Determinants of Neuronal Susceptibility to Mitochondrial Disease
Share This Topical Collection
Editor
 Dr. Albert Quintana
Dr. Albert Quintana
Dr. Albert Quintana
E-Mail
Website
Collection Editor
1. Department of Cellular Biology, Physiology and Immunology, University Autonomous of Barcelona, 08193 Bellaterra, Barcelona, Spain
2. Institute of Neurosciences, Autonomous University of Barcelona, 08193 Bellaterra, Barcelona, Spain
Interests: mitochondrial disease; neuroinflammation; neurodegeneration; animal models; cell type-specific; transcriptomic
Topical Collection Information
Dear Colleagues,
Mutations that render mitochondria unable to generate energy cause a group of rare and usually fatal pathologies collectively known as mitochondrial disease. It has been estimated that 1 in 5000 children will develop a mitochondrial disease. Currently, there is no cure for mitochondrial disease and the treatments available are mostly ineffective. Energy-demanding cells such as neurons are especially sensitive to mitochondrial disease, and they account for most of the clinical signs and symptoms observed in humans, such as hypotonia, ataxia, seizures and early death. However, even if every single cell in the body carries the mutation, only specific brain areas seem to be affected by the deficiency. The advent of novel approaches at the genetic, metabolic and proteomic level with cell-type and single-cell resolution are furthering our knowledge of the molecular underpinnings of neuronal fate in the context of mitochondrial dysfunction.
Our Topical Collection focuses on collecting research on the neuronal populations susceptible to mitochondrial disease and the mechanisms which cause these neurons to die. This knowledge is essential in order to understand and fight these incurable diseases.
Dr. Albert Quintana
Collection Editor
Manuscript Submission Information
Manuscripts should be submitted online at www.mdpi.com by registering and logging in to this website. Once you are registered, click here to go to the submission form. Manuscripts can be submitted until the deadline. All submissions that pass pre-check are peer-reviewed. Accepted papers will be published continuously in the journal (as soon as accepted) and will be listed together on the collection website. Research articles, review articles as well as short communications are invited. For planned papers, a title and short abstract (about 100 words) can be sent to the Editorial Office for announcement on this website.
Submitted manuscripts should not have been published previously, nor be under consideration for publication elsewhere (except conference proceedings papers). All manuscripts are thoroughly refereed through a single-blind peer-review process. A guide for authors and other relevant information for submission of manuscripts is available on the Instructions for Authors page. Cells is an international peer-reviewed open access semimonthly journal published by MDPI.
Please visit the Instructions for Authors page before submitting a manuscript.
The Article Processing Charge (APC) for publication in this open access journal is 2700 CHF (Swiss Francs).
Submitted papers should be well formatted and use good English. Authors may use MDPI's
English editing service prior to publication or during author revisions.
Keywords
- mitochondrial disease
- neuropathology
- cell-type-specific
- single cell
Published Papers (8 papers)
Open AccessArticle
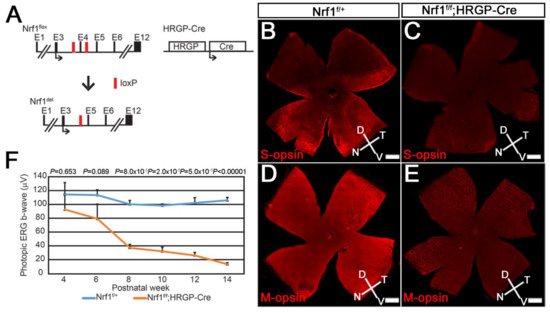
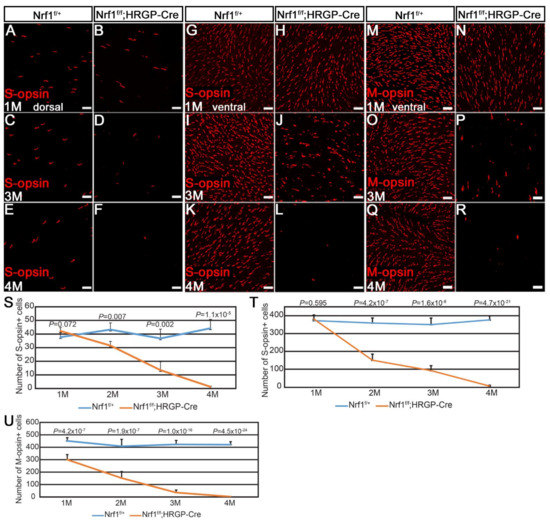
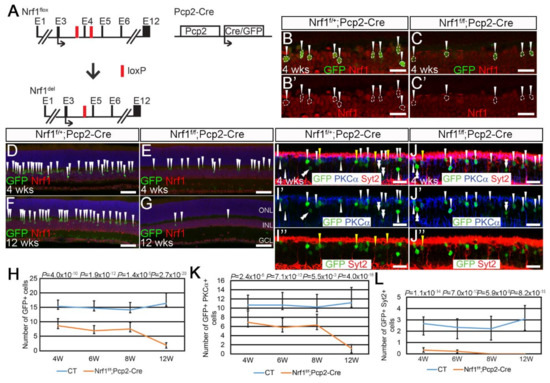
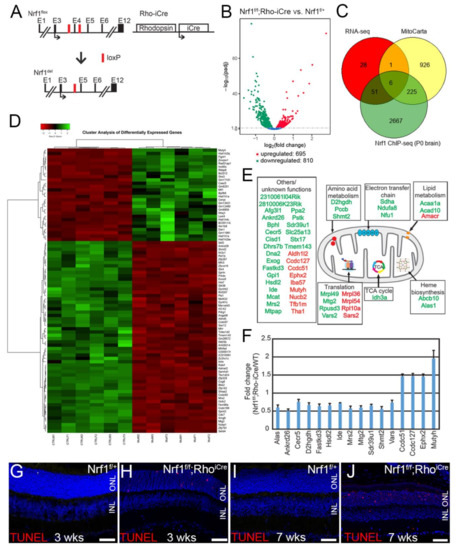
Differential Susceptibility of Retinal Neurons to the Loss of Mitochondrial Biogenesis Factor Nrf1
by
Takae Kiyama, Ching-Kang Chen, Annie Zhang and Chai-An Mao
Cited by 1 | Viewed by 1922
Abstract
The retina, the accessible part of the central nervous system, has served as a model system to study the relationship between energy utilization and metabolite supply. When the metabolite supply cannot match the energy demand, retinal neurons are at risk of death. As
[...] Read more.
The retina, the accessible part of the central nervous system, has served as a model system to study the relationship between energy utilization and metabolite supply. When the metabolite supply cannot match the energy demand, retinal neurons are at risk of death. As the powerhouse of eukaryotic cells, mitochondria play a pivotal role in generating ATP, produce precursors for macromolecules, maintain the redox homeostasis, and function as waste management centers for various types of metabolic intermediates. Mitochondrial dysfunction has been implicated in the pathologies of a number of degenerative retinal diseases. It is well known that photoreceptors are particularly vulnerable to mutations affecting mitochondrial function due to their high energy demand and susceptibility to oxidative stress. However, it is unclear how defective mitochondria affect other retinal neurons. Nuclear respiratory factor 1 (Nrf1) is the major transcriptional regulator of mitochondrial biogenesis, and loss of
Nrf1 leads to defective mitochondria biogenesis and eventually cell death. Here, we investigated how different retinal neurons respond to the loss of
Nrf1. We provide in vivo evidence that the disruption of
Nrf1-mediated mitochondrial biogenesis results in a slow, progressive degeneration of all retinal cell types examined, although they present different sensitivity to the deletion of
Nrf1, which implicates differential energy demand and utilization, as well as tolerance to mitochondria defects in different neuronal cells. Furthermore, transcriptome analysis on rod-specific
Nrf1 deletion uncovered a previously unknown role of Nrf1 in maintaining genome stability.
Full article
►▼
Show Figures
Open AccessCommunication
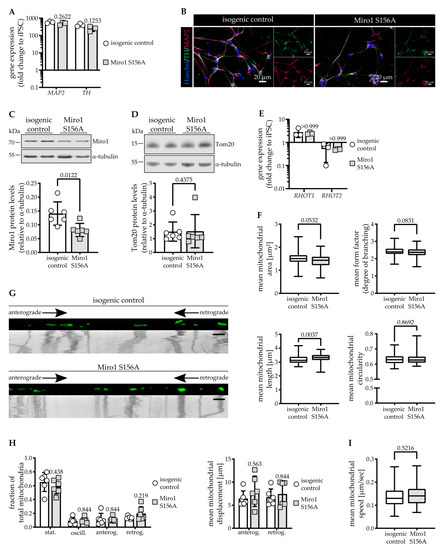
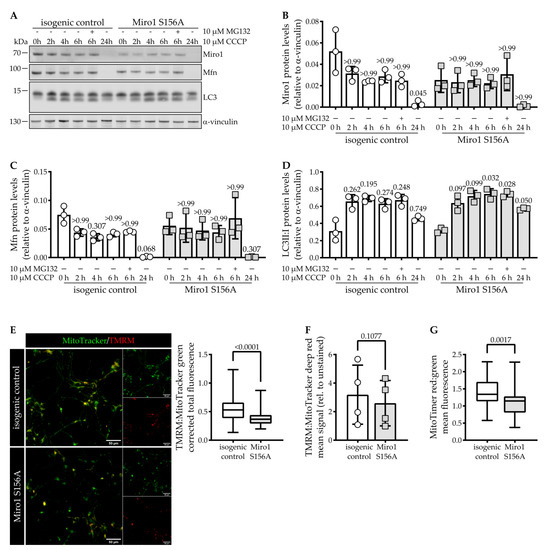
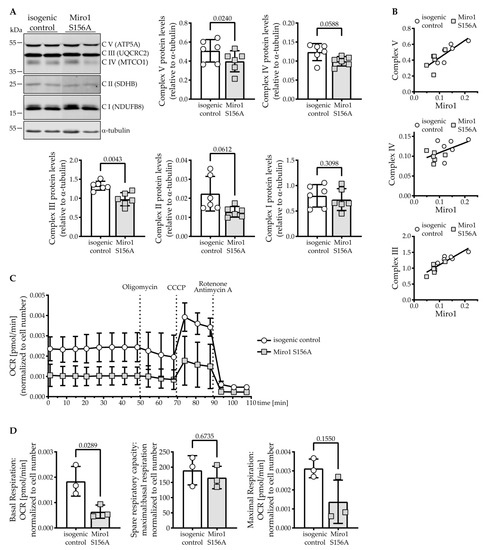
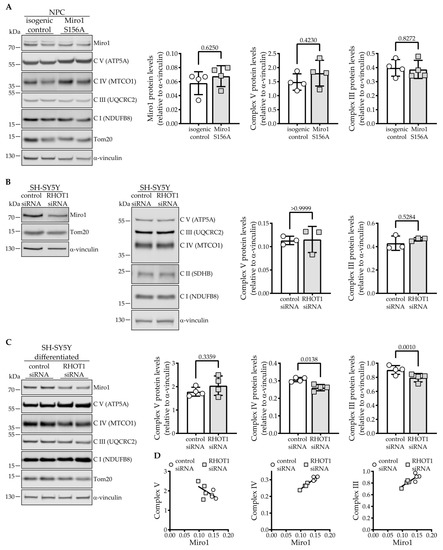
Steady-State Levels of Miro1 Linked to Phosphorylation at Serine 156 and Mitochondrial Respiration in Dopaminergic Neurons
by
Lisa Schwarz and Julia C. Fitzgerald
Cited by 4 | Viewed by 2400
Abstract
Miro1 has emerged as an interesting target to study Parkinson’s disease-relevant pathways since it is a target of PINK1 and Parkin. Miro1 is a mitochondrial GTPase with the primary function of facilitating mitochondrial movement, and its knockout in mice is postnatally lethal. Here,
[...] Read more.
Miro1 has emerged as an interesting target to study Parkinson’s disease-relevant pathways since it is a target of PINK1 and Parkin. Miro1 is a mitochondrial GTPase with the primary function of facilitating mitochondrial movement, and its knockout in mice is postnatally lethal. Here, we investigated the effect of the artificial
RHOT1/Miro1 S156A mutation since it is a putative PINK1 phosphorylation site shown to be involved in Miro1 degradation and mitochondrial arrest during mitophagy. We gene-edited a homozygous phospho-null Miro1 S156A mutation in induced pluripotent stem cells to study the mutation in human dopaminergic neurons. This mutation causes a significant depletion of Miro1 steady-state protein levels and impairs further Miro1 degradation upon CCCP-induced mitophagy. However, mitochondrial mass measured by Tom20 protein levels, as well as mitochondrial area, are not affected in Miro1 S156A neurons. The mitochondria are slightly lengthened, which is in line with their increased turnover. Under basal conditions, we found no discernable effect of the mutation on mitochondrial movement in neurites. Interestingly, the S156A mutation leads to a significant reduction of mitochondrial oxygen consumption, which is accompanied by a depletion of OXPHOS complexes III and V. These effects are not mirrored by Miro1 knockdown in neuroblastoma cells, but they are observed upon differentiation. Undifferentiated Miro1 S156A neural precursor cells do not have decreased Miro1 levels nor OXPHOS complexes, suggesting that the effect of the mutation is tied to development. In mature dopaminergic neurons, the inhibition of Miro1 Ser156 phosphorylation elicits a mild loss of mitochondrial quality involving reduced mitochondrial membrane potential, which is sufficient to induce compensatory events involving OXPHOS. We suggest that the mechanism governing Miro1 steady-state levels depends on differentiation state and metabolic demand, thus underscoring the importance of this pathway in the pathobiology of Parkinson’s disease.
Full article
►▼
Show Figures
Open AccessArticle
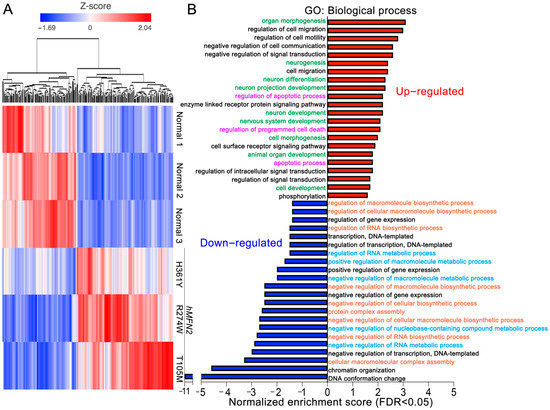
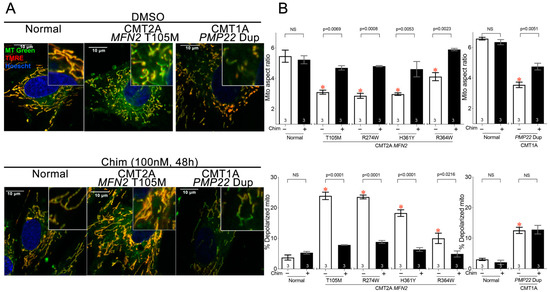
Mitochondrial Phenotypes in Genetically Diverse Neurodegenerative Diseases and Their Response to Mitofusin Activation
by
Xiawei Dang, Emily K. Walton, Barbara Zablocka, Robert H. Baloh, Michael E. Shy and Gerald W. Dorn II
Cited by 10 | Viewed by 3260
Abstract
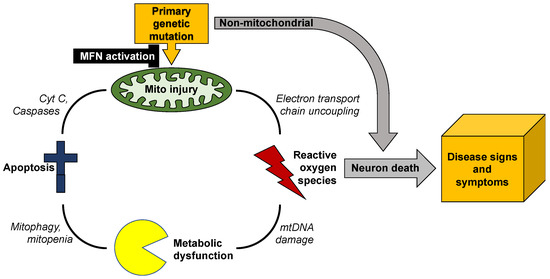
Mitochondrial fusion is essential to mitochondrial fitness and cellular health. Neurons of patients with genetic neurodegenerative diseases often exhibit mitochondrial fragmentation, reflecting an imbalance in mitochondrial fusion and fission (mitochondrial dysdynamism). Charcot–Marie–Tooth (CMT) disease type 2A is the prototypical disorder of impaired mitochondrial
[...] Read more.
Mitochondrial fusion is essential to mitochondrial fitness and cellular health. Neurons of patients with genetic neurodegenerative diseases often exhibit mitochondrial fragmentation, reflecting an imbalance in mitochondrial fusion and fission (mitochondrial dysdynamism). Charcot–Marie–Tooth (CMT) disease type 2A is the prototypical disorder of impaired mitochondrial fusion caused by mutations in the fusion protein mitofusin (MFN)2. Yet, cultured CMT2A patient fibroblast mitochondria are often reported as morphologically normal. Metabolic stress might evoke pathological mitochondrial phenotypes in cultured patient fibroblasts, providing a platform for the pre-clinical individualized evaluation of investigational therapeutics. Here, substitution of galactose for glucose in culture media was used to redirect CMT2A patient fibroblasts (MFN2 T105M, R274W, H361Y, R364W) from glycolytic metabolism to mitochondrial oxidative phosphorylation, which provoked characteristic mitochondrial fragmentation and depolarization and induced a distinct transcriptional signature. Pharmacological MFN activation of metabolically reprogrammed fibroblasts partially reversed the mitochondrial abnormalities in CMT2A and CMT1 and a subset of Parkinson’s and Alzheimer’s disease patients, implicating addressable mitochondrial dysdynamism in these illnesses.
Full article
►▼
Show Figures
Open AccessReview
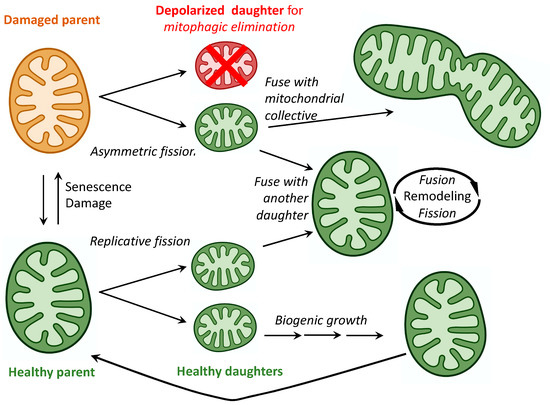
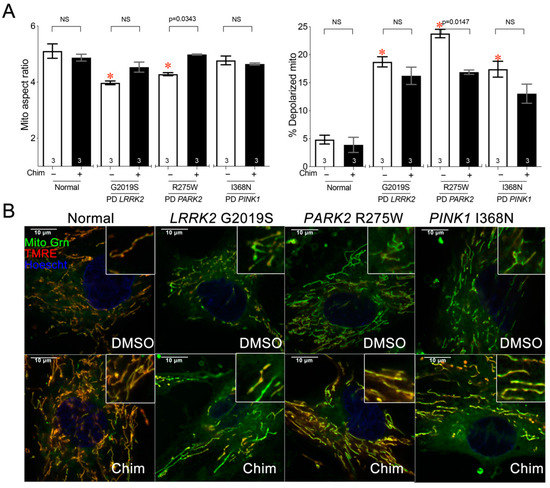
Predicting Mitochondrial Dynamic Behavior in Genetically Defined Neurodegenerative Diseases
by
Gerald W. Dorn II and Xiawei Dang
Cited by 10 | Viewed by 5177
Abstract
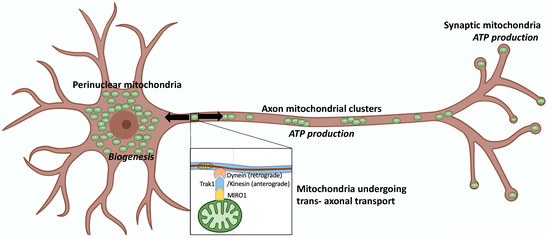
Mitochondrial dynamics encompass mitochondrial fusion, fission, and movement. Mitochondrial fission and fusion are seemingly ubiquitous, whereas mitochondrial movement is especially important for organelle transport through neuronal axons. Here, we review the roles of different mitochondrial dynamic processes in mitochondrial quantity and quality control,
[...] Read more.
Mitochondrial dynamics encompass mitochondrial fusion, fission, and movement. Mitochondrial fission and fusion are seemingly ubiquitous, whereas mitochondrial movement is especially important for organelle transport through neuronal axons. Here, we review the roles of different mitochondrial dynamic processes in mitochondrial quantity and quality control, emphasizing their impact on the neurological system in Charcot–Marie–Tooth disease type 2A, amyotrophic lateral sclerosis, Friedrich’s ataxia, dominant optic atrophy, and Alzheimer’s, Huntington’s, and Parkinson’s diseases. In addition to mechanisms and concepts, we explore in detail different technical approaches for measuring mitochondrial dynamic dysfunction in vitro, describe how results from tissue culture studies may be applied to a better understanding of mitochondrial dysdynamism in human neurodegenerative diseases, and suggest how this experimental platform can be used to evaluate candidate therapeutics in different diseases or in individual patients sharing the same clinical diagnosis.
Full article
►▼
Show Figures
Open AccessArticle
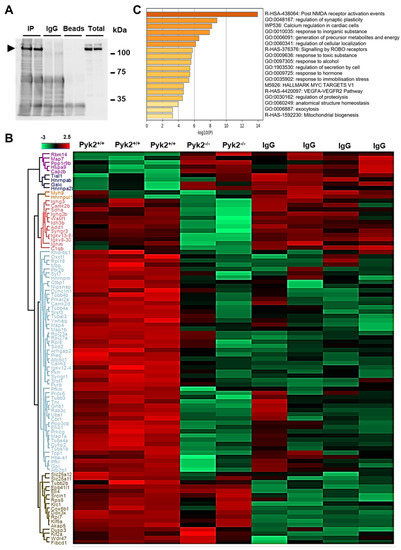
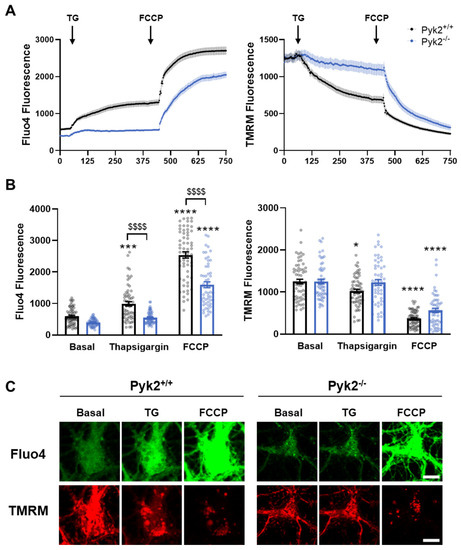
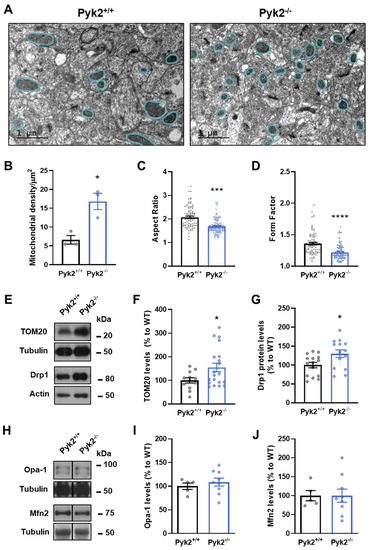
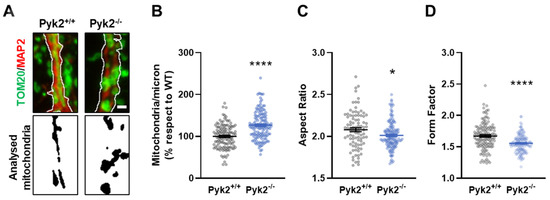
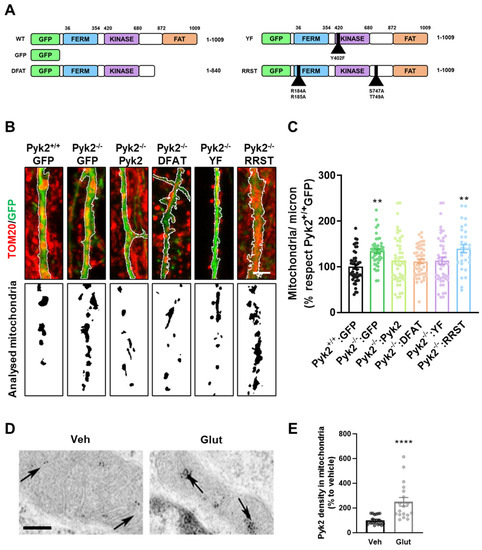
Pyk2 Regulates MAMs and Mitochondrial Dynamics in Hippocampal Neurons
by
Laura López-Molina, Joaquín Fernández-Irigoyen, Carmen Cifuentes-Díaz, Jordi Alberch, Jean-Antoine Girault, Enrique Santamaría, Silvia Ginés and Albert Giralt
Cited by 4 | Viewed by 3302
Abstract
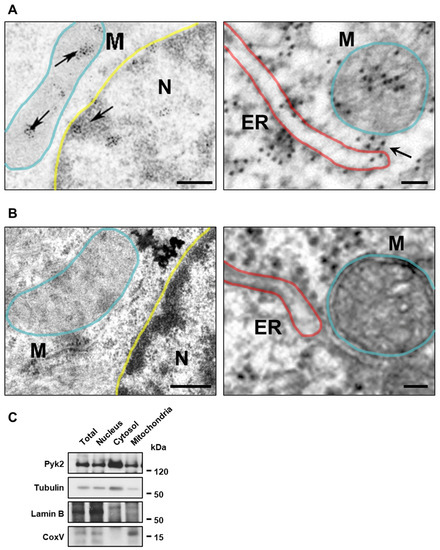
Pyk2 is a non-receptor tyrosine kinase enriched in hippocampal neurons, which can be activated by calcium-dependent mechanisms. In neurons, Pyk2 is mostly localised in the cytosol and dendritic shafts but can translocate to spines and/or to the nucleus. Here, we explore the function
[...] Read more.
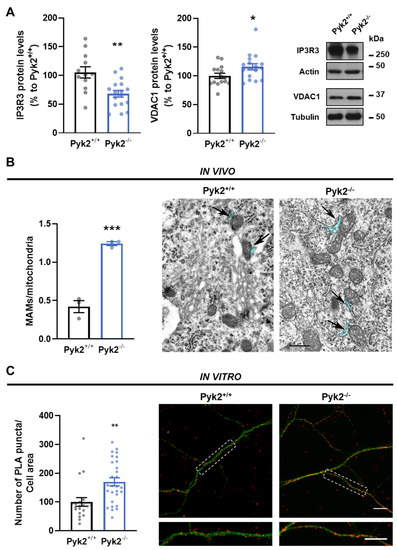
Pyk2 is a non-receptor tyrosine kinase enriched in hippocampal neurons, which can be activated by calcium-dependent mechanisms. In neurons, Pyk2 is mostly localised in the cytosol and dendritic shafts but can translocate to spines and/or to the nucleus. Here, we explore the function of a new localisation of Pyk2 in mitochondria-associated membranes (MAMs), a subdomain of ER-mitochondria surface that acts as a signalling hub in calcium regulation. To test the role of Pyk2 in MAMs’ calcium transport, we used full Pyk2 knockout mice (Pyk2
−/−) for in vivo and in vitro studies. Here we report that Pyk2
−/− hippocampal neurons present increased ER-mitochondrial contacts along with defective calcium homeostasis. We also show how the absence of Pyk2 modulates mitochondrial dynamics and morphology. Taken all together, our results point out that Pyk2 could be highly relevant in the modulation of ER-mitochondria calcium efflux, affecting in turn mitochondrial function.
Full article
►▼
Show Figures
Open AccessArticle
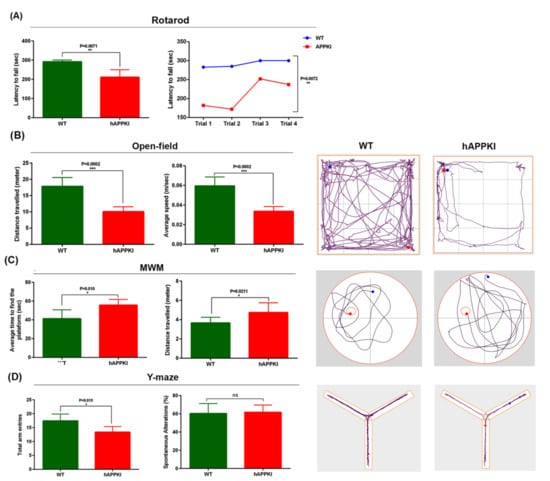
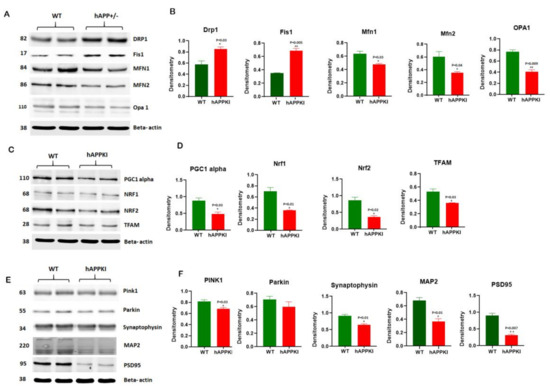
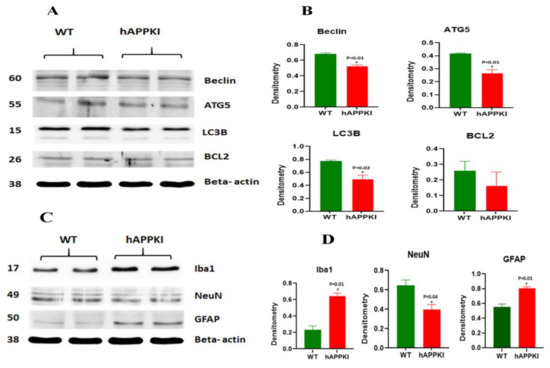
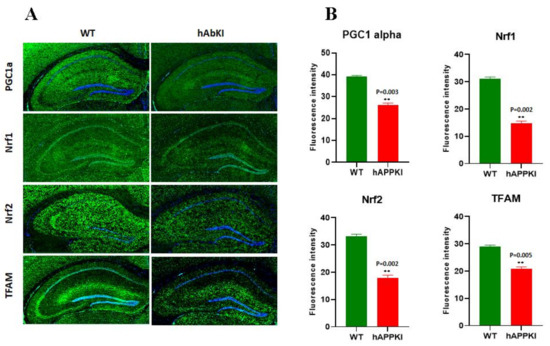
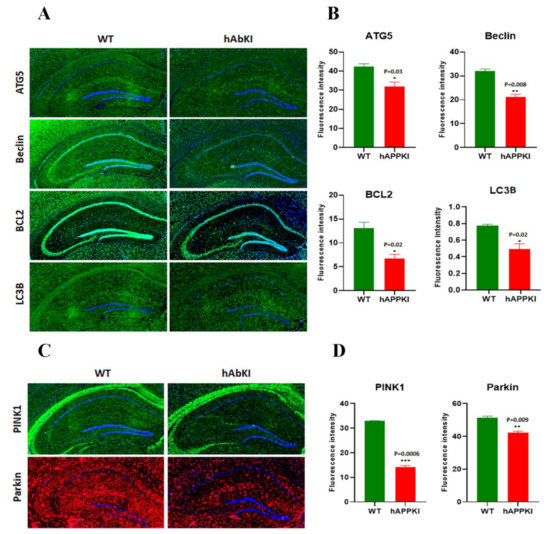
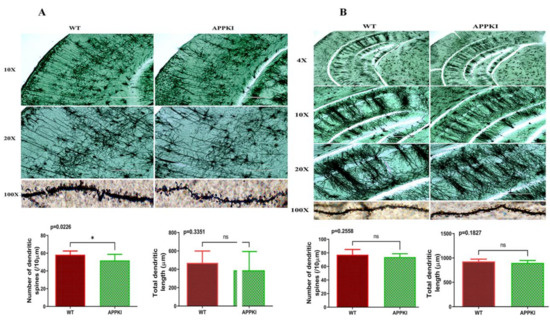
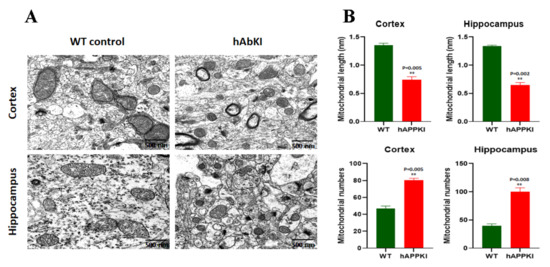
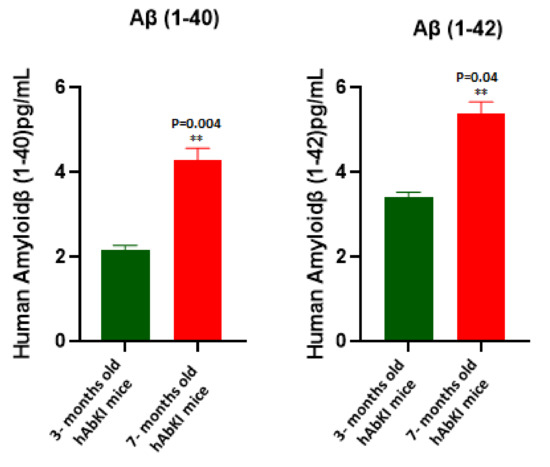
Early Cellular, Molecular, Morphological and Behavioral Changes in the Humanized Amyloid-Beta-Knock-In Mouse Model of Late-Onset Alzheimer’s Disease
by
Sudhir Kshirsagar, Rainier Vladlen Alvir, Ashly Hindle, Subodh Kumar, Murali Vijayan, Jangampalli Adi Pradeepkiran, Arubala P. Reddy, Bhagavathi Ramasubramanian and P. Hemachandra Reddy
Cited by 10 | Viewed by 4030
Abstract
The purpose of our study is to investigate early cellular, molecular, morphological and behavioral changes in humanized amyloid-beta-knock-in (hAbKI) mice. Using seven-month-old homozygous hAbKI mice, we studied behavioral phenotype parameters, including spatial learning and memory (Morris Water Maze), locomotor activity (open field), working
[...] Read more.
The purpose of our study is to investigate early cellular, molecular, morphological and behavioral changes in humanized amyloid-beta-knock-in (hAbKI) mice. Using seven-month-old homozygous hAbKI mice, we studied behavioral phenotype parameters, including spatial learning and memory (Morris Water Maze), locomotor activity (open field), working memory (Y-maze) and motor coordination (rotarod); mRNA abundance, protein levels, soluble amyloid-beta 40 and 42 levels and regional immunoreactivities of key markers of mitochondrial dynamics, mitochondrial biogenesis, synaptic health, mitophagy and autophagy; mitochondrial function and using transmission electron microscopy & Golgi–Cox staining, we assessed mitochondrial morphology and dendritic spines. Our extensive behavioral analysis revealed that seven-month-old hAbKI mice showed impairments in motor coordination, reduced locomotor and exploration activities, impairments in working memory and spatial learning and memory. Our mRNA and protein analyses revealed the increased expression of mitochondrial-fission genes and reduced expression of mitochondrial-fusion, mitochondrial-biogenesis, synaptic, autophagy and mitophagy genes in seven-month-old hAbKI mice. An immunofluorescence analysis revealed altered immunoreactivities and agreed with the immunoblot results. Transmission-electron-microscopy data revealed increased mitochondrial fragmentation and reduced mitochondrial length in both hippocampal and cortical tissues of seven-month-old hAbKI mice and mitochondrial function defective. A Golgi–Cox-staining analysis revealed reduced dendritic spines in both cerebral cortices and hippocampi of hAbKI mice. Soluble amyloid-beta (1–40 and 1–42) were detected in three-month-old hAbKI mice and progressively increased in seven-month-old mice. These observations suggest that the human amyloid-beta peptide is sufficient to cause behavioral, mitochondrial, synaptic and ultrastructural changes in seven-month-old hAbKI mice. Our study findings also suggest that hAbKI mice might serve as a model for preclinical studies of preventive therapies.
Full article
►▼
Show Figures
Open AccessReview
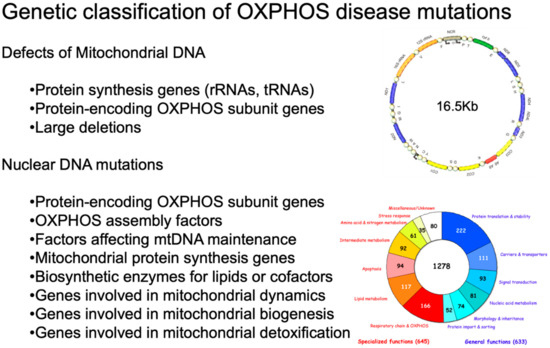
Mitochondrial Neurodegeneration
by
Massimo Zeviani and Carlo Viscomi
Cited by 27 | Viewed by 5418
Abstract
Mitochondria are cytoplasmic organelles, which generate energy as heat and ATP, the universal energy currency of the cell. This process is carried out by coupling electron stripping through oxidation of nutrient substrates with the formation of a proton-based electrochemical gradient across the inner
[...] Read more.
Mitochondria are cytoplasmic organelles, which generate energy as heat and ATP, the universal energy currency of the cell. This process is carried out by coupling electron stripping through oxidation of nutrient substrates with the formation of a proton-based electrochemical gradient across the inner mitochondrial membrane. Controlled dissipation of the gradient can lead to production of heat as well as ATP, via ADP phosphorylation. This process is known as oxidative phosphorylation, and is carried out by four multiheteromeric complexes (from I to IV) of the mitochondrial respiratory chain, carrying out the electron flow whose energy is stored as a proton-based electrochemical gradient. This gradient sustains a second reaction, operated by the mitochondrial ATP synthase, or complex V, which condensates ADP and Pi into ATP. Four complexes (CI, CIII, CIV, and CV) are composed of proteins encoded by genes present in two separate compartments: the nuclear genome and a small circular DNA found in mitochondria themselves, and are termed mitochondrial DNA (mtDNA). Mutations striking either genome can lead to mitochondrial impairment, determining infantile, childhood or adult neurodegeneration. Mitochondrial disorders are complex neurological syndromes, and are often part of a multisystem disorder. In this paper, we divide the diseases into those caused by mtDNA defects and those that are due to mutations involving nuclear genes; from a clinical point of view, we discuss pediatric disorders in comparison to juvenile or adult-onset conditions. The complementary genetic contributions controlling organellar function and the complexity of the biochemical pathways present in the mitochondria justify the extreme genetic and phenotypic heterogeneity of this new area of inborn errors of metabolism known as ‘mitochondrial medicine’.
Full article
►▼
Show Figures
Open AccessFeature PaperEditor’s ChoiceReview
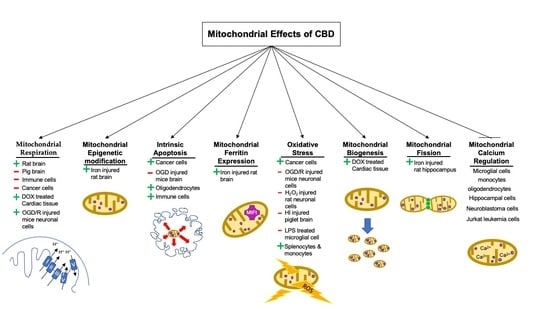
Regulatory Effects of Cannabidiol on Mitochondrial Functions: A Review
by
John Zewen Chan and Robin Elaine Duncan
Cited by 37 | Viewed by 4498
Abstract
Cannabidiol (CBD) is part of a group of phytocannabinoids derived from
Cannabissativa. Initial work on CBD presumed the compound was inactive, but it was later found to exhibit antipsychotic, anti-depressive, anxiolytic, and antiepileptic effects. In recent decades, evidence has indicated a role
[...] Read more.
Cannabidiol (CBD) is part of a group of phytocannabinoids derived from
Cannabissativa. Initial work on CBD presumed the compound was inactive, but it was later found to exhibit antipsychotic, anti-depressive, anxiolytic, and antiepileptic effects. In recent decades, evidence has indicated a role for CBD in the modulation of mitochondrial processes, including respiration and bioenergetics, mitochondrial DNA epigenetics, intrinsic apoptosis, the regulation of mitochondrial and intracellular calcium concentrations, mitochondrial fission, fusion and biogenesis, and mitochondrial ferritin concentration and mitochondrial monoamine oxidase activity regulation. Despite these advances, current data demonstrate contradictory findings with regard to not only the magnitude of effects mediated by CBD, but also to the direction of effects. For example, there are data indicating that CBD treatment can increase, decrease, or have no significant effect on intrinsic apoptosis. Differences between studies in cell type, cell-specific response to CBD, and, in some cases, dose of CBD may help to explain differences in outcomes. Most studies on CBD and mitochondria have utilized treatment concentrations that exceed the highest recorded plasma concentrations in humans, suggesting that future studies should focus on CBD treatments within a range observed in pharmacokinetic studies. This review focuses on understanding the mechanisms of CBD-mediated regulation of mitochondrial functions, with an emphasis on findings in neural cells and tissues and therapeutic relevance based on human pharmacokinetics.
Full article
►▼
Show Figures




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}