
ALDH1-Mediated Autophagy Sensitizes Glioblastoma Cells to Ferroptosis
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture, Reagents, Antibodies, and Plasmid
2.2. CRISPR/Cas9 Knockout
2.3. Glioblastoma Primary Cell Lines
2.4. Cell Viability Assays
2.5. LDH Release Assay
2.6. Lipid Peroxidation Assays
2.7. Immunoprecipitation Assays
2.8. Sphere Formation Assays
2.9. ALDH1a3 Stable Transfection
2.10. Statistics
3. Results
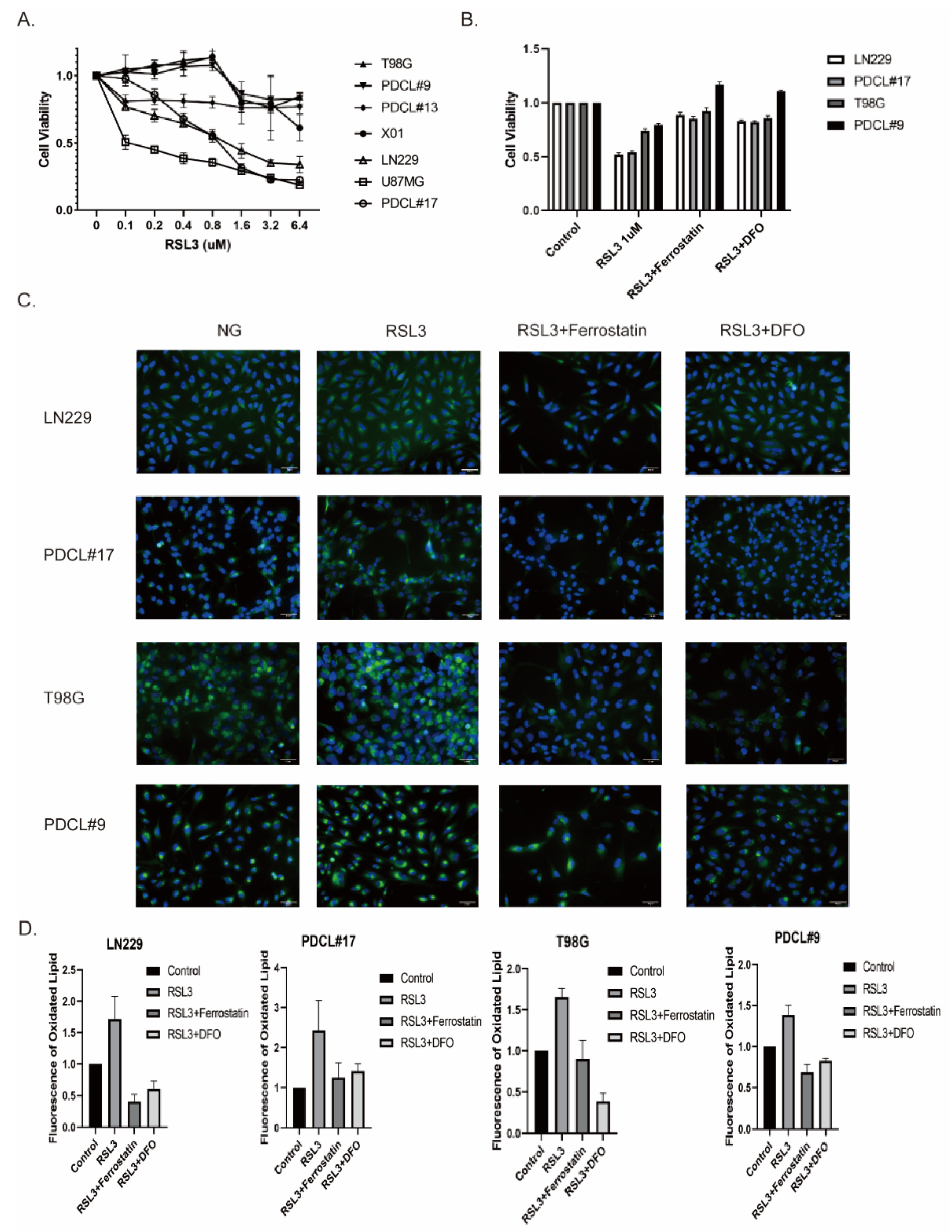
3.1. RSL3-Induced Ferroptosis-Like Cell Death in Glioblastoma Cell Lines
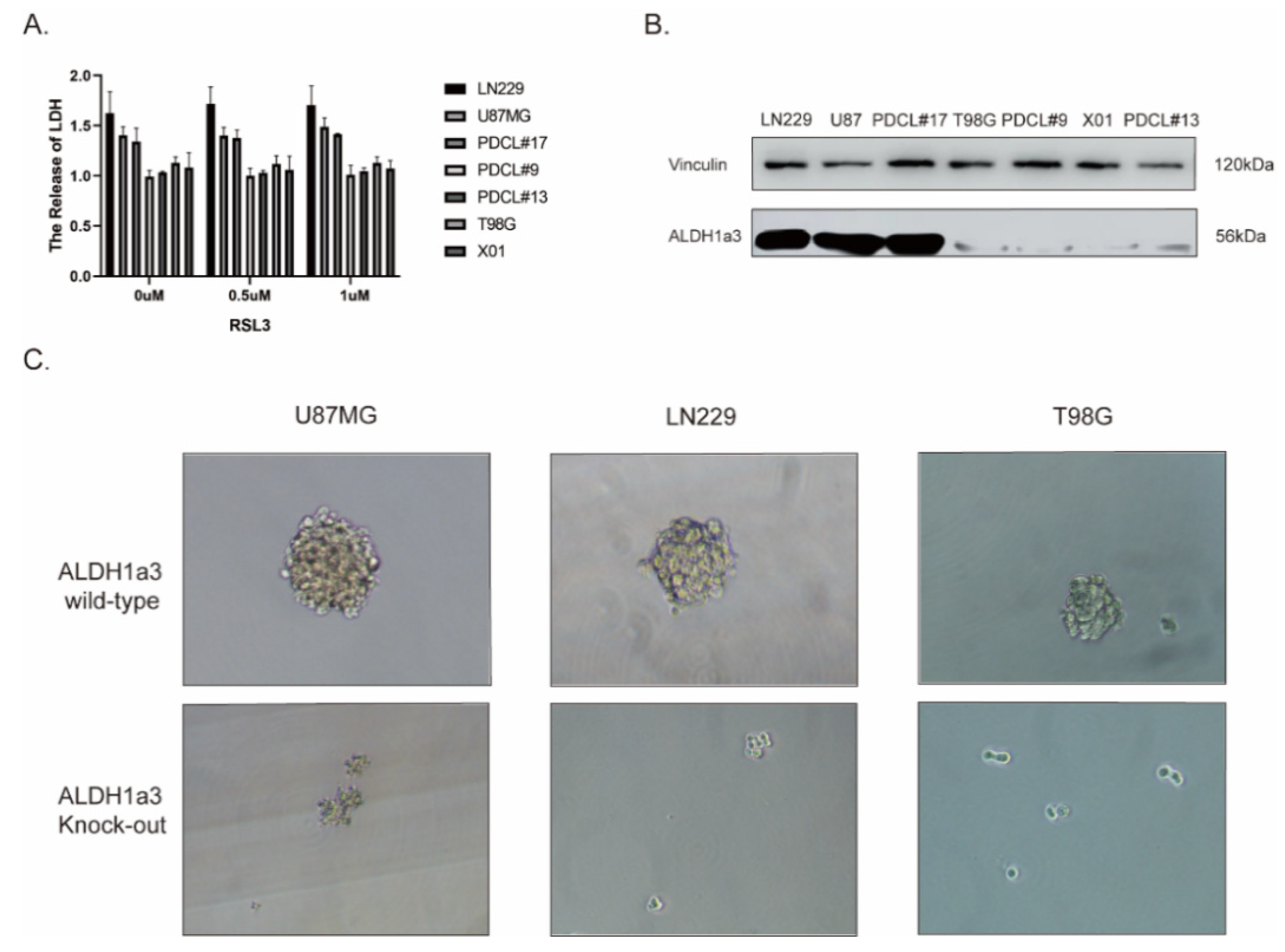
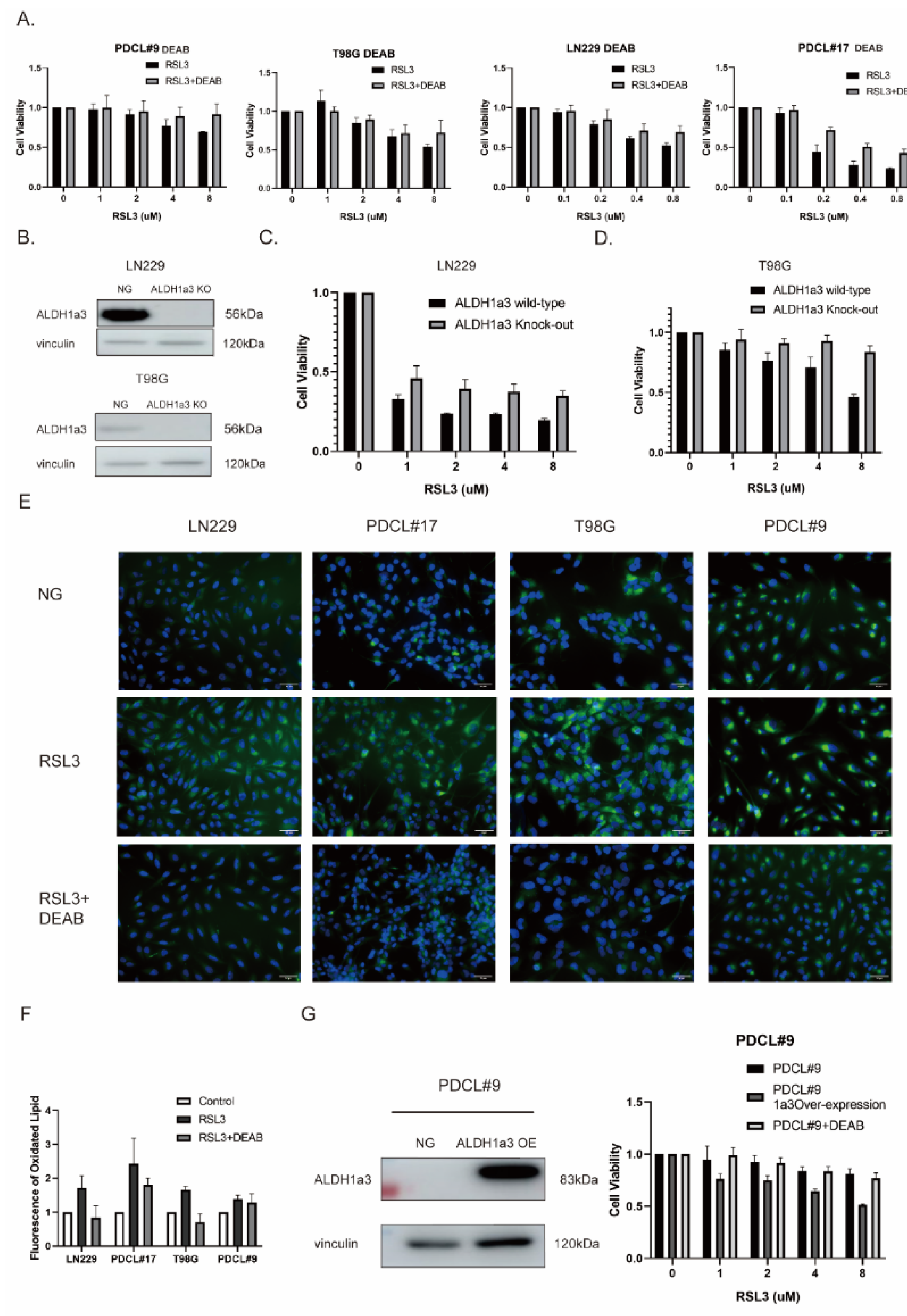
3.2. The Sensitivity to RSL3-Induced Ferroptotic Cell Death in Glioblastoma Correlates with the Expression of ALDH1a3
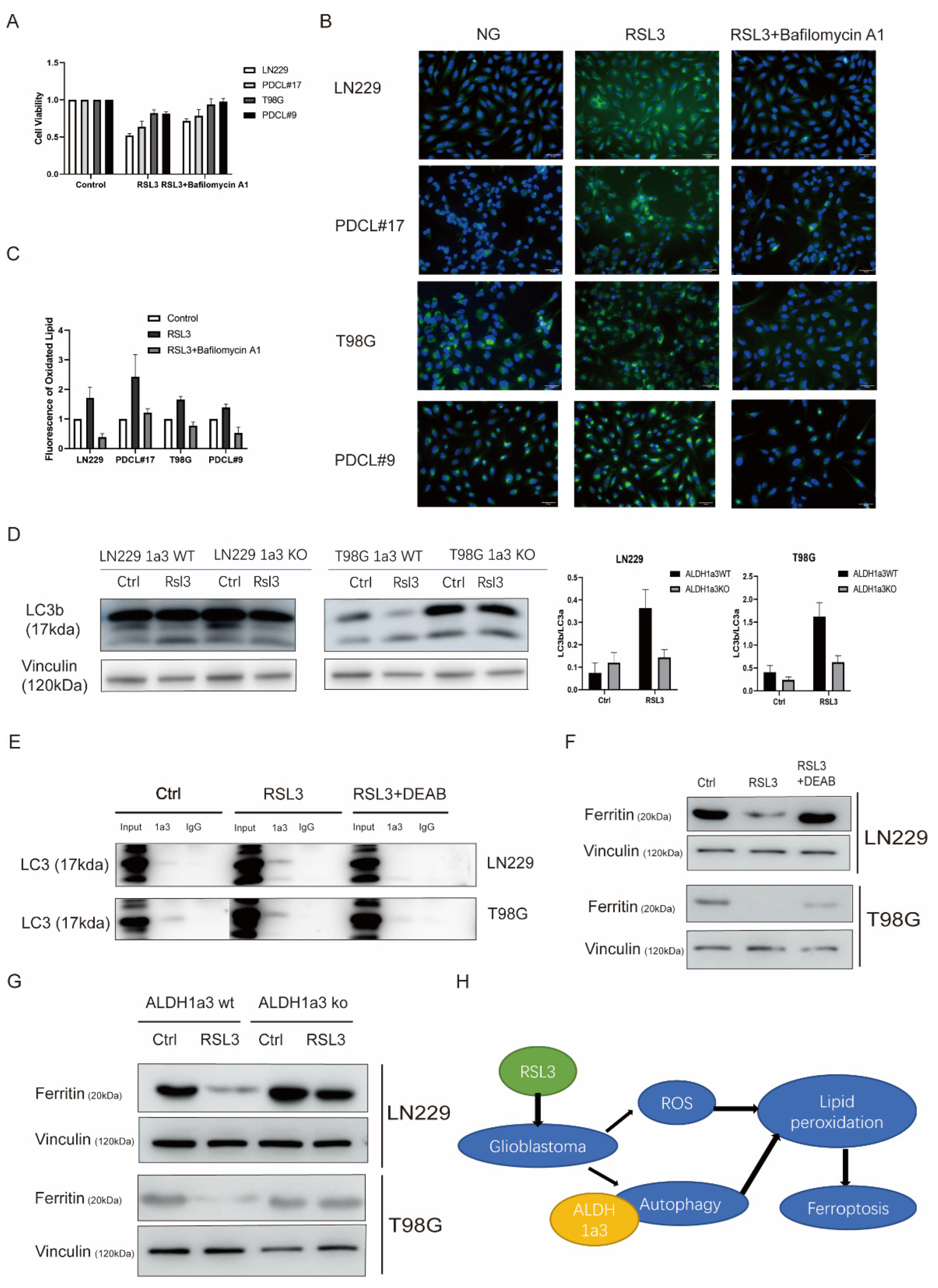
3.3. Autophagy Seems to Be Necessary for RSL3-Induced Cell Death
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Cojoc, M.; Mabert, K.; Muders, M.H.; Dubrovska, A. A role for cancer stem cells in therapy resistance: Cellular and molecular mechanisms. Semin Cancer Biol. 2015, 31, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Marcato, P.; Dean, C.A.; Giacomantonio, C.A.; Lee, P.W. Aldehyde dehydrogenase: Its role as a cancer stem cell marker comes down to the specific isoform. Cell Cycle. 2011, 10, 1378–1384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasper, M.; Schafer, A.; Piontek, G.; Teufel, J.; Brockhoff, G.; Ringel, F.; Heindl, S.; Zimmer, C.; Schlegel, J. Aldehyde dehydrogenase 1 positive glioblastoma cells show brain tumor stem cell capacity. Neuro Oncol. 2010, 12, 1024–1033. [Google Scholar] [CrossRef] [PubMed]
- Schafer, A.; Teufel, J.; Ringel, F.; Bettstetter, M.; Hoepner, I.; Rasper, M.; Gempt, J.; Koeritzer, J.; Schmidt-Graf, F.; Meyer, B.; et al. Aldehyde dehydrogenase 1A1--a new mediator of resistance to temozolomide in glioblastoma. Neuro Oncol. 2012, 14, 1452–1464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.; Wu, Y.; Mayer, K.; von Rosenstiel, C.; Schecker, J.; Baur, S.; Wurstle, S.; Liesche-Starnecker, F.; Gempt, J.; Schlegel, J. Lipid Peroxidation Plays an Important Role in Chemotherapeutic Effects of Temozolomide and the Development of Therapy Resistance in Human Glioblastoma. Transl. Oncol. 2020, 13, 100748. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef]
- Seibt, T.M.; Proneth, B.; Conrad, M. Role of GPX4 in ferroptosis and its pharmacological implication. Free Radic. Biol. Med. 2019, 133, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Li, Y.; Zhang, R.; Wang, F.; Wang, T.; Jiao, Y. The Role of Erastin in Ferroptosis and Its Prospects in Cancer Therapy. Onco Targets Ther. 2020, 13, 5429–5441. [Google Scholar] [CrossRef] [PubMed]
- Soeda, A.; Park, M.; Lee, D.; Mintz, A.; Androutsellis-Theotokis, A.; McKay, R.D.; Engh, J.; Iwama, T.; Kunisada, T.; Kassam, A.B.; et al. Hypoxia promotes expansion of the CD133-positive glioma stem cells through activation of HIF-1alpha. Oncogene 2009, 28, 3949–3959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.; Schecker, J.; Wurstle, S.; Schneider, F.; Schonfelder, M.; Schlegel, J. Aldehyde dehydrogenase 1A3 (ALDH1A3) is regulated by autophagy in human glioblastoma cells. Cancer Lett. 2018, 417, 112–123. [Google Scholar] [CrossRef]
- Berger, T.R.; Wen, P.Y.; Lang-Orsini, M.; Chukwueke, U.N. World Health Organization 2021 Classification of Central Nervous System Tumors and Implications for Therapy for Adult-Type Gliomas: A Review. JAMA Oncol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdalla, F.C.; Abeliovich, H.; Abraham, R.T.; Acevedo-Arozena, A.; Adeli, K.; Agholme, L.; Agnello, M.; Agostinis, P.; Aguirre-Ghiso, J.A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012, 8, 445–544. [Google Scholar] [CrossRef] [Green Version]
- Imai, H.; Matsuoka, M.; Kumagai, T.; Sakamoto, T.; Koumura, T. Lipid Peroxidation-Dependent Cell Death Regulated by GPx4 and Ferroptosis. Curr. Top. Microbiol. Immunol. 2017, 403, 143–170. [Google Scholar]
- Yang, F.C.; Wang, C.; Zhu, J.; Gai, Q.J.; Mao, M.; He, J.; Qin, Y.; Yao, X.X.; Wang, Y.X.; Lu, H.M.; et al. Inhibitory effects of temozolomide on glioma cells is sensitized by RSL3-induced ferroptosis but negatively correlated with expression of ferritin heavy chain 1 and ferritin light chain. Lab Investig. 2022, 102, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kong, Y.; Ma, Y.; Ni, S.; Wikerholmen, T.; Xi, K.; Zhao, F.; Zhao, Z.; Wang, J.; Huang, B.; et al. Loss of COPZ1 induces NCOA4 mediated autophagy and ferroptosis in glioblastoma cell lines. Oncogene 2021, 40, 1425–1439. [Google Scholar] [CrossRef]
- Liu, J.; Kuang, F.; Kroemer, G.; Klionsky, D.J.; Kang, R.; Tang, D. Autophagy-Dependent Ferroptosis: Machinery and Regulation. Cell Chem. Biol. 2020, 27, 420–435. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Chen, X.; Tan, Q.; Zhou, H.; Xu, J.; Gu, Q. Inhibiting Ferroptosis through Disrupting the NCOA4-FTH1 Interaction: A New Mechanism of Action. ACS Cent. Sci. 2021, 7, 980–989. [Google Scholar] [CrossRef] [PubMed]
- Kram, H.; Prokop, G.; Haller, B.; Gempt, J.; Wu, Y.; Schmidt-Graf, F.; Schlegel, J.; Conrad, M.; Liesche-Starnecker, F. Glioblastoma Relapses Show Increased Markers of Vulnerability to Ferroptosis. Front. Oncol. 2022, 12, 841418. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Y.; Kram, H.; Gempt, J.; Liesche-Starnecker, F.; Wu, W.; Schlegel, J. ALDH1-Mediated Autophagy Sensitizes Glioblastoma Cells to Ferroptosis. Cells 2022, 11, 4015. https://doi.org/10.3390/cells11244015
Wu Y, Kram H, Gempt J, Liesche-Starnecker F, Wu W, Schlegel J. ALDH1-Mediated Autophagy Sensitizes Glioblastoma Cells to Ferroptosis. Cells. 2022; 11(24):4015. https://doi.org/10.3390/cells11244015
Chicago/Turabian StyleWu, Yang, Helena Kram, Jens Gempt, Friederike Liesche-Starnecker, Wei Wu, and Jürgen Schlegel. 2022. "ALDH1-Mediated Autophagy Sensitizes Glioblastoma Cells to Ferroptosis" Cells 11, no. 24: 4015. https://doi.org/10.3390/cells11244015