradation. J. Biochem. 2022, 171, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Luo, Z.-Q. Post-translational regulation of ubiquitin signaling. J. Cell Biol. 2019, 218, 1776–1786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yau, R.; Rape, M. The increasing complexity of the ubiquitin code. Nature 2016, 18, 579–586. [Google Scholar] [CrossRef]
- Budenholzer, L.; Cheng, C.L.; Li, Y.; Hochstrasser, M. Proteasome Structure and Assembly. J. Mol. Biol. 2017, 429, 3500–3524. [Google Scholar] [CrossRef]
- Tomko, R.J., Jr.; Hochstrasser, M. Molecular architecture and assembly of the eukaryotic proteasome. Annu. Rev. Biochem. 2013, 82, 415–445. [Google Scholar] [CrossRef] [Green Version]
- Sahu, I.; Glickman, M. Structural Insights into Substrate Recognition and Processing by the 20S Proteasome. Biomolecules 2021, 11, 148. [Google Scholar] [CrossRef]
- Kim, K.; Brush, J.M.; Watson, P.A.; Cacalano, N.A.; Iwamoto, K.S.; McBride, W.H. Epidermal Growth Factor Receptor vIII Expression in U87 Glioblastoma Cells Alters Their Proteasome Composition, Function, and Response to Irradiation. Mol. Cancer Res. 2008, 6, 426–434. [Google Scholar] [CrossRef] [Green Version]
- Fabre, B.; Lambour, T.; Garrigues, L.; Ducoux-Petit, M.; Amalric, F.; Monsarrat, B.; Burlet-Schiltz, O.; Bousquet-Dubouch, M.-P. Label-Free Quantitative Proteomics Reveals the Dynamics of Proteasome Complexes Composition and Stoichiometry in a Wide Range of Human Cell Lines. J. Proteome Res. 2014, 13, 3027–3037. [Google Scholar] [CrossRef]
- Greene, E.R.; Dong, K.C.; Martin, A. Understanding the 26S proteasome molecular machine from a structural and conformational dynamics perspective. Curr. Opin. Struct. Biol. 2020, 61, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Sahu, I.; Glickman, M.H. Proteasome in action: Substrate degradation by the 26S proteasome. Biochem. Soc. Trans. 2021, 49, 629–644. [Google Scholar] [CrossRef] [PubMed]
- de Poot, S.A.H.; Tian, G.; Finley, D. Meddling with Fate: The Proteasomal Deubiquitinating Enzymes. J. Mol. Biol. 2017, 429, 3525–3545. [Google Scholar] [CrossRef]
- Muli, C.S.; Tian, W.; Trader, D.J. Small-Molecule Inhibitors of the Proteasome’s Regulatory Particle. ChemBioChem 2019, 20, 1739–1753. [Google Scholar] [CrossRef]
- Sijts, E.J.A.M.; Kloetzel, P.-M. The role of the proteasome in the generation of MHC class I ligands and immune responses. Cell Mol. Life Sci. 2011, 68, 1491–1502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vigneron, N.; Eynde, B.J.V.D. Proteasome Subtypes and Regulators in the Processing of Antigenic Peptides Presented by Class I Molecules of the Major Histocompatibility Complex. Biomolecules 2014, 4, 994–1025. [Google Scholar] [CrossRef] [Green Version]
- Piccinini, M.; Rinaudo, M.T.; Anselmino, A.; Ramondetti, C.; Buccinnà, B.; Fiano, V.; Schiffer, D. Characterization of the 20S proteasome in human glioblastomas. Anticancer Res. 2005, 25, 3203–3210. [Google Scholar]
- Pickering, A.M.; Davies, K.J.A. Differential roles of proteasome and immunoproteasome regulators Pa28αβ, Pa28γ and Pa200 in the degradation of oxidized proteins. Arch. Biochem. Biophys. 2012, 523, 181–190. [Google Scholar] [CrossRef] [Green Version]
- Sijts, A.; Sun, Y.; Janek, K.; Kral, S.; Paschen, A.; Schadendorf, D.; Kloetzel, P.-M. The role of the proteasome activator PA28 in MHC class I antigen processing. Mol. Immunol. 2002, 39, 165–169. [Google Scholar] [CrossRef]
- Ustrell, V.; Hoffman, L.; Pratt, G.; Rechsteiner, M. PA200, a nuclear proteasome activator involved in DNA repair. EMBO J. 2002, 21, 3516–3525. [Google Scholar] [CrossRef] [Green Version]
- Blickwedehl, J.; Agarwal, M.; Seong, C.; Pandita, R.K.; Melendy, T.; Sung, P.; Bangia, N. Role for proteasome activator PA200 and post-glutamyl proteasome activity in genomic stability. Proc. Natl. Acad. Sci. USA 2008, 105, 16165–16170. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.-X.; Ma, S.; Han, X.; Luo, Z.-Y.; Zhu, Q.-Q.; Chiba, T.; Xie, W.; Lin, K.; Qiu, X.-B. Proteasome activator PA200 maintains stability of histone marks during transcription and aging. Theranostics 2021, 11, 1458–1472. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Trivedi, A.K. Regulation of apoptosis by E3 ubiquitin ligases in ubiquitin proteasome system. Cell Biol. Int. 2020, 44, 721–734. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Zhai, X.; Liang, P.; Cui, H. Overcoming TRAIL Resistance for Glioblastoma Treatment. Biomolecules 2021, 11, 572. [Google Scholar] [CrossRef] [PubMed]
- Lospinoso Severini, L.; Bufalieri, F.; Infante, P.; Di Marcotullio, L. Proteolysis-Targeting Chimera (PROTAC): Is the Technology Looking at the Treatment of Brain Tumors? Front. Cell Dev. Biol. 2022, 10, 854352. [Google Scholar] [CrossRef]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef]
- Melia, T.J.; Lystad, A.H.; Simonsen, A. Autophagosome biogenesis: From membrane growth to closure. J. Cell Biol. 2020, 219, e202002085. [Google Scholar] [CrossRef]
- Zhou, F.; Wu, Z.; Zhao, M.; Murtazina, R.; Cai, J.; Zhang, A.; Li, R.; Sun, D.; Li, W.; Zhao, L.; et al. Rab5-dependent autophagosome closure by ESCRT. J. Cell Biol. 2019, 218, 1908–1927. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.G.; Zhang, H. Autophagosome maturation: An epic journey from the ER to lysosomes. J. Cell Biol. 2019, 218, 757–770. [Google Scholar] [CrossRef]
- Manea, A.J.; Ray, S.K. Regulation of autophagy as a therapeutic option in glioblastoma. Apoptosis 2021, 26, 574–599. [Google Scholar] [CrossRef]
- Zhang, X.; Deibert, C.P.; Kim, W.-J.; Jaman, E.; Rao, A.V.; Lotze, M.T.; Amankulor, N.M. Autophagy inhibition is the next step in the treatment of glioblastoma patients following the Stupp era. Cancer Gene Ther. 2021, 28, 971–983. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Niu, L.; Bai, Y.; Le, W. Glioblastoma: Targeting the autophagy in tumorigenesis. Brain Res. Bull. 2019, 153, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.; Baig, M.; Mahfooz, S.; Rahim, M.; Karacam, B.; Elbasan, E.B.; Ulasov, I.; Dong, J.-J.; Hatiboglu, M.A. Deciphering the Role of Autophagy in Treatment of Resistance Mechanisms in Glioblastoma. Int. J. Mol. Sci. 2021, 22, 1318. [Google Scholar] [CrossRef]
- Tamrakar, S.; Yashiro, M.; Kawashima, T.; Uda, T.; Terakawa, Y.; Kuwae, Y.; Ohata, K. Clinicopathological Significance of Autopha-gy-related Proteins and its Association with Genetic Alterations in Gliomas. Anticancer Res. 2019, 39, 1233–1242. [Google Scholar] [CrossRef] [PubMed]
- Colella, B.; Faienza, F.; Di Bartolomeo, S. EMT Regulation by Autophagy: A New Perspective in Glioblastoma Biology. Cancers 2019, 11, 312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cecconi, F.; Levine, B. The Role of Autophagy in Mammalian Development: Cell Makeover Rather than Cell Death. Dev. Cell 2008, 15, 344–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, E.Y.; Xu, N.; O’Prey, J.; Lao, L.Y.; Joshi, S.; Long, J.S.; O’Prey, M.; Croft, D.R.; Beaumatin, F.; Baudot, A.D.; et al. Loss of autophagy causes a synthetic lethal deficiency in DNA repair. Proc. Natl. Acad. Sci. USA 2015, 112, 773–778. [Google Scholar] [CrossRef] [Green Version]
- Denton, D.; Kumar, S. Autophagy-dependent cell death. Cell Death Differ. 2019, 26, 605–616. [Google Scholar] [CrossRef] [Green Version]
- Chien, C.-H.; Hsueh, W.-T.; Chuang, J.-Y.; Chang, K.-Y. Role of autophagy in therapeutic resistance of glioblastoma. J. Cancer Metastasis Treat. 2019, 5, 66. [Google Scholar] [CrossRef] [Green Version]
- Noonan, J.; Zarrer, J.; Murphy, B.M. Targeting Autophagy in Glioblastoma. Crit. Rev. Oncog. 2016, 21, 241–252. [Google Scholar] [CrossRef]
- Wang, F.; Ning, S.; Yu, B.; Wang, Y. USP14: Structure, Function, and Target Inhibition. Front. Pharmacol. 2021, 12, 801328. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zou, S.; Yin, D.; Zhao, L.; Finley, D.; Wu, Z.; Mao, Y. USP14-regulated allostery of the human proteasome by time-resolved cryo-EM. Nature 2022, 605, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, M.; Hirata, N.; Suizu, F. The links between AKT and two intracellular proteolytic cascades: Ubiquitination and au-tophagy. Biochim. Biophys. Acta 2014, 1846, 342–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, D.; Shan, B.; Lee, B.-H.; Zhu, K.; Zhang, T.; Sun, H.; Liu, M.; Shi, L.; Liang, W.; Qian, L.; et al. Phosphorylation and activation of ubiquitin-specific protease-14 by Akt regulates the ubiquitin-proteasome system. eLife 2015, 4, e10510. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zhang, C.; Gu, C.; Li, Q.; Wu, N. Function of Deubiquitinating Enzyme USP14 as Oncogene in Different Types of Cancer. Cell. Physiol. Biochem. 2016, 38, 993–1002. [Google Scholar] [CrossRef]
- Han, K.H.; Kwak, M.; Lee, T.H.; Park, M.-S.; Jeong, I.-H.; Kim, M.J.; Jin, J.-O.; Lee, P.C.-W. USP14 Inhibition Regulates Tumorigenesis by Inducing Autophagy in Lung Cancer In Vitro. Int. J. Mol. Sci. 2019, 20, 5300. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Wang, J.; Yuan, X.; Yang, S.; Xu, X.; Li, K.; Tian, Y. IU1 suppresses proliferation of cervical cancer cells through MDM2 deg-radation. Int. J. Biol. Sci. 2020, 16, 2951–2963. [Google Scholar] [CrossRef]
- Xie, W.; Xu, L. Ubiquitin-specific protease 14 promotes radio-resistance and suppresses autophagy in oral squamous cell car-cinoma. Exp. Cell Res. 2021, 398, 112385. [Google Scholar] [CrossRef]
- Wu, T.; Li, C.; Zhou, C.; Niu, X.; Li, G.; Zhou, Y.; Gu, X.; Cui, H. Inhibition of USP14 enhances anti-tumor effect in vemurafenib-resistant melanoma by regulation of Skp2. Cell Biol. Toxicol. 2022. ahead of print. [Google Scholar] [CrossRef]
- Donovan, P.; Cato, K.; Legaie, R.; Jayalath, R.; Olsson, G.; Hall, B.; Olson, S.; Boros, S.; Reynolds, B.A.; Harding, A. Hyperdiploid tumor cells increase phenotypic heterogeneity within Glioblastoma tumors. Mol. BioSyst. 2014, 10, 741–758. [Google Scholar] [CrossRef]
- Liang, W.; Fang, J.; Zhou, S.; Hu, W.; Yang, Z.; Li, Z.; Dai, L.; Tao, Y.; Fu, X.; Wang, X. The role of ubiquitin-specific peptidases in glioma progression. Biomed. Pharmacother. 2022, 146, 112585. [Google Scholar] [CrossRef] [PubMed]
- Minchenko, O.H.; Tsymbal, D.O.; Minchenko, D.O.; Riabovol, O.O.; Halkin, O.V.; Ratushna, O.O. IRE-1α regulates expression of ubiquitin specific peptidases during hypoxic response in U87 glioma cells. Endoplasmic Reticulum Stress Dis. 2016, 3, 50–62. [Google Scholar] [CrossRef] [Green Version]
- Tsai, Y.; Xia, C.; Sun, Z. The Inhibitory Effect of 6-Gingerol on Ubiquitin-Specific Peptidase 14 Enhances Autophagy-Dependent Ferroptosis and Anti-Tumor in vivo and in vitro. Front. Pharmacol. 2020, 11, 598555. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Park, S.; Lee, J.H.; Mun, J.Y.; Choi, W.H.; Yun, Y.; Lee, J.; Kim, J.H.; Kang, M.-J.; Lee, M.J. Dual Function of USP14 Deubiquitinase in Cellular Proteasomal Activity and Autophagic Flux. Cell Rep. 2018, 24, 732–743. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Shan, B.; Sun, H.; Xiao, J.; Zhu, K.; Xie, X.; Li, X.; Liang, W.; Lu, X.; Qian, L.; et al. USP14 regulates autophagy by suppressing K63 ubiquitination of Beclin 1. Genes Dev. 2016, 30, 1718–1730. [Google Scholar] [CrossRef] [Green Version]
- Min, Y.; Lee, S.; Kim, M.-J.; Chun, E.; Lee, K.-Y. Ubiquitin-Specific Protease 14 Negatively Regulates Toll-Like Receptor 4-Mediated Signaling and Autophagy Induction by Inhibiting Ubiquitination of TAK1-Binding Protein 2 and Beclin 1. Front. Immunol. 2017, 8, 1827. [Google Scholar] [CrossRef] [Green Version]
- Pavel, M.; Tanasa, R.; Park, S.J.; Rubinsztein, D.C. The complexity of biological control systems: An autophagy case study. BioEssays 2022, 44, e2100224. [Google Scholar] [CrossRef]
- Sharma, A.; Alswillah, T.; Singh, K.; Chatterjee, P.; Willard, B.; Venere, M.; Summers, M.K.; Almasan, A. USP14 regulates DNA damage repair by targeting RNF168-dependent ubiquitination. Autophagy 2018, 14, 1976–1990. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Alswillah, T.; Kapoor, I.; Debjani, P.; Willard, B.; Summers, M.K.; Gong, Z.; Almasan, A. USP14 is a deubiquitinase for Ku70 and critical determinant of non-homologous end joining repair in autophagy and PTEN-deficient cells. Nucleic Acids Res. 2020, 48, 736–747. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.G.; Zhao, H.; Miao, L.; Wang, L.; Sun, F.; Zhang, H. The p53-induced Gene Ei24 Is an Essential Component of the Basal Autophagy Pathway. J. Biol. Chem. 2012, 287, 42053–42063. [Google Scholar] [CrossRef] [Green Version]
- Zang, Y.; Zhu, L.; Li, T.; Wang, Q.; Li, J.; Qian, Y.; Wei, L.; Xie, M.; Tang, W.-H.; Liu, X.; et al. EI24 Suppresses Tumorigenesis in Pancreatic Cancer via Regulating c-Myc. Gastroenterol. Res. Pract. 2018, 2018, 2626545. [Google Scholar] [CrossRef] [PubMed]
- Mork, C.N.; Faller, D.V.; Spanjaard, R.A. Loss of putative tumor suppressor EI24/PIG8 confers resistance to etoposide. FEBS Lett. 2007, 581, 5440–5444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, L.; Ma, J.; Yang, W.; Cao, L.; Wang, X.; Niu, L.; Li, Y.; Zhou, W.; Zhang, Y.; Liu, J.; et al. EI24 Inhibits Cell Proliferation and Drug Resistance of Esophageal Squamous Cell Carcinoma. Front. Oncol. 2020, 10, 1570. [Google Scholar] [CrossRef] [PubMed]
- Devkota, S.; Sung, Y.H.; Choi, J.-M.; Lee, J.; Ha, N.Y.; Kim, H.; Cho, B.C.; Song, J.; Lee, H.-W. Ei24-deficiency attenuates protein kinase Cα signaling and skin carcinogenesis in mice. Int. J. Biochem. Cell Biol. 2012, 44, 1887–1896. [Google Scholar] [CrossRef]
- Nam, T.W.; Park, S.Y.; Lee, J.H.; Roh, J.I.; Lee, H.-W. Effect of EI24 expression on the tumorigenesis of ApcMin/+ colorectal cancer mouse model. Biochem. Biophys. Res. Commun. 2019, 514, 1087–1092. [Google Scholar] [CrossRef]
- Devkota, S.; Jeong, H.; Kim, Y.; Ali, M.; Roh, J.-I.; Hwang, D.; Lee, H.-W. Functional characterization of EI24-induced autophagy in the degradation of RING-domain E3 ligases. Autophagy 2016, 12, 2038–2053. [Google Scholar] [CrossRef]
- Zhang, W.; Sun, Y.; Liu, L.; Li, Z. Prognostic Significance of TNFR-Associated Factor 1 and 2 (TRAF1 and TRAF2) in Glioblastoma. J. Pharmacol. Exp. Ther. 2017, 23, 4506–4512. [Google Scholar] [CrossRef] [Green Version]
- Fortin Ensign, S.P.; Mathews, I.T.; Eschbacher, J.M.; Loftus, J.C.; Symons, M.H.; Tran, N.L. The Src homology 3 domain-containing guanine nucleotide exchange factor is overexpressed in high-grade gliomas and promotes tumor necrosis factor-like weak inducer of apoptosis-fibroblast growth factor-inducible 14-induced cell migration and invasion via tumor necrosis factor receptor-associated factor 2. J. Biol. Chem. 2013, 288, 21887–21897. [Google Scholar]
- Lin, J.-C.; Kuo, C.-Y.; Tsai, J.-T.; Liu, W.-H. miR-671-5p Inhibition by MSI1 Promotes Glioblastoma Tumorigenesis via Radiore-sistance, Tumor Motility and Cancer Stem-like Cell Properties. Biomedicines 2021, 10, 21. [Google Scholar] [CrossRef]
- Zheng, M.; Morgan-Lappe, S.E.; Yang, J.; Bockbrader, K.M.; Pamarthy, D.; Thomas, D.; Fesik, S.W.; Sun, Y. Growth Inhibition and Radiosensitization of Glioblastoma and Lung Cancer Cells by Small Interfering RNA Silencing of Tumor Necrosis Factor Receptor–Associated Factor 2. Cancer Res. 2008, 68, 7570–7578. [Google Scholar] [CrossRef] [Green Version]
- Ohtsu, N.; Nakatani, Y.; Yamashita, D.; Ohue, S.; Ohnishi, T.; Kondo, T. Eva1 Maintains the Stem-like Character of Glioblasto-ma-Initiating Cells by Activating the Noncanonical NF-κB Signaling Pathway. Cancer Res. 2016, 76, 171–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, V.; Tewari, R.; Sk, U.H.; Joseph, C.; Sen, E. Ebselen sensitizes glioblastoma cells to Tumor Necrosis Factor (TNFα)-induced apoptosis through two distinct pathways involving NF-κB downregulation and Fas-mediated formation of death inducing signaling complex. Int. J. Cancer 2008, 123, 2204–2212. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.-M.; NI, J.-D.; Song, D.; Ding, M.; Huang, J. Interplay between unfolded protein response and autophagy promotes tumor drug resistance. Oncol. Lett. 2015, 10, 1959–1969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wade, M.; Li, Y.-C.; Wahl, G.M. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat. Rev. Cancer 2013, 13, 83–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnoff, T.E.; El-Deiry, W.S. MDM2/MDM4 amplification and CDKN2A deletion in metastatic melanoma and glioblastoma multiforme may have implications for targeted therapeutics and immunotherapy. Am. J. Cancer Res. 2022, 12, 2102–2117. [Google Scholar]
- Qi, S.M.; Cheng, G.; Cheng, X.D.; Xu, Z.; Xu, B.; Zhang, W.D.; Qin, J.J. Targeting USP7-Mediated Deubiquitination of MDM2/MDMX-p53 Pathway for Cancer Therapy: Are We There Yet? Front. Cell Dev. Biol. 2020, 8, 233. [Google Scholar] [CrossRef] [Green Version]
- Cheng, C.; Niu, C.; Yang, Y.; Wang, Y.; Lu, M. Expression of HAUSP in gliomas correlates with disease progression and survival of patients. Oncol. Rep. 2013, 29, 1730–1736. [Google Scholar] [CrossRef] [Green Version]
- Ko, C.-Y.; Lin, C.-H.; Chuang, J.-Y.; Chang, W.-C.; Hsu, T.-I. MDM2 Degrades Deacetylated Nucleolin through Ubiquitination to Promote Glioma Stem-Like Cell Enrichment for Chemotherapeutic Resistance. Mol. Neurobiol. 2018, 55, 3211–3223. [Google Scholar] [CrossRef]
- Bohlman, S.; Manfredi, J.J. p53-Independent Effects of Mdm2. Subcell. Biochem. 2014, 85, 235–246. [Google Scholar] [CrossRef] [Green Version]
- Miles, X.; Vandevoorde, C.; Hunter, A.; Bolcaen, J. MDM2/X Inhibitors as Radiosensitizers for Glioblastoma Targeted Therapy. Front. Oncol. 2021, 11, 703442. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.-L.; DeLay, M.; Jahangiri, A.; Molinaro, A.M.; Rose, S.D.; Carbonell, W.S.; Aghi, M.K. Hypoxia-Induced Autophagy Promotes Tumor Cell Survival and Adaptation to Antiangiogenic Treatment in Glioblastoma. Cancer Res 2012, 72, 1773–1783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, K.; Unger, E. Overcoming tumor hypoxia as a barrier to radiotherapy, chemotherapy and immunotherapy in cancer treatment. Int. J. Nanomed. 2018, 13, 6049–6058. [Google Scholar] [CrossRef]
- Domènech, M.; Hernández, A.; Plaja, A.; Martínez-Balibrea, E.; Balañà, C. Hypoxia: The Cornerstone of Glioblastoma. Int. J. Mol. Sci. 2021, 22, 12608. [Google Scholar] [CrossRef] [PubMed]
- Womeldorff, M.; Gillespie, D.; Jensen, R.L. Hypoxia-inducible factor–1 and associated upstream and downstream proteins in the pathophysiology and management of glioblastoma. Neurosurg. Focus 2014, 37, E8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazure, N.M.; Pouysségur, J. Hypoxia-induced autophagy: Cell death or cell survival? Curr. Opin. Cell Biol. 2010, 22, 177–180. [Google Scholar] [CrossRef]
- Daskalaki, I.; Gkikas, I.; Tavernarakis, N. Hypoxia and Selective Autophagy in Cancer Development and Therapy. Front. Cell Dev. Biol. 2018, 6, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouysségur, J.; Mazure, N.M. Hypoxia-Induced Autophagy Is Mediated through Hypoxia-Inducible Factor Induction of BNIP3 and BNIP3L via Their BH3 Domains. Mol. Cell. Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef] [Green Version]
- Zhang, A.; Huang, Z.; Tao, W.; Zhai, K.; Wu, Q.; Rich, J.N.; Bao, S. USP33 deubiquitinates and stabilizes HIF-2alpha to promote hy-poxia response in glioma stem cells. EMBO J. 2022, 41, e109187. [Google Scholar] [CrossRef]
- Li, Z.; Wang, D.; Messing, E.M.; Wu, G. VHL protein-interacting deubiquitinating enzyme 2 deubiquitinates and stabilizes HIF-1α. EMBO Rep. 2005, 6, 373–378. [Google Scholar] [CrossRef] [Green Version]
- Du, S.-C.; Zhu, L.; Wang, Y.-X.; Liu, J.; Zhang, D.; Chen, Y.-L.; Peng, Q.; Liu, W.; Liu, B. SENP1-mediated deSUMOylation of USP28 regulated HIF-1α accumulation and activation during hypoxia response. Cancer Cell Int. 2019, 19, 4. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, K.; Frew, I.; Hagensen, M.K.; Skals, M.; Habelhah, H.; Bhoumik, A.; Kadoya, T.; Erdjument-Bromage, H.; Tempst, P.; Frappell, P.B.; et al. Siah2 Regulates Stability of Prolyl-Hydroxylases, Controls HIF1α Abundance, and Modulates Physiological Responses to Hypoxia. Cell 2004, 117, 941–952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, H.; Zheng, B.; Wu, Y.; Tang, Y.; Wang, L.; Gao, Y.; Gong, H.; DU, J.; Yu, R. Ubiquitin ligase Siah1 promotes the migration and invasion of human glioma cells by regulating HIF-1α signaling under hypoxia. Oncol. Rep. 2015, 33, 1185–1190. [Google Scholar] [CrossRef] [PubMed]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef] [Green Version]
- Kunadis, E.; Lakiotaki, E.; Korkolopoulou, P.; Piperi, C. Targeting post-translational histone modifying enzymes in glioblastoma. Pharmacol. Ther. 2021, 220, 107721. [Google Scholar] [CrossRef]
- Ramaiah, M.J.; Tangutur, A.D.; Manyam, R.R. Epigenetic modulation and understanding of HDAC inhibitors in cancer therapy. Life Sci. 2021, 277, 119504. [Google Scholar] [CrossRef]
- Seto, E.; Yoshida, M. Erasers of Histone Acetylation: The Histone Deacetylase Enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef] [Green Version]
- Ho, T.C.S.; Chan, A.H.Y.; Ganesan, A. Thirty Years of HDAC Inhibitors: 2020 Insight and Hindsight. J. Med. Chem. 2020, 63, 12460–12484. [Google Scholar] [CrossRef]
- Li, T.; Zhang, C.; Hassan, S.; Liu, X.; Song, F.; Chen, K.; Yang, J. Histone deacetylase 6 in cancer. J. Hematol. Oncol. 2018, 11, 111. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.B.; Hsu, C.C.; Hsu, T.I.; Liou, J.P.; Chang, K.Y.; Chen, P.Y.; Chuang, J.Y. Increased activation of HDAC1/2/6 and Sp1 underlies therapeutic resistance and tumor growth in glioblastoma. Neuro Oncol. 2020, 22, 1439–1451. [Google Scholar] [CrossRef]
- Kim, G.W.; Lee, D.H.; Yeon, S.-K.; Jeon, Y.H.; Yoo, J.; Lee, S.W.; Kwon, S.H. Temozolomide-resistant Glioblastoma Depends on HDAC6 Activity through Regulation of DNA Mismatch Repair. Anticancer Res. 2019, 39, 6731–6741. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Hu, P.; Tang, F.; Lian, H.; Chen, X.; Zhang, Y.; Xie, C. HDAC6 promotes cell proliferation and confers resistance to te-mozolomide in glioblastoma. Cancer Lett. 2016, 379, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Xia, Y.; Hu, K.; Zeng, S.; Wu, L.; Liu, S.; Zhi, C.; Lai, M.; Chen, D.; Xie, L.; et al. Histone deacetylase 6 promotes growth of glioblastoma through the MKK7/JNK/c-Jun signaling pathway. J. Neurochem. 2020, 152, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Urdiciain, A.; Erausquin, E.; Zelaya, M.; Zazpe, I.; Lanciego, J.; Meléndez, B.; Rey, J.; Idoate, M.; Galdo, N.R.-D.; Castresana, J. Silencing of Histone Deacetylase 6 Decreases Cellular Malignancy and Contributes to Primary Cilium Restoration, Epithelial-to-Mesenchymal Transition Reversion, and Autophagy Inhibition in Glioblastoma Cell Lines. Biology 2021, 10, 467. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.W.; Chen, M.T.; Lin, L.T.; Huang, P.I.; Lo, W.L.; Yang, Y.P.; Wu, C.W. TDP-43/HDAC6 axis promoted tumor progression and regulated nutrient deprivation-induced autophagy in glioblastoma. Oncotarget 2017, 8, 56612–56625. [Google Scholar] [CrossRef] [Green Version]
- Scotter, E.L.; Vance, C.; Nishimura, A.L.; Lee, Y.-B.; Chen, H.-J.; Urwin, H.; Sardone, V.; Mitchell, J.C.; Rogelj, B.; Rubinsztein, D.C.; et al. Differential roles of the ubiquitin proteasome system (UPS) and autophagy in the clearance of soluble and aggregated TDP-43 species. J. Cell Sci. 2014, 127, 1263–1278. [Google Scholar] [CrossRef] [Green Version]
- Jo, M.; Lee, S.; Kim, K.; Lee, S.; Kim, S.R.; Kim, H.-J. Inhibition of MEK5 suppresses TDP-43 toxicity via the mTOR-independent activation of the autophagy-lysosome pathway. Biochem. Biophys. Res. Commun. 2019, 513, 925–932. [Google Scholar] [CrossRef]
- Salemi, L.M.; Almawi, A.W.; Lefebvre, K.J.; Schild-Poulter, C. Aggresome formation is regulated by RanBPM through an interac-tion with HDAC6. Biol. Open. 2014, 3, 418–430. [Google Scholar] [CrossRef] [Green Version]
- Boyault, C.; Gilquin, B.; Zhang, Y.; Rybin, V.; Garman, E.; Meyer-Klaucke, W.; Matthias, P.; Müller, C.; Khochbin, S. HDAC6–p97/VCP controlled polyubiquitin chain turnover. EMBO J. 2006, 25, 3357–3366. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.Y.; Zhang, C.; Zhang, Y.; Chen, L.; Chen, B.D.; Li, Q.Z.; Li, W.P. A novel HDAC6 inhibitor Tubastatin A: Controls HDAC6-p97/VCP-mediated ubiquitination-autophagy turnover and reverses Temozolomide-induced ER stress-tolerance in GBM cells. Cancer Lett. 2017, 391, 89–99. [Google Scholar] [CrossRef]
- Strickland, M.; Stoll, E.A. Metabolic Reprogramming in Glioma. Front. Cell Dev. Biol. 2017, 5, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, K.M.; Shen, H.; McKelvey, K.J.; Gee, H.E.; Hau, E. Targeting Glucose Metabolism of Cancer Cells with Dichloroacetate to Radiosensitize High-Grade Gliomas. Int. J. Mol. Sci. 2021, 22, 7265. [Google Scholar] [CrossRef] [PubMed]
- Duraj, T.; García-Romero, N.; Carrión-Navarro, J.; Madurga, R.; Mendivil, A.; Prat-Acin, R.; Garcia-Cañamaque, L.; Ayuso-Sacido, A. Beyond the Warburg Effect: Oxidative and Glycolytic Phenotypes Coexist within the Metabolic Heterogeneity of Glioblastoma. Cells 2021, 10, 202. [Google Scholar] [CrossRef] [PubMed]
- Agnihotri, S.; Golbourn, B.; Huang, X.; Remke, M.; Younger, S.; Cairns, R.A.; Chalil, A.; Smith, C.A.; Krumholtz, S.-L.; Mackenzie, D.; et al. PINK1 Is a Negative Regulator of Growth and the Warburg Effect in Glioblastoma. Cancer Res 2016, 76, 4708–4719. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Zhu, P.; Huang, R.; Wang, C.; Sun, L.; Lan, B.; He, Y.; Zhao, H.; Gao, Y. PINK1: The guard of mitochondria. Life Sci. 2020, 259, 118247. [Google Scholar] [CrossRef]
- Qin, S.; Ye, L.; Zheng, Y.; Gao, J. Cytosolic PINK1 orchestrates protein translation during proteasomal stress by phosphorylating the translation elongation factor eEF1A1. FEBS Lett. 2021, 595, 507–520. [Google Scholar] [CrossRef]
- Okatsu, K.; Uno, M.; Koyano, F.; Go, E.; Kimura, M.; Oka, T.; Matsuda, N. A dimeric PINK1-containing complex on depolarized mito-chondria stimulates Parkin recruitment. J. Biol. Chem. 2013, 288, 36372–36384. [Google Scholar] [CrossRef] [Green Version]
- Gonçalves, F.; Morais, V. PINK1: A Bridge between Mitochondria and Parkinson’s Disease. Life 2021, 11, 371. [Google Scholar] [CrossRef]
- Yeo, C.W.; Ng, F.S.; Chai, C.; Tan, J.M.; Koh, G.R.; Chong, Y.K.; Lim, K.L. Parkin pathway activation mitigates glioma cell prolifera-tion and predicts patient survival. Cancer Res. 2012, 72, 2543–2553. [Google Scholar] [CrossRef] [Green Version]
- Lin, D.-C.; Xu, L.; Chen, Y.; Yan, H.; Hazawa, M.; Doan, N.; Said, J.W.; Ding, L.-W.; Liu, L.-Z.; Yang, H.; et al. Genomic and Functional Analysis of the E3 Ligase PARK2 in Glioma. Cancer Res. 2015, 75, 1815–1827. [Google Scholar] [CrossRef] [Green Version]
- Heo, J.-M.; Ordureau, A.; Paulo, J.A.; Rinehart, J.; Harper, J.W. The PINK1-PARKIN Mitochondrial Ubiquitylation Pathway Drives a Program of OPTN/NDP52 Recruitment and TBK1 Activation to Promote Mitophagy. Mol. Cell 2015, 60, 7–20. [Google Scholar] [CrossRef] [Green Version]
- Bernardini, J.P.; Lazarou, M.; Dewson, G. Parkin and mitophagy in cancer. Oncogene 2017, 36, 1315–1327. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, C.; Zhao, Y.; Yue, X.; Wu, H.; Huang, S.; Chen, J.; Tomsky, K.; Xie, H.; Khella, C.A.; et al. Parkin targets HIF-1α for ubiquitination and degradation to inhibit breast tumor progression. Nat. Commun. 2017, 8, 1823. [Google Scholar] [CrossRef] [PubMed]
- Maugeri, G.; D’Amico, A.G.; Reitano, R.; Saccone, S.; Federico, C.; Cavallaro, S.; D’Agata, V. Parkin modulates expression of HIF-1α and HIF-3α during hypoxia in gliobastoma-derived cell lines in vitro. Cell Tissue Res. 2016, 364, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Veeriah, S.; Morris, L.G.; Solit, D.; Chan, T.A. The familial Parkinson Disease gene PARK2 is a multisite tumor suppressor on chromosome 6q25.2-27 that regulates cyclin E. Cell Cycle 2010, 9, 1451–1452. [Google Scholar] [CrossRef] [Green Version]
- Tay, S.-P.; Yeo, C.W.; Chai, C.; Chua, P.-J.; Tan, H.-M.; Ang, A.X.; Yip, D.L.; Sung, J.-X.; Tan, P.H.; Bay, B.-H.; et al. Parkin Enhances the Expression of Cyclin-dependent Kinase 6 and Negatively Regulates the Proliferation of Breast Cancer Cells. J. Biol. Chem. 2010, 285, 29231–29238. [Google Scholar] [CrossRef] [Green Version]
- Durcan, T.M.; Fon, E.A. USP8 and PARK2/parkin-mediated mitophagy. Autophagy 2015, 11, 428–429. [Google Scholar] [CrossRef] [Green Version]
- Cornelissen, T.; Haddad, D.; Wauters, F.; Van Humbeeck, C.; Mandemakers, W.; Koentjoro, B.; Sue, C.; Gevaert, K.; De Strooper, B.; Verstreken, P.; et al. The deubiquitinase USP15 antagonizes Parkin-mediated mitochondrial ubiquitination and mitophagy. Hum. Mol. Genet. 2014, 23, 5227–5242. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, C.N.; Baughman, J.M.; Phu, L.; Tea, J.S.; Yu, C.; Coons, M.; Kirkpatrick, D.S.; Bingol, B.; Corn, J.E. USP30 and parkin homeostatically regulate atypical ubiquitin chains on mitochondria. Nat. Cell Biol. 2015, 17, 160–169. [Google Scholar] [CrossRef]
- Rouland, L.; Duplan, E.; dos Santos, L.R.; Bernardin, A.; Katula, K.S.; Manfioletti, G.; Idbaih, A.; Checler, F.; da Costa, C.A. Therapeutic potential of parkin as a tumor suppressor via transcriptional control of cyclins in glioblastoma cell and animal models. Theranostics 2021, 11, 10047–10063. [Google Scholar] [CrossRef]
- Jonas, S.; Izaurralde, E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat. Rev. Genet. 2015, 16, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Saliminejad, K.; Khorram Khorshid, H.R.; Soleymani Fard, S.; Ghaffari, S.H. An overview of microRNAs: Biology, functions, therapeutics, and analysis methods. J. Cell Physiol. 2019, 234, 5451–5465. [Google Scholar] [CrossRef] [PubMed]
- Juźwik, C.A.; Drake, S.S.; Zhang, Y.; Paradis-Isler, N.; Sylvester, A.; Amar-Zifkin, A.; Fournier, A.E. microRNA dysregulation in neuro-degenerative diseases: A systematic review. Prog. Neurobiol. 2019, 182, 101664. [Google Scholar] [CrossRef]
- Ali Syeda, Z.; Langden, S.S.S.; Munkhzul, C.; Lee, M.; Song, S.J. Regulatory Mechanism of MicroRNA Expression in Cancer. Int. J. Mol. Sci. 2020, 21, 1723. [Google Scholar] [CrossRef] [Green Version]
- Kinser, H.E.; Pincus, Z. MicroRNAs as modulators of longevity and the aging process. Hum. Genet. 2020, 139, 291–308. [Google Scholar] [CrossRef] [PubMed]
- Siasos, G.; Bletsa, E.; Stampouloglou, P.K.; Oikonomou, E.; Tsigkou, V.; Paschou, S.A.; Vlasis, K.; Marinos, G.; Vavuranakis, M.; Stefanadis, C.; et al. MicroRNAs in cardiovascular disease. Hell. J. Cardiol. 2020, 61, 165–173. [Google Scholar] [CrossRef]
- Wang, Y.; Hou, J.; He, D.; Sun, M.; Zhang, P.; Yu, Y.; Chen, Y. The Emerging Function and Mechanism of ceRNAs in Cancer. Trends Genet. 2016, 32, 211–224. [Google Scholar] [CrossRef] [Green Version]
- Friedman, R.C.; Farh, K.K.-H.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef] [Green Version]
- Smith, E.S.; Whitty, E.; Yoo, B.; Moore, A.; Sempere, L.F.; Medarova, Z. Clinical Applications of Short Non-Coding RNA-Based Therapies in the Era of Precision Medicine. Cancers 2022, 14, 1588. [Google Scholar] [CrossRef]
- Zogg, H.; Singh, R.; Ro, S. Current Advances in RNA Therapeutics for Human Diseases. Int. J. Mol. Sci. 2022, 23, 2736. [Google Scholar] [CrossRef]
- Henriksen, M.; Johnsen, K.B.; Andersen, H.H.; Pilgaard, L.; Duroux, M. MicroRNA Expression Signatures Determine Prognosis and Survival in Glioblastoma Multiforme—A Systematic Overview. Mol. Neurobiol. 2014, 50, 896–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piwecka, M.; Rolle, K.; Belter, A.; Barciszewska, A.M.; Żywicki, M.; Michalak, M.; Barciszewski, J. Comprehensive analysis of microRNA ex-pression profile in malignant glioma tissues. Mol. Oncol. 2015, 9, 1324–1340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pottoo, F.H.; Javed, N.; Rahman, J.U.; Abu-Izneid, T.; Alam Khan, F. Targeted delivery of miRNA based therapeuticals in the clinical management of Glioblastoma Multiforme. Semin. Cancer Biol. 2021, 69, 391–398. [Google Scholar] [CrossRef]
- Akers, J.C.; Ramakrishnan, V.; Kim, R.; Skog, J.; Nakano, I.; Pingle, S.; Chen, C.C. MiR-21 in the extracellular vesicles (EVs) of cerebrospinal fluid (CSF): A platform for glioblastoma biomarker development. PLoS ONE. 2013, 8, e78115. [Google Scholar] [CrossRef]
- Zottel, A.; Šamec, N.; Kump, A.; Dall’Olio, L.R.R.; Dominkuš, P.P.; Romih, R.; Hudoklin, S.; Mlakar, J.; Nikitin, D.; Sorokin, M.; et al. Analysis of miR-9-5p, miR-124-3p, miR-21-5p, miR-138-5p, and miR-1-3p in Glioblastoma Cell Lines and Extracellular Vesicles. Int. J. Mol. Sci. 2020, 21, 8491. [Google Scholar] [CrossRef]
- Masoudi, M.S.; Mehrabian, E.; Mirzaei, H. MiR-21: A key player in glioblastoma pathogenesis. J. Cell Biochem. 2018, 119, 1285–1290. [Google Scholar] [CrossRef]
- Gwak, H.-S.; Kim, T.H.; Jo, G.H.; Kim, Y.-J.; Kwak, H.-J.; Kim, J.H.; Yin, J.; Yoo, H.; Lee, S.H.; Park, J.B. Silencing of MicroRNA-21 Confers Radio-Sensitivity through Inhibition of the PI3K/AKT Pathway and Enhancing Autophagy in Malignant Glioma Cell Lines. PLoS ONE 2012, 7, e47449. [Google Scholar] [CrossRef] [PubMed]
- Harmalkar, M.N.; Upraity, S.; Kazi, S.; Shirsat, N.V. Tamoxifen-Induced Cell Death of Malignant Glioma Cells Is Brought about by Oxidative-Stress-Mediated Alterations in the Expression of BCL2 Family Members and Is Enhanced on miR-21 Inhibition. J. Mol. Neurosci. 2015, 57, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Li, R.; Shi, W.; Jiang, T.; Wang, Y.; Li, C.; Qu, X. Silencing of MicroRNA-21 confers the sensitivity to tamoxifen and fulvestrant by enhancing autophagic cell death through inhibition of the PI3K-AKT-mTOR pathway in breast cancer cells. Biomed Pharmacother. 2016, 77, 37–44. [Google Scholar] [CrossRef]
- Zheng, P.; Guo, H.; Li, G.; Han, S.; Luo, F.; Liu, Y. PSMB4 promotes multiple myeloma cell growth by activating NF-κB-miR-21 signaling. Biochem. Biophys. Res. Commun. 2015, 458, 328–333. [Google Scholar] [CrossRef]
- Chen, J.-C.; Hsieh, Y.-Y.; Lo, H.-L.; Li, A.; Chou, C.-J.; Yang, P.-M. In Vitro and In Silico Mechanistic Insights into miR-21-5p-Mediated Topoisomerase Drug Resistance in Human Colorectal Cancer Cells. Biomolecules 2019, 9, 467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Jiang, Z.; Li, Y.; Wang, K.; Chen, X.; Liu, G. Downregulation of miR-21 inhibits the malignant phenotype of pancreatic cancer cells by targeting VHL. OncoTargets Ther. 2019, 12, 7215–7226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Møller, H.G.; Rasmussen, A.P.; Andersen, H.H.; Johnsen, K.B.; Henriksen, M.; Duroux, M. A Systematic Review of MicroRNA in Glioblastoma Multiforme: Micro-modulators in the Mesenchymal Mode of Migration and Invasion. Mol. Neurobiol. 2013, 47, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Wan, X.; Alvarez, A.A.; James, C.D.; Song, X.; Yang, Y.; Sastry, N.; Nakano, I.; Sulman, E.P.; Hu, B.; et al. MIR93 (microRNA-93) regulates tumorigenicity and therapy response of glioblastoma by targeting autophagy. Autophagy 2019, 15, 1100–1111. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, X.; Chen, X.; Liu, J.; Gu, H.; Fan, R.; Ge, H. Long non-coding RNA HOTAIR knockdown enhances radiosensitivity through regulating microRNA-93/ATG12 axis in colorectal cancer. Cell Death Dis. 2020, 11, 175. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Yang, Y.; Ba, Z.; Li, S.; Chen, H.; Hou, X.; Ma, L.; He, P.; Jiang, L.; Li, L.; et al. MicroRNA-93 Regulates Hypoxia-Induced Autophagy by Targeting ULK1. Oxidative Med. Cell Longev. 2017, 2017, 2709053. [Google Scholar] [CrossRef]
- Zhang, C.; Nie, P.; Zhou, C.; Hu, Y.; Duan, S.; Gu, M.; Jiang, D.; Wang, Y.; Deng, Z.; Chen, J.; et al. Oxidative stress-induced mitophagy is suppressed by the miR-106b-93-25 cluster in a protective manner. Cell Death Dis. 2021, 12, 209. [Google Scholar] [CrossRef]
- He, S.; Deng, J.; Li, G.; Wang, B.; Cao, Y.; Tu, Y. Down-regulation of Nedd4L is Associated with the Aggressive Progression and Worse Prognosis of Malignant Glioma. Jpn. J. Clin. Oncol. 2012, 42, 196–201. [Google Scholar] [CrossRef] [Green Version]
- Guarnieri, A.L.; Towers, C.G.; Drasin, D.J.; Oliphant, M.U.J.; Andrysik, Z.; Hotz, T.J.; Vartuli, R.L.; Linklater, E.S.; Pandey, A.; Khanal, S.; et al. The miR-106b-25 cluster mediates breast tumor initiation through activation of NOTCH1 via direct repression of NEDD4L. Oncogene 2018, 37, 3879–3893. [Google Scholar] [CrossRef]
- Bazzoni, R.; Bentivegna, A. Role of Notch Signaling Pathway in Glioblastoma Pathogenesis. Cancers 2019, 11, 292. [Google Scholar] [CrossRef] [Green Version]
- Janjua, T.I.; Rewatkar, P.; Ahmed-Cox, A.; Saeed, I.; Mansfeld, F.M.; Kulshreshtha, R.; Kumeria, T.; Ziegler, D.S.; Kavallaris, M.; Mazzieri, R.; et al. Frontiers in the treatment of glioblastoma: Past, present and emerging. Adv. Drug Deliv. Rev. 2021, 171, 108–138. [Google Scholar] [CrossRef]
- Bagley, S.J.; Kothari, S.; Rahman, R.; Lee, E.Q.; Dunn, G.P.; Galanis, E.; Chang, S.M.; Nabors, L.B.; Ahluwalia, M.S.; Stupp, R.; et al. Glioblastoma Clinical Trials: Current Landscape and Opportunities for Improvement. Clin. Cancer Res. 2022, 28, 594–602. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HDAC6 Inhibitor | Downstream Target(s) | Effects on GBM | References |
|---|---|---|---|
| MPT0B291 | Sp1 | Decreased transcription of BMI1, hTERT, and cell cycle genes | [130] |
| A452 | EGFR MGMT MSH2 MSH6 mutp53 | Increased apoptosis when combined with TMZ | [131] |
| ACY-1215 | EGFR MGMT MSH2 MSH6 mutp53 MKK7/JNK/c-Jun | Increased apoptosis when combined with TMZ causing decrease in tumor growth | [131,132,133] |
| CAY 10603 | EGFR MGMT MSH2 MSH6 mutp53 MKK7/JNK/c-Jun | Increased apoptosis when combined with TMZ causing decrease in tumor growth | [131,132,133] |
| Tubastatin A | MSH2 MSH6 mutp53 p97/VCP | Increased apoptosis when combined with TMZ | [131,132] |
| siRNA (SASI_Hs01_ 00048982 and SASI_Hs02_0034079) | α-Tubulin Slug Snail Sonic Hedgehog | Decreased proliferation and migration, and reverted EMT phenotype | [134] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Visintin, R.; Ray, S.K. Intersections of Ubiquitin-Proteosome System and Autophagy in Promoting Growth of Glioblastoma Multiforme: Challenges and Opportunities. Cells 2022, 11, 4063. https://doi.org/10.3390/cells11244063
Visintin R, Ray SK. Intersections of Ubiquitin-Proteosome System and Autophagy in Promoting Growth of Glioblastoma Multiforme: Challenges and Opportunities. Cells. 2022; 11(24):4063. https://doi.org/10.3390/cells11244063
Chicago/Turabian StyleVisintin, Rhett, and Swapan K. Ray. 2022. "Intersections of Ubiquitin-Proteosome System and Autophagy in Promoting Growth of Glioblastoma Multiforme: Challenges and Opportunities" Cells 11, no. 24: 4063. https://doi.org/10.3390/cells11244063