



Cells 2023, 12(8), 1177; https://doi.org/10.3390/cells12081177 - 18 Apr 2023
Cited by 1 | Viewed by 1979
Abstract
►
Show Figures
Microcephaly with pontine and cerebellar hypoplasia (MICPCH) syndrome is a neurodevelopmental disorder caused by the deficiency of the X-chromosomal gene CASK. However, the molecular mechanisms by which CASK deficiency causes cerebellar hypoplasia in this syndrome remain elusive. In this study, we used CASK
[...] Read more.
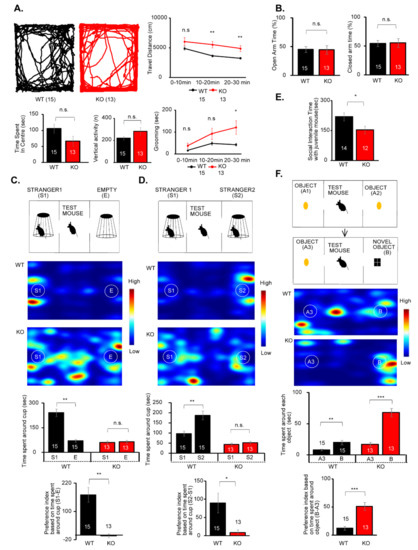
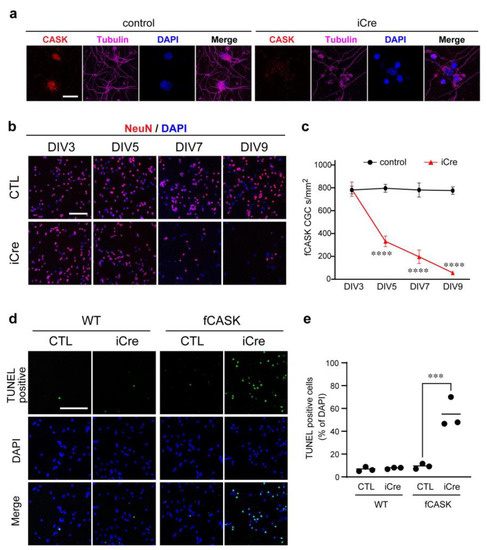
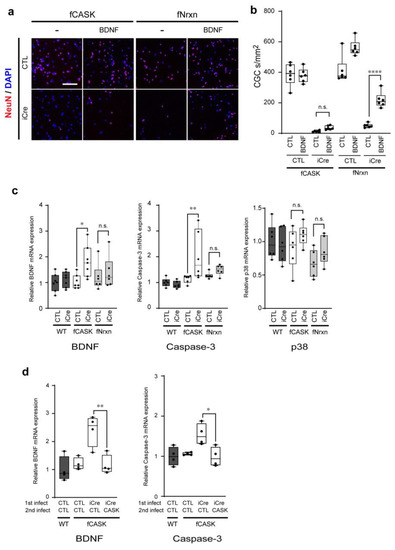
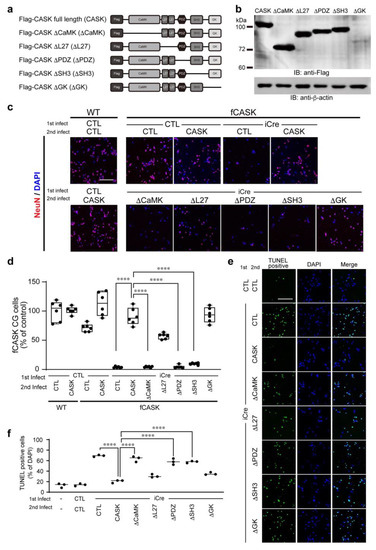
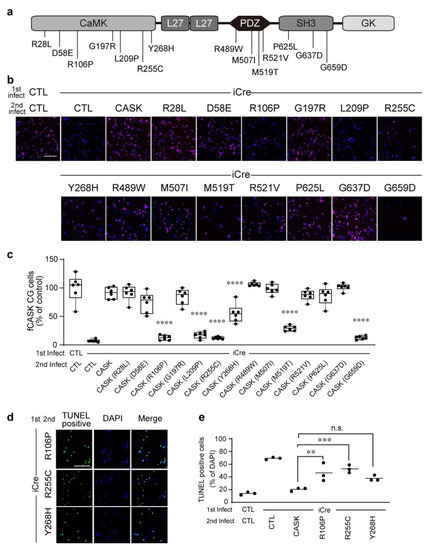
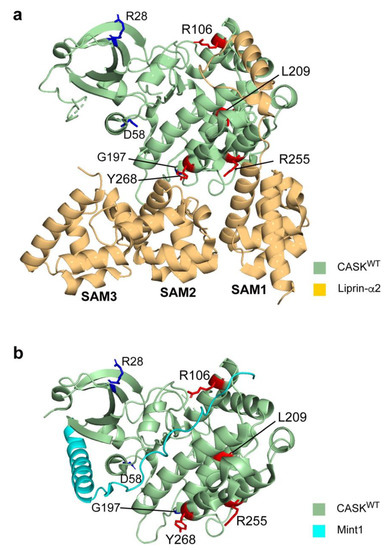
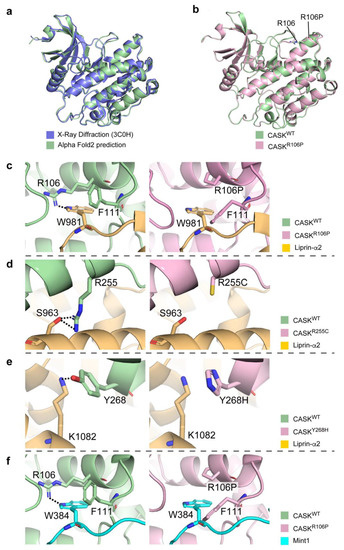
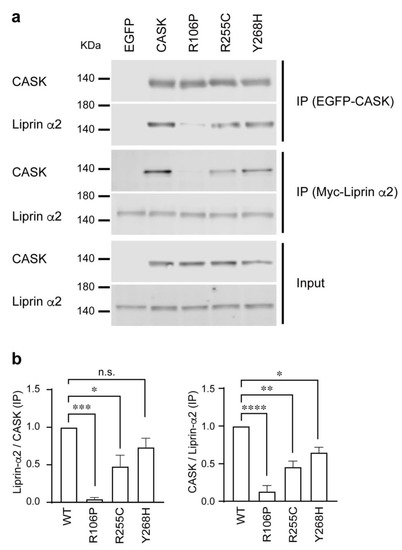
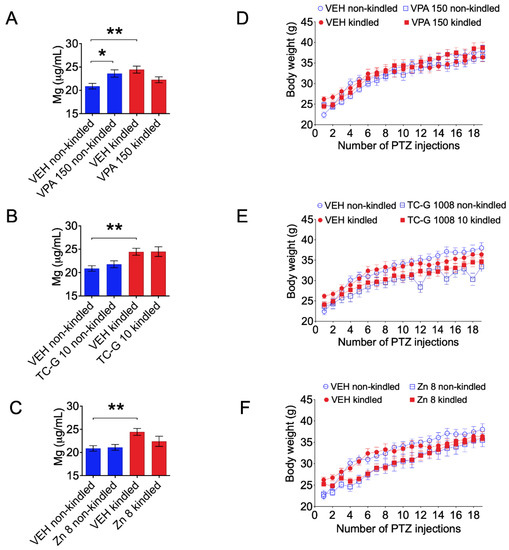
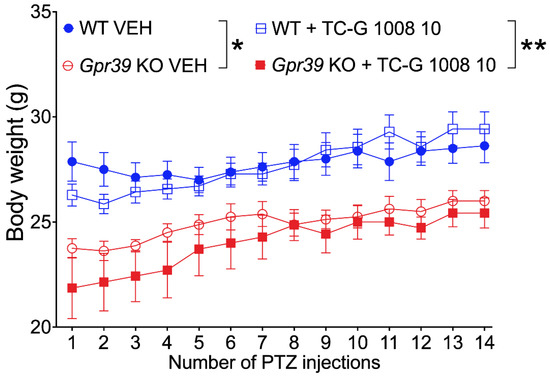
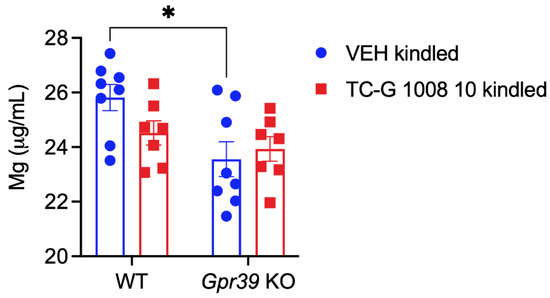
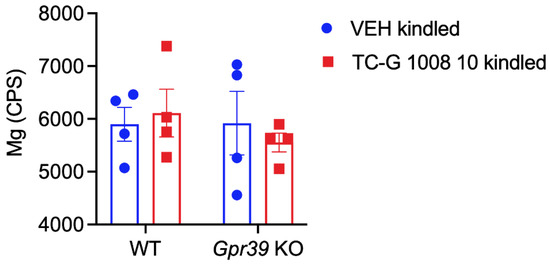
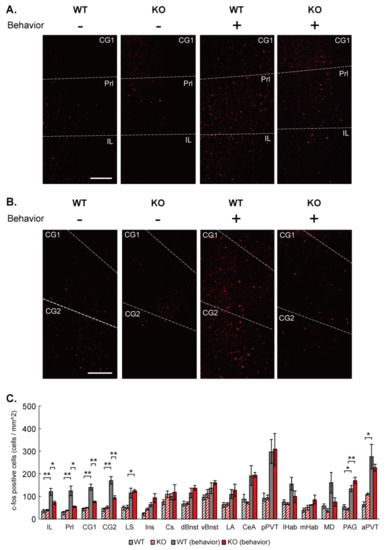
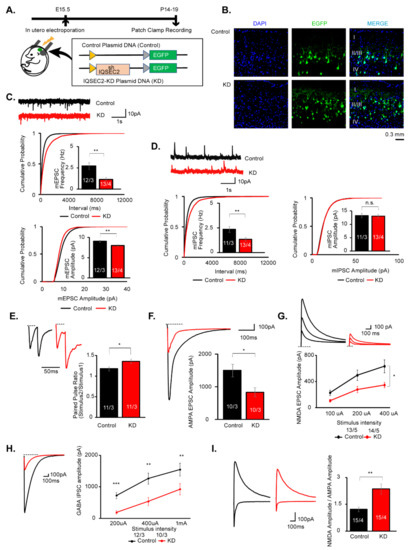
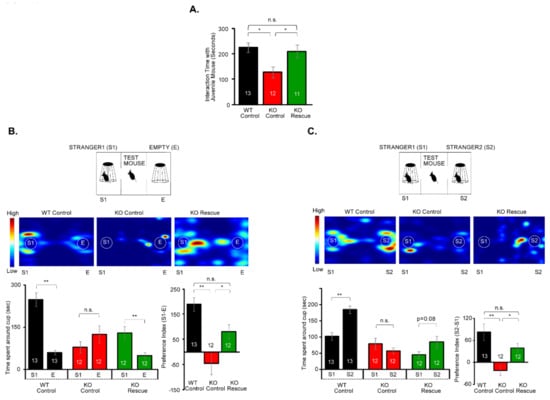
Microcephaly with pontine and cerebellar hypoplasia (MICPCH) syndrome is a neurodevelopmental disorder caused by the deficiency of the X-chromosomal gene CASK. However, the molecular mechanisms by which CASK deficiency causes cerebellar hypoplasia in this syndrome remain elusive. In this study, we used CASK knockout (KO) mice as models for MICPCH syndrome and investigated the effect of CASK mutants. Female CASK heterozygote KO mice replicate the progressive cerebellar hypoplasia observed in MICPCH syndrome. CASK KO cultured cerebellar granule (CG) cells show progressive cell death that can be rescued by co-infection with lentivirus expressing wild-type CASK. Rescue experiments with CASK deletion mutants identify that the CaMK, PDZ, and SH3, but not L27 and guanylate kinase domains of CASK are required for the survival of CG cells. We identify missense mutations in the CaMK domain of CASK derived from human patients that fail to rescue the cell death of cultured CASK KO CG cells. Machine learning-based structural analysis using AlphaFold 2.2 predicts that these mutations disrupt the structure of the binding interface with Liprin-α2. These results suggest that the interaction with Liprin-α2 via the CaMK domain of CASK may be involved in the pathophysiology of cerebellar hypoplasia in MICPCH syndrome.
Full article
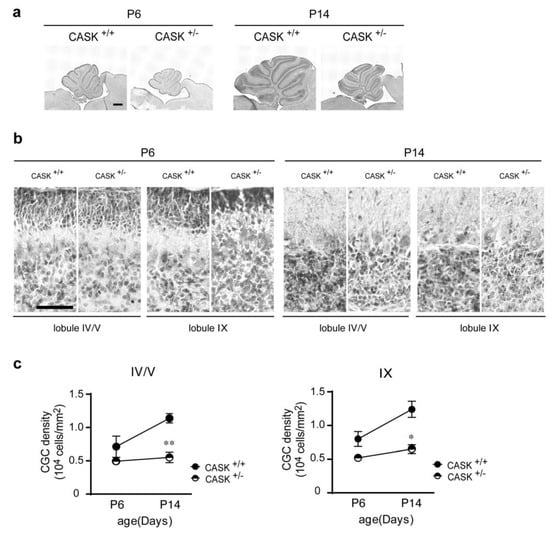
Figure 1
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}