

Autosomal Dominant Hypocalcemia Type 1 and Neonatal Focal Seizures

 , ,
, ,
Abstract
:1. Introduction
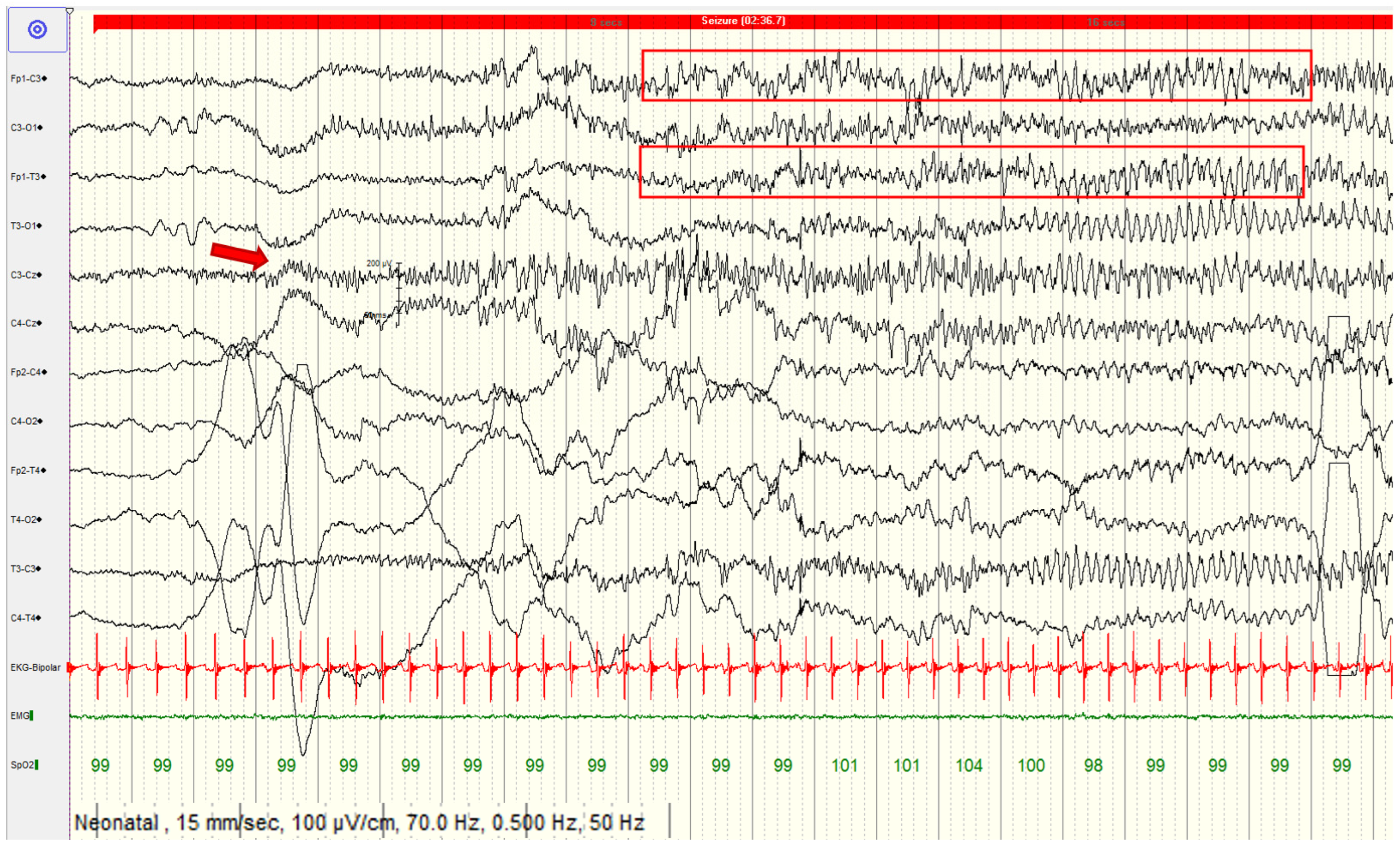
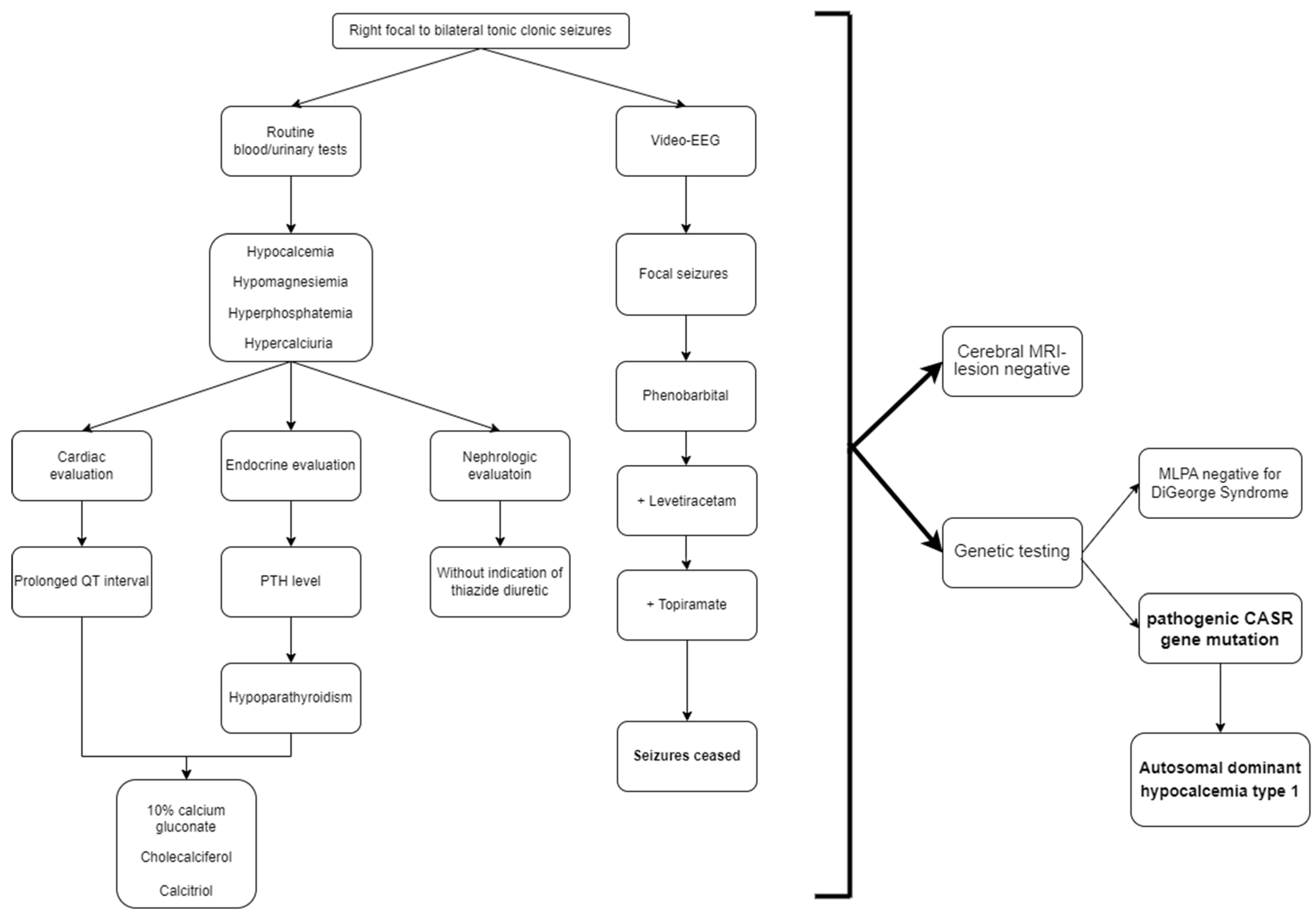
2. Case Report
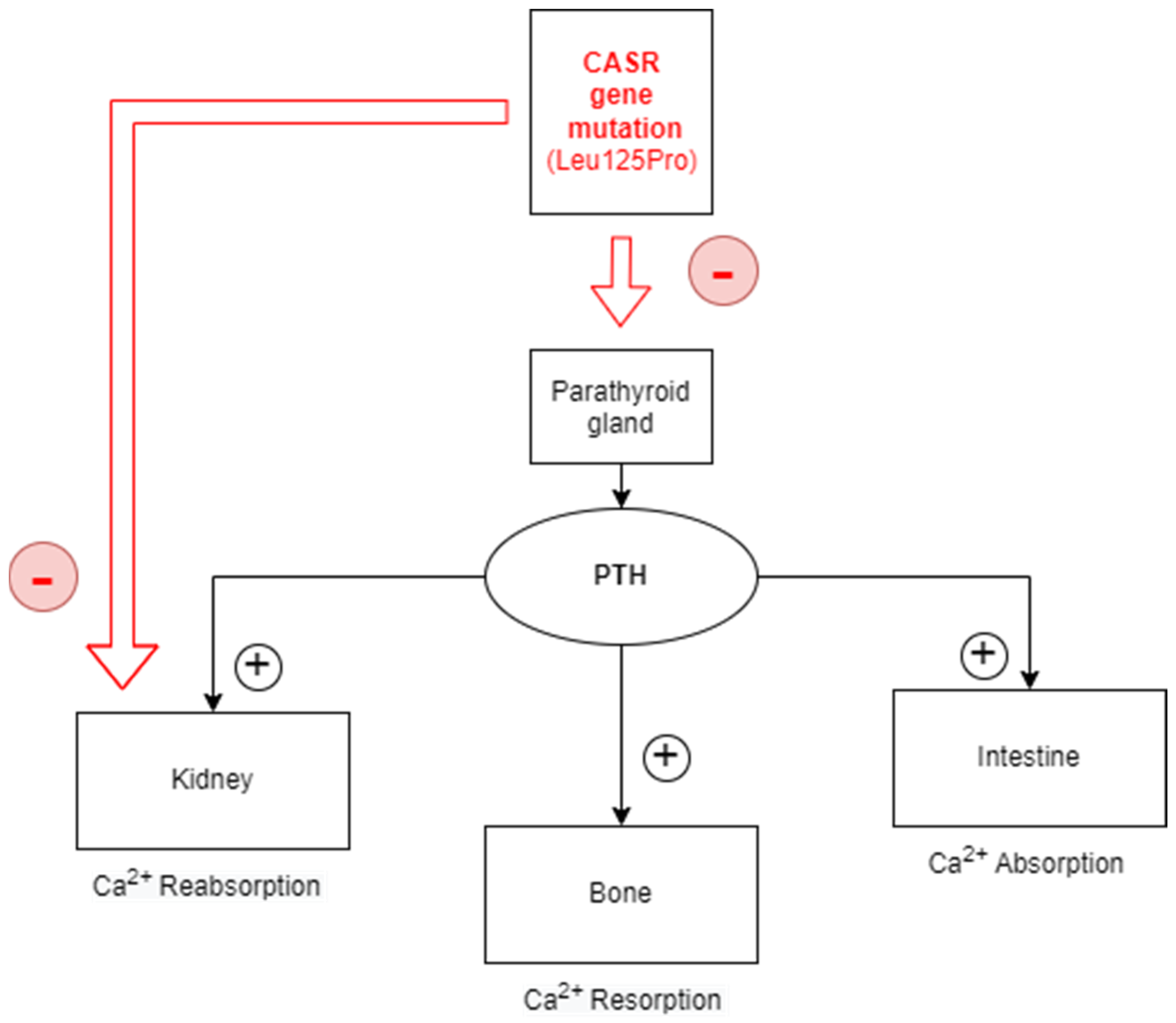
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cooper, M.S.; Gittoes, N.J.L. Diagnosis and Management of Hypocalcaemia. BMJ 2008, 336, 1298–1302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vahe, C.; Benomar, K.; Espiard, S.; Coppin, L.; Jannin, A.; Odou, M.F.; Vantyghem, M.C. Diseases Associated with Calcium-Sensing Receptor. Orphanet J. Rare Dis. 2017, 12, 19. [Google Scholar] [CrossRef] [Green Version]
- Roszko, K.L.; Bi, R.D.; Mannstadt, M. Autosomal Dominant Hypocalcemia (Hypoparathyroidism) Types 1 and 2. Front. Physiol. 2016, 7, 458. [Google Scholar] [CrossRef] [Green Version]
- Tsuji, T.; Hiroyuki, A.; Uraki, S.; Doi, A.; Morita, S.; Iwakura, H.; Nishi, M.; Furuta, H.; Akamizu, T. Autosomal Dominant Hypocalcemia with Atypical Urine Findings Accompanied by Novel CaSR Gene Mutation and VitD Deficiency. J. Endocr. Soc. 2021, 5, bvaa190. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, A.Q.; Jørgensen, N.R.; Schwarz, P. Identification and Functional Characterization of a Novel Mutation in the Human Calcium-Sensing Receptor That Co-Segregates with Autosomal-Dominant Hypocalcemia. Front. Endocrinol. 2018, 9, 200. [Google Scholar] [CrossRef] [Green Version]
- Roszko, K.L.; Stapleton Smith, L.M.; Sridhar, A.V.; Roberts, M.S.; Hartley, I.R.; Gafni, R.I.; Collins, M.T.; Fox, J.C.; Nemeth, E.F. Autosomal Dominant Hypocalcemia Type 1: A Systematic Review. J. Bone Miner. Res. 2022, 37, 1926–1935. [Google Scholar] [CrossRef] [PubMed]
- Elston, M.S.; Elajnaf, T.; Hannan, F.M.; Thakker, R.V. Autosomal Dominant Hypocalcemia Type 1 (ADH1) Associated with Myoclonus and Intracerebral Calcifications. J. Endocr. Soc. 2022, 6, bvac042. [Google Scholar] [CrossRef]
- Gomes, V.; Silvestre, C.; Ferreira, F.; Bugalho, M.J.G.M. Autosomal Dominant Hypocalcaemia: Identification of Two Novel Variants of CASR Gene. BMJ Case Rep. 2020, 13, e234391. [Google Scholar] [CrossRef]
- Wu, Y.; Zhang, C.; Huang, X.; Cao, L.; Liu, S.; Zhong, P. Autosomal Dominant Hypocalcemia with a Novel CASR Mutation: A Case Study and Literature Review. J. Int. Med. Res. 2022, 50, 03000605221110489. [Google Scholar] [CrossRef]
- Moon, J.-E.; Lee, S.-J.; Park, S.-H.; Kim, J.; Jin, D.-K.; Ko, C.W. De Novo a Novel Variant of CaSR Gene in a Neonate with Congenital Hypoparathyroidism. Ann. Pediatr. Endocrinol. Metab. 2018, 23, 107–111. [Google Scholar] [CrossRef] [Green Version]
- Rossi, G.C.; Patterson, A.L.; McGregor, A.L.; Wheless, J.W. Intractable Generalized Epilepsy and Autosomal Dominant Hypocalcemia: A Case Report. Child Neurol. Open 2019, 6, 2329048X19876199. [Google Scholar] [CrossRef] [Green Version]
- Ryu, H.K.; Park, S.U.; Shim, J.W.; Shim, J.Y.; Jung, H.L.; Park, M.S.; Kim, D.S. The Ictal Electroencephalographic Findings in a Neonate with Hypocalcemia Seizure. J. Korean Child Neurol. Soc. 2007, 15, 102–105. [Google Scholar]
- Patel, B.A.; Chakor, R.T.; Kothari, K.V.; Nadaf, S. Reversible Central Neural Hyperexcitability: An Electroencephalographic Clue to Hypocalcaemia. BMJ Case Rep. 2017, 2017, bcr2017220994. [Google Scholar] [CrossRef] [PubMed]
- Vargas-Poussou, R.; Huang, C.; Hulin, P.; Houillier, P.; Jeunemaître, X.; Paillard, M.; Planelles, G.; Déchaux, M.; Miller, R.T.; Antignac, C. Functional Characterization of a Calcium-Sensing Receptor Mutation in Severe Autosomal Dominant Hypocalcemia with a Bartter-Like Syndrome. J. Am. Soc. Nephrol. 2002, 13, 2259–2266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapoor, A.; Satishchandra, P.; Ratnapriya, R.; Reddy, R.; Kadandale, J.; Shankar, S.K.; Anand, A. An Idiopathic Epilepsy Syndrome Linked to 3q13.3-Q21 and Missense Mutations in the Extracellular Calcium Sensing Receptor Gene. Ann. Neurol. 2008, 64, 158–167. [Google Scholar] [CrossRef]
- Shoback, D. Clinical Practice. Hypoparathyroidism. N. Engl. J. Med. 2008, 359, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Vuralli, D. Clinical Approach to Hypocalcemia in Newborn Period and Infancy: Who Should Be Treated? Int. J. Pediatr. 2019, 2019, 4318075. [Google Scholar] [CrossRef] [Green Version]
- Pressler, R.M.; Cilio, M.R.; Mizrahi, E.M.; Moshé, S.L.; Nunes, M.L.; Plouin, P.; Vanhatalo, S.; Yozawitz, E.; de Vries, L.S.; Puthenveettil Vinayan, K.; et al. The ILAE Classification of Seizures and the Epilepsies: Modification for Seizures in the Neonate. Position Paper by the ILAE Task Force on Neonatal Seizures. Epilepsia 2021, 62, 615–628. [Google Scholar] [CrossRef]
- Nakajima, K.; Yamazaki, K.; Kimura, H.; Takano, K.; Miyoshi, H.; Sato, K. Novel Gain of Function Mutations of the Calcium-Sensing Receptor in Two Patients with PTH-Deficient Hypocalcemia. Intern. Med. Tokyo Jpn. 2009, 48, 1951–1956. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, T.; Bai, M.; Lane, C.R.; Matsumoto, S.; Minamitani, K.; Minagawa, M.; Niimi, H.; Brown, E.M.; Yasuda, T. Familial Hypoparathyroidism: Identification of a Novel Gain of Function Mutation in Transmembrane Domain 5 of the Calcium-Sensing Receptor1. J. Clin. Endocrinol. Metab. 1998, 83, 2497–2502. [Google Scholar] [CrossRef] [Green Version]
- Sato, K.; Hasegawa, Y.; Nakae, J.; Nanao, K.; Takahashi, I.; Tajima, T.; Shinohara, N.; Fujieda, K. Hydrochlorothiazide Effectively Reduces Urinary Calcium Excretion in Two Japanese Patients with Gain-of-Function Mutations of the Calcium-Sensing Receptor Gene. J. Clin. Endocrinol. Metab. 2002, 87, 3068–3073. [Google Scholar] [CrossRef]
- Boros, E.; Heinrichs, C.; Brachet, C. Autosomal Dominant Hypocalcemia Type I: About Difficult Management Despite 1-34 Recombinant PTH Treatment in an Infant. Ann. Case Rep. 2022, 7, 1062. [Google Scholar]
- Puliani, G.; Hasenmajer, V.; Simonelli, I.; Sada, V.; Pofi, R.; Minnetti, M.; Cozzolino, A.; Napoli, N.; Pasqualetti, P.; Gianfrilli, D.; et al. Safety and Efficacy of PTH 1–34 and 1–84 Therapy in Chronic Hypoparathyroidism: A Meta—Analysis of Prospective Trials. J. Bone Miner. Res. 2022, 37, 1233–1250. [Google Scholar] [CrossRef]
- Vokes, T.J.; Mannstadt, M.; Levine, M.A.; Clarke, B.L.; Lakatos, P.; Chen, K.; Piccolo, R.; Krasner, A.; Shoback, D.M.; Bilezikian, J.P. Recombinant Human Parathyroid Hormone Effect on Health-Related Quality of Life in Adults with Chronic Hypoparathyroidism. J. Clin. Endocrinol. Metab. 2017, 103, 722–731. [Google Scholar] [CrossRef] [PubMed]
- Shulman, D. Subcutaneous Infusion of RhPTH1-34 during Pregnancy and Nursing in a Woman with Autosomal Dominant Hypoparathyroidism 1. J. Endocr. Soc. 2022, 6, bvac031. [Google Scholar] [CrossRef] [PubMed]
- Fox, A.; Gilbert, R. Use of Teriparatide in a Four Year Old Patient with Autosomal Dominant Hypocalcaemia. Arch. Dis. Child. 2016, 101, e2. [Google Scholar] [CrossRef]
- Dayal, D.; Gupta, A.; Gupta, S.; Dutta, A. Recombinant Parathyroid Hormone for Hypoparathyroidism in Children: A Narrative Review. Pediatr. Endocrinol. Diabetes Metab. 2019, 25, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Roberts, M.S.; Gafni, R.I.; Brillante, B.; Guthrie, L.C.; Streit, J.; Gash, D.; Gelb, J.; Krusinska, E.; Brennan, S.C.; Schepelmann, M.; et al. Treatment of Autosomal Dominant Hypocalcemia Type 1 with the Calcilytic NPSP795 (SHP635). J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2019, 34, 1609–1618. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Serum Electrolytes | At Admission | 7 Days after Treatment Initiation | 10 Days after Treatment Initiation |
|---|---|---|---|
| Calcium | 5.94 mg/dL | 7.94 mg/dL | 9.69 mg/dL |
| Magnesium | 1.41 mg/dL | 1.56 mg/dL | 1.72 mg/dL |
| Phosphorus | 10.76 mg/dL | 6.93 mg/dL | 6.37 mg/dL |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teleanu, R.I.; Sarman, M.A.; Epure, D.A.; Matei, M.; Roşca, I.; Roza, E. Autosomal Dominant Hypocalcemia Type 1 and Neonatal Focal Seizures. Children 2023, 10, 1011. https://doi.org/10.3390/children10061011
Teleanu RI, Sarman MA, Epure DA, Matei M, Roşca I, Roza E. Autosomal Dominant Hypocalcemia Type 1 and Neonatal Focal Seizures. Children. 2023; 10(6):1011. https://doi.org/10.3390/children10061011
Chicago/Turabian StyleTeleanu, Raluca Ioana, Marlene Alexandra Sarman, Diana Anamaria Epure, Margarita Matei, Ioana Roşca, and Eugenia Roza. 2023. "Autosomal Dominant Hypocalcemia Type 1 and Neonatal Focal Seizures" Children 10, no. 6: 1011. https://doi.org/10.3390/children10061011