
TP53 Alterations in Myelodysplastic Syndromes and Acute Myeloid Leukemia

Abstract
:1. Introduction
2. Characteristics of TP53 Mutations in MDS/AML
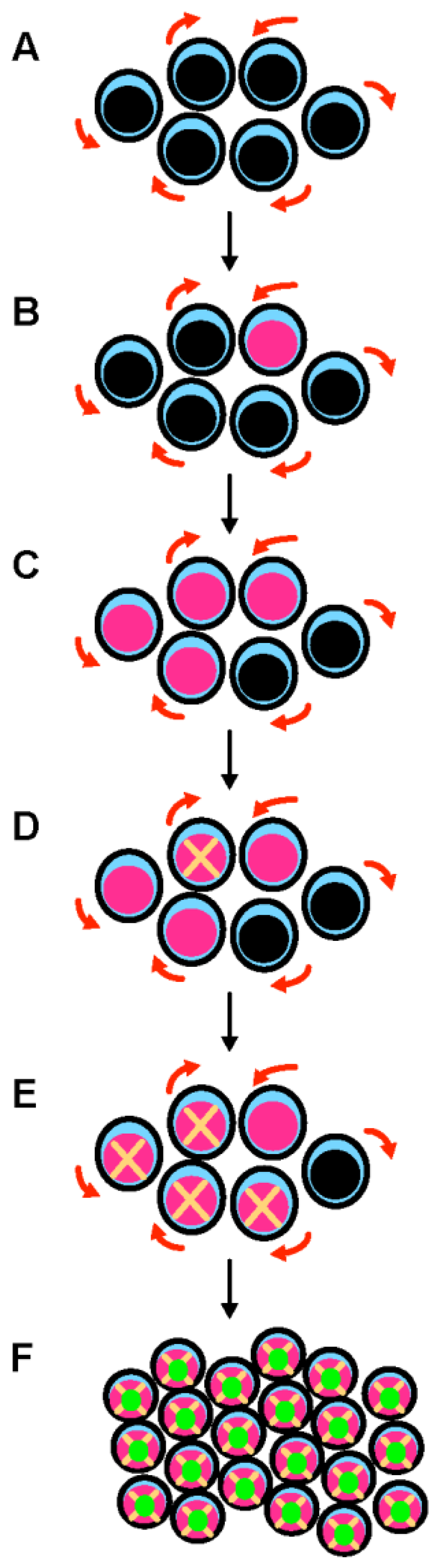
3. When Do TP53 Mutations Arise in MDS and AML Cells?
4. How Can p53 Mutants Drive Leukemia Development and Maintain the Leukemic State?
5. Treatment of TP53 Mutated MDS/AML
5.1. The Current Disappointing Situation
5.2. How Can Treatment of MDS/AMLs with TP53 Mutation Be Improved?
5.3. Drugs Currently Used in Clinical Trials
5.4. Drugs so Far Used Only in Preclinical Models

{kind=link}
{kind=link}
| Drug | Mechanism of Action | Trial | Cancer Type | Results | Reference |
|---|---|---|---|---|---|
| APR-246 | Reconforming agent | NCT04383938 | Advanced solid tumor (bladder, gastric, NSCLC, urothelial) | Phase Ib Combination with Pembrolizumab | Park H et al. [143] |
| NCT03072043 | MDS/AML | Phase II Plus Azacitidine Response rates: MDS 73% Oligoblastic AML 64% | Sallman D et al. [109] | ||
| NCT03588078 | MDS/AML | Phase II Plus Azacitidine Response rates: MDS 62% AML 33% | Cluzeau T et al. [110] | ||
| NCT03931291 | MDS/AML | Phase II Plus Azacitidine Maintenance therapy following transplant 1-year survival 78.8% | Mishra A et al. [144] | ||
| NCT03745716 | MDS/AML | Phase III Plus Azacitidine vs. Azacitidine No significant difference for primary endpoint | Press release | ||
| NCT04214860 | MDS/AML | Phase I Plus Azacitidine & Venetoclax | Trial completed Not reported | ||
| Arsenic trioxide | Reconforming agent | NCT03855371 | MDS/AML | Phase I Plus Decitabine | Currently enrolling |
| Ganetespib (STA-9090) | HSP90 inhibitor Mutant p53 degradation | NCT02012192 | High-grade platinum-resistant ovarian cancer | Phase I/II Combined with Paclitaxel Safe use of the combination | Ray-Coquard et al. [145] |
| Atorvastatin | Mutant p53 degradation | NCT03560882 | MDS/AML Solid tumors | Pilot trial | Currently enrolling |
| Vorinostat (SAHA) | HDAC inhibitor Mutant p53 degradation | NCT02042989 | Metastatic solid tumors | Phase I Plus Ixazomib (proteasome inhibitor) Median survival 7.3 months | Wang Y et al. [146] |
| NCT01339871 | Metastatic solid tumors | Phase I Plus Pazopanib (VEGF inhibitor) Median survival 12.7 months | Wang Y et al. [146] | ||
| Adavosertib (AZD1775/ MK-1775) | Wee1 inhibitor Synthetic lethality | NCT01164995 | Refractory ovarian cancer | Phase II Plus Carboplatin Median survival 12.6 months | Leijen S et al. [138] |
| NCT03668340 | Recurrent uterineserous carcinoma | Phase II Monotherapy Median PFS 6.1 months Median response 9.0 months | Liu et al. [147] |
6. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vousden, K.H.; Prives, C. Blinded by the Light: The Growing Complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pant, V.; Quintás-Cardama, A.; Lozano, G. The p53 pathway in hematopoiesis: Lessons from mouse models, implications for humans. Blood 2012, 120, 5118–5127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aloni-Grinstein, R.; Shetzer, Y.; Kaufman, T.; Rotter, V. p53: The barrier to cancer stem cell formation. FEBS Lett. 2014, 588, 2580–2589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bykov, V.J.N.; Issaeva, N.; Shilov, A.; Hultcrantz, M.; Pugacheva, E.; Chumakov, P.; Bergman, J.; Wiman, K.G.; Selivanova, G. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat. Med. 2002, 8, 282–288. [Google Scholar] [CrossRef]
- Junttila, M.R.; Karnezis, A.N.; Garcia, D.; Madriles, F.; Kortlever, R.M.; Rostker, F.; Brown Swigart, L.; Pham, D.M.; Seo, Y.; Evan, G.I.; et al. Selective activation of p53-mediated tumour suppression in high-grade tumours. Nature 2010, 468, 567–571. [Google Scholar] [CrossRef] [Green Version]
- Feldser, D.M.; Kostova, K.K.; Winslow, M.M.; Taylor, S.E.; Cashman, C.; Whittaker, C.A.; Sanchez-Rivera, F.J.; Resnick, R.; Bronson, R.; Hemann, M.T.; et al. Stage-specific sensitivity to p53 restoration during lung cancer progression. Nature 2010, 468, 572–575. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Suh, Y.-A.; Fuller, M.Y.; Jackson, J.G.; Xiong, S.; Terzian, T.; Quintás-Cardama, A.; Bankson, J.A.; El-Naggar, A.K.; Lozano, G. Restoring expression of wild-type p53 suppresses tumor growth but does not cause tumor regression in mice with a p53 missense mutation. J. Clin. Investig. 2011, 121, 893–904. [Google Scholar] [CrossRef]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef]
- Toledo, F.; Wahl, G.M. Regulating the p53 pathway: In vitro hypotheses, in vivo veritas. Nat. Rev. Cancer 2006, 6, 909–923. [Google Scholar] [CrossRef]
- Wasylishen, A.R.; Lozano, G. Attenuating the p53 Pathway in Human Cancers: Many Means to the Same End. Cold Spring Harb. Perspect. Med. 2016, 6, a026211. [Google Scholar] [CrossRef] [PubMed]
- Vieler, M.; Sanyal, S. p53 Isoforms and Their Implications in Cancer. Cancers 2018, 10, 288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ånensen, N.; Hjelle, S.M.; Van Belle, W.; Haaland, I.; Silden, E.; Bourdon, J.-C.; Hovland, R.; Taskén, K.; Knappskog, S.; Lønning, P.E.; et al. Correlation analysis of p53 protein isoforms with NPM1/FLT3 mutations and therapy response in acute myeloid leukemia. Oncogene 2012, 31, 1533–1545. [Google Scholar] [CrossRef] [Green Version]
- Bergh, J.; Norberg, T.; Sjögren, S.; Lindgren, A.; Holmberg, L. Complete sequencing of the p53 gene provides prognostic information in breast cancer patients, particularly in relation to adjuvant systemic therapy and radiotherapy. Nat. Med. 1995, 1, 1029–1034. [Google Scholar] [CrossRef] [PubMed]
- Aas, T.; Børresen, A.L.; Geisler, S.; Smith-Sørensen, B.; Johnsen, H.; Varhaug, J.E.; Akslen, L.A.; Lønning, P.E. Specific P53 mutations are associated with de novo resistance to doxorubicin in breast cancer patients. Nat. Med. 1996, 2, 811–814. [Google Scholar] [CrossRef] [PubMed]
- Shelling, A.N. Role of p53 in drug resistance in ovarian cancer. Lancet 1997, 349, 744–745. [Google Scholar] [CrossRef]
- Petty, R.; Evans, A.; Duncan, I.; Kurbacher, C.; Cree, I. Drug resistance in ovarian cancer—The role of p53. Pathol. Oncol. Res. 1998, 4, 97–102. [Google Scholar] [CrossRef]
- Horio, Y.; Takahashi, T.; Kuroishi, T.; Hibi, K.; Suyama, M.; Niimi, T.; Shimokata, K.; Yamakawa, K.; Nakamura, Y.; Ueda, R. Prognostic significance of p53 mutations and 3p deletions in primary resected non-small cell lung cancer. Cancer Res. 1993, 53, 1–4. [Google Scholar]
- Hamada, M.; Fujiwara, T.; Hizuta, A.; Gochi, A.; Naomoto, Y.; Takakura, N.; Takahashi, K.; Roth, J.A.; Tanaka, N.; Orita, K. The p53 gene is a potent determinant of chemosensitivity and radiosensitivity in gastric and colorectal cancers. J. Cancer Res. Clin. Oncol. 1996, 122, 360–365. [Google Scholar] [CrossRef]
- Wattel, E.; Preudhomme, C.; Hecquet, B.; Vanrumbeke, M.; Quesnel, B.; Dervite, I.; Morel, P.; Fenaux, P. p53 mutations are associated with resistance to chemotherapy and short survival in hematologic malignancies. Blood 1994, 84, 3148–3157. [Google Scholar] [CrossRef] [Green Version]
- Wilson, W.H.; Teruya-Feldstein, J.; Fest, T.; Harris, C.; Steinberg, S.M.; Jaffe, E.S.; Raffeld, M. Relationship of p53, bcl-2, and tumor proliferation to clinical drug resistance in non-Hodgkin’s lymphomas. Blood 1997, 89, 601–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, W.M.; Brennan, J.A.; Zahurak, M.; Goodman, S.N.; Westra, W.H.; Schwab, D.; Yoo, G.H.; Lee, D.J.; Forastiere, A.A.; Sidransky, D. p53 mutation and locoregional treatment failure in head and neck squamous cell carcinoma. J. Natl. Cancer Inst. 1996, 88, 1580–1586. [Google Scholar] [CrossRef] [PubMed]
- Fenaux, P.; Platzbecker, U.; Ades, L. How we manage adults with myelodysplastic syndrome. Br. J. Haematol. 2020, 189, 1016–1027. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Döhner, H.; Campbell, P.J. Genomic Classification in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 375, 900–901. [Google Scholar] [CrossRef]
- Slingerland, J.M.; Minden, M.D.; Benchimol, S. Mutation of the p53 gene in human acute myelogenous leukemia. Blood 1991, 77, 1500–1507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fenaux, P.; Jonveaux, P.; Quiquandon, I.; Laï, J.L.; Pignon, J.M.; Loucheux-Lefebvre, M.H.; Bauters, F.; Berger, R.; Kerckaert, J.P. P53 gene mutations in acute myeloid leukemia with 17p monosomy. Blood 1991, 78, 1652–1657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fenaux, P.; Preudhomme, C.; Quiquandon, I.; Jonveaux, P.; Laï, J.L.; Vanrumbeke, M.; Loucheux-Lefebvre, M.H.; Bauters, F.; Berger, R.; Kerckaert, J.P. Mutations of the P53 gene in acute myeloid leukaemia. Br. J. Haematol. 1992, 80, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Ok, C.Y.; Patel, K.P.; Garcia-Manero, G.; Routbort, M.J.; Peng, J.; Tang, G.; Goswami, M.; Young, K.H.; Singh, R.; Medeiros, L.J.; et al. TP53 mutation characteristics in therapy-related myelodysplastic syndromes and acute myeloid leukemia is similar to de novo diseases. J. Hematol. Oncol. 2015, 8, 45. [Google Scholar] [CrossRef] [Green Version]
- Haase, D.; Stevenson, K.E.; Neuberg, D.; Maciejewski, J.P.; Nazha, A.; Sekeres, M.A.; Ebert, B.L.; Garcia-Manero, G.; Haferlach, C.; Haferlach, T.; et al. TP53 mutation status divides myelodysplastic syndromes with complex karyotypes into distinct prognostic subgroups. Leukemia 2019, 3, 1747–1758. [Google Scholar] [CrossRef] [Green Version]
- Quintás-Cardama, A.; Hu, C.; Qutub, A.; Qiu, Y.H.; Zhang, X.; Post, S.M.; Zhang, N.; Coombes, K.; Kornblau, S.M. p53 pathway dysfunction is highly prevalent in acute myeloid leukemia independent of TP53 mutational status. Leukemia 2017, 31, 1296–1305. [Google Scholar] [CrossRef]
- Bueso-Ramos, C.E.; Yang, Y.; deLeon, E.; McCown, P.; Stass, S.A.; Albitar, M. The human MDM-2 oncogene is overexpressed in leukemias. Blood 1993, 82, 2617–2623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faderl, S.; Kantarjian, H.M.; Estey, E.; Manshouri, T.; Chan, C.Y.; Rahman Elsaied, A.; Kornblau, S.M.; Cortes, J.; Thomas, D.A.; Pierce, S.; et al. The prognostic significance of p16(INK4a)/p14(ARF) locus deletion and MDM-2 protein expression in adult acute myelogenous leukemia. Cancer 2000, 89, 1976–1982. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.; Tergaonkar, V.; Lane, D.P. Double-edged swords as cancer therapeutics: Simultaneously targeting p53 and NF-kappaB pathways. Nat. Rev. Drug Discov. 2008, 7, 1031–1040. [Google Scholar] [CrossRef]
- Wade, M.; Li, Y.-C.; Wahl, G.M. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat. Rev. Cancer 2013, 13, 83–96. [Google Scholar] [CrossRef] [Green Version]
- Khurana, A.; Shafer, D.A. MDM2 antagonists as a novel treatment option for acute myeloid leukemia: Perspectives on the therapeutic potential of idasanutlin (RG7388). OncoTargets Ther. 2019, 12, 2903–2910. [Google Scholar] [CrossRef] [Green Version]
- Müller-Tidow, C.; Metzelder, S.K.; Buerger, H.; Packeisen, J.; Ganser, A.; Heil, G.; Kügler, K.; Adigüzel, G.; Schwäble, J.; Steffen, B.; et al. Expression of the p14ARF tumor suppressor predicts survival in acute myeloid leukemia. Leukemia 2004, 18, 720–726. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Kon, N.; Zhong, J.; Zhang, P.; Yu, L.; Gu, W. Differential effects on ARF stability by normal versus oncogenic levels of c-Myc expression. Mol. Cell 2013, 51, 46–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, J.L.; Preudhomme, C.; Zandecki, M.; Flactif, M.; Vanrumbeke, M.; Lepelley, P.; Wattel, E.; Fenaux, P. Myelodysplastic syndromes and acute myeloid leukemia with 17p deletion. An entity characterized by specific dysgranulopoïesis and a high incidence of P53 mutations. Leukemia 1995, 9, 370–381. [Google Scholar]
- Soenen, V.; Preudhomme, C.; Roumier, C.; Daudignon, A.; Laï, J.L.; Fenaux, P. 17p Deletion in acute myeloid leukemia and myelodysplastic syndrome. Analysis of breakpoints and deleted segments by fluorescence in situ. Blood 1998, 91, 1008–1015. [Google Scholar] [CrossRef] [Green Version]
- Sabapathy, K.; Lane, D.P. Therapeutic targeting of p53: All mutants are equal, but some mutants are more equal than others. Nat. Rev. Clin. Oncol. 2018, 15, 13–30. [Google Scholar] [CrossRef]
- Prochazka, K.T.; Pregartner, G.; Rücker, F.G.; Heitzer, E.; Pabst, G.; Wölfler, A.; Zebisch, A.; Berghold, A.; Döhner, K.; Sill, H. Clinical implications of subclonal TP53 mutations in acute myeloid leukemia. Haematologica 2019, 104, 516–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernard, E.; Nannya, Y.; Hasserjian, R.P.; Devlin, S.M.; Tuechler, H.; Medina-Martinez, J.S.; Yoshizato, T.; Shiozawa, Y.; Saiki, R.; Malcovati, L.; et al. Implications of TP53 allelic state for genome stability, clinical presentation and outcomes in myelodysplastic syndromes. Nat. Med. 2020, 26, 1549–1556. [Google Scholar] [CrossRef] [PubMed]
- Jädersten, M.; Saft, L.; Smith, A.; Kulasekararaj, A.; Pomplun, S.; Göhring, G.; Hedlund, A.; Hast, R.; Schlegelberger, B.; Porwit, A.; et al. TP53 mutations in low-risk myelodysplastic syndromes with del(5q) predict disease progression. J. Clin. Oncol. 2011, 29, 1971–1979. [Google Scholar] [CrossRef]
- Sebaa, A.; Ades, L.; Baran-Marzack, F.; Mozziconacci, M.-J.; Penther, D.; Dobbelstein, S.; Stamatoullas, A.; Récher, C.; Prebet, T.; Moulessehoul, S.; et al. Incidence of 17p deletions and TP53 mutation in myelodysplastic syndrome and acute myeloid leukemia with 5q deletion. Genes Chromosomes Cancer 2012, 51, 1086–1092. [Google Scholar] [CrossRef] [PubMed]
- Scharenberg, C.; Giai, V.; Pellagatti, A.; Saft, L.; Dimitriou, M.; Jansson, M.; Jädersten, M.; Grandien, A.; Douagi, I.; Neuberg, D.S.; et al. Progression in patients with low- and intermediate-1-risk del(5q) myelodysplastic syndromes is predicted by a limited subset of mutations. Haematologica 2017, 102, 498–508. [Google Scholar] [CrossRef] [Green Version]
- Lodé, L.; Ménard, A.; Flet, L.; Richebourg, S.; Loirat, M.; Eveillard, M.; Le Bris, Y.; Godon, C.; Theisen, O.; Gagez, A.-L.; et al. Emergence and evolution of TP53 mutations are key features of disease progression in myelodysplastic patients with lower-risk del(5q) treated with lenalidomide. Haematologica 2018, 103, e143–e146. [Google Scholar] [CrossRef] [Green Version]
- Volkert, S.; Kohlmann, A.; Schnittger, S.; Kern, W.; Haferlach, T.; Haferlach, C. Association of the type of 5q loss with complex karyotype, clonal evolution, TP53 mutation status, and prognosis in acute myeloid leukemia and myelodysplastic syndrome. Genes Chromosomes Cancer 2014, 53, 402–410. [Google Scholar] [CrossRef]
- Forment, J.V.; Kaidi, A.; Jackson, S.P. Chromothripsis and cancer: Causes and consequences of chromosome shattering. Nat. Rev. Cancer 2012, 12, 663–670. [Google Scholar] [CrossRef]
- Rücker, F.G.; Dolnik, A.; Blätte, T.J.; Teleanu, V.; Ernst, A.; Thol, F.; Heuser, M.; Ganser, A.; Döhner, H.; Döhner, K.; et al. Chromothripsis is linked to TP53 alteration, cell cycle impairment, and dismal outcome in acute myeloid leukemia with complex karyotype. Haematologica 2018, 103, e17–e20. [Google Scholar] [CrossRef] [Green Version]
- Bejar, R.; Stevenson, K.E.; Caughey, B.; Lindsley, R.C.; Mar, B.G.; Stojanov, P.; Getz, G.; Steensma, D.P.; Ritz, J.; Soiffer, R.; et al. Somatic mutations predict poor outcome in patients with myelodysplastic syndrome after hematopoietic stem-cell transplantation. J. Clin. Oncol. 2014, 32, 2691–2698. [Google Scholar] [CrossRef] [Green Version]
- Bally, C.; Adès, L.; Renneville, A.; Sebert, M.; Eclache, V.; Preudhomme, C.; Mozziconacci, M.-J.; de The, H.; Lehmann-Che, J.; Fenaux, P. Prognostic value of TP53 gene mutations in myelodysplastic syndromes and acute myeloid leukemia treated with azacitidine. Leuk. Res. 2014, 38, 751–755. [Google Scholar] [CrossRef] [PubMed]
- Desoutter, J.; Gay, J.; Berthon, C.; Ades, L.; Gruson, B.; Geffroy, S.; Plantier, I.; Marceau, A.; Helevaut, N.; Fernandes, J.; et al. Molecular prognostic factors in acute myeloid leukemia receiving first-line therapy with azacitidine. Leukemia 2016, 30, 1416–1418. [Google Scholar] [CrossRef] [PubMed]
- Welch, J.S.; Petti, A.A.; Miller, C.A.; Fronick, C.C.; O’Laughlin, M.; Fulton, R.S.; Wilson, R.K.; Baty, J.D.; Duncavage, E.J.; Tandon, B.; et al. TP53 and Decitabine in Acute Myeloid Leukemia and Myelodysplastic Syndromes. N. Engl. J. Med. 2016, 375, 2023–2036. [Google Scholar] [CrossRef]
- Martinez-Høyer, S.; Deng, Y.; Parker, J.; Jiang, J.; Mo, A.; Docking, T.R.; Gharaee, N.; Li, J.; Umlandt, P.; Fuller, M.; et al. Loss of lenalidomide-induced megakaryocytic differentiation leads to therapy resistance in del(5q) myelodysplastic syndrome. Nat. Cell Biol. 2020, 22, 526–533. [Google Scholar] [CrossRef]
- Wong, T.N.; Ramsingh, G.; Young, A.L.; Miller, C.A.; Touma, W.; Welch, J.S.; Lamprecht, T.L.; Shen, D.; Hundal, J.; Fulton, R.S.; et al. Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature 2015, 518, 552–555. [Google Scholar] [CrossRef] [Green Version]
- Lindsley, R.C.; Saber, W.; Mar, B.G.; Redd, R.; Wang, T.; Haagenson, M.D.; Grauman, P.V.; Hu, Z.-H.; Spellman, S.R.; Lee, S.J.; et al. Prognostic Mutations in Myelodysplastic Syndrome after Stem-Cell Transplantation. N. Engl. J. Med. 2017, 376, 536–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genovese, G.; Kähler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef] [Green Version]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [Green Version]
- Coombs, C.C.; Zehir, A.; Devlin, S.M.; Kishtagari, A.; Syed, A.; Jonsson, P.; Hyman, D.M.; Solit, D.B.; Robson, M.E.; Baselga, J.; et al. Therapy-Related Clonal Hematopoiesis in Patients with Non-hematologic Cancers Is Common and Associated with Adverse Clinical Outcomes. Cell Stem Cell 2017, 21, 374–382.e4. [Google Scholar] [CrossRef] [Green Version]
- Wong, T.N.; Miller, C.A.; Jotte, M.R.M.; Bagegni, N.; Baty, J.D.; Schmidt, A.P.; Cashen, A.F.; Duncavage, E.J.; Helton, N.M.; Fiala, M.; et al. Cellular stressors contribute to the expansion of hematopoietic clones of varying leukemic potential. Nat. Commun. 2018, 9, 455. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Gao, R.; Yao, C.; Kobayashi, M.; Liu, S.Z.; Yoder, M.C.; Broxmeyer, H.; Kapur, R.; Boswell, H.S.; Mayo, L.D.; et al. Genotoxic stresses promote clonal expansion of hematopoietic stem cells expressing mutant p53. Leukemia 2018, 32, 850–854. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Wang, Q.; Yu, H.; Capitano, M.L.; Vemula, S.; Nabinger, S.C.; Gao, R.; Yao, C.; Kobayashi, M.; Geng, Z.; et al. Mutant p53 drives clonal hematopoiesis through modulating epigenetic pathway. Nat. Commun. 2019, 10, 5649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Liu, Y. p53 involvement in clonal hematopoiesis of indeterminate potential. Curr. Opin. Hematol. 2019, 26, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Miller, P.G.; Steensma, D.P. Implications of Clonal Hematopoiesis for Precision Oncology. JCO Precis. Oncol. 2020, 4, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Haferlach, C.; Dicker, F.; Herholz, H.; Schnittger, S.; Kern, W.; Haferlach, T. Mutations of the TP53 gene in acute myeloid leukemia are strongly associated with a complex aberrant karyotype. Leukemia 2008, 22, 1539–1541. [Google Scholar] [CrossRef]
- Bowen, D.; Groves, M.J.; Burnett, A.K.; Patel, Y.; Allen, C.; Green, C.; Gale, R.E.; Hills, R.; Linch, D.C. TP53 gene mutation is frequent in patients with acute myeloid leukemia and complex karyotype, and is associated with very poor prognosis. Leukemia 2009, 23, 203–206. [Google Scholar] [CrossRef] [Green Version]
- Stoddart, A.; Fernald, A.A.; Wang, J.; Davis, E.M.; Karrison, T.; Anastasi, J.; Le Beau, M.M. Haploinsufficiency of del(5q) genes, Egr1 and Apc, cooperate with Tp53 loss to induce acute myeloid leukemia in mice. Blood 2014, 123, 1069–1078. [Google Scholar] [CrossRef] [Green Version]
- Prokocimer, M.; Molchadsky, A.; Rotter, V. Dysfunctional diversity of p53 proteins in adult acute myeloid leukemia: Projections on diagnostic workup and therapy. Blood 2017, 130, 699–712. [Google Scholar] [CrossRef] [Green Version]
- Harutyunyan, A.; Klampfl, T.; Cazzola, M.; Kralovics, R. p53 lesions in leukemic transformation. N. Engl. J. Med. 2011, 364, 488–490. [Google Scholar] [CrossRef] [Green Version]
- Rampal, R.; Ahn, J.; Abdel-Wahab, O.; Nahas, M.; Wang, K.; Lipson, D.; Otto, G.A.; Yelensky, R.; Hricik, T.; McKenney, A.S.; et al. Genomic and functional analysis of leukemic transformation of myeloproliferative neoplasms. Proc. Natl. Acad. Sci. USA 2014, 111, E5401–E5410. [Google Scholar] [CrossRef] [Green Version]
- Ohgami, R.S.; Ma, L.; Merker, J.D.; Gotlib, J.R.; Schrijver, I.; Zehnder, J.L.; Arber, D.A. Next-generation sequencing of acute myeloid leukemia identifies the significance of TP53, U2AF1, ASXL1, and TET2 mutations. Mod. Pathol. 2015, 28, 706–714. [Google Scholar] [CrossRef] [Green Version]
- Courtier, F.; Carbuccia, N.; Garnier, S.; Guille, A.; Adélaïde, J.; Cervera, N.; Gelsi-Boyer, V.; Mozziconacci, M.-J.; Rey, J.; Vey, N.; et al. Genomic analysis of myeloproliferative neoplasms in chronic and acute phases. Haematologica 2017, 102, e11–e14. [Google Scholar] [CrossRef] [PubMed]
- Kubesova, B.; Pavlova, S.; Malcikova, J.; Kabathova, J.; Radova, L.; Tom, N.; Tichy, B.; Plevova, K.; Kantorova, B.; Fiedorova, K.; et al. Low-burden TP53 mutations in chronic phase of myeloproliferative neoplasms: Association with age, hydroxyurea administration, disease type and JAK2 mutational status. Leukemia 2018, 32, 450–461. [Google Scholar] [CrossRef] [PubMed]
- Shlush, L.I.; Zandi, S.; Mitchell, A.; Chen, W.C.; Brandwein, J.M.; Gupta, V.; Kennedy, J.A.; Schimmer, A.D.; Schuh, A.C.; Yee, K.W.; et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 2014, 506, 328–333. [Google Scholar] [CrossRef] [Green Version]
- Corces-Zimmerman, M.R.; Hong, W.-J.; Weissman, I.L.; Medeiros, B.C.; Majeti, R. Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proc. Natl. Acad. Sci. USA 2014, 111, 2548–2553. [Google Scholar] [CrossRef] [Green Version]
- Muller, P.A.J.; Vousden, K.H. Mutant p53 in cancer: New functions and therapeutic opportunities. Cancer Cell 2014, 25, 304–317. [Google Scholar] [CrossRef] [Green Version]
- Heuser, M. Therapy-related myeloid neoplasms: Does knowing the origin help to guide treatment? Hematol. Am. Soc. Hematol. Educ. Program 2016, 2016, 24–32. [Google Scholar] [CrossRef]
- Lal, R.; Lind, K.; Heitzer, E.; Ulz, P.; Aubell, K.; Kashofer, K.; Middeke, J.M.; Thiede, C.; Schulz, E.; Rosenberger, A.; et al. Somatic TP53 mutations characterize preleukemic stem cells in acute myeloid leukemia. Blood 2017, 129, 2587–2591. [Google Scholar] [CrossRef] [Green Version]
- Rausch, T.; Jones, D.T.W.; Zapatka, M.; Stütz, A.M.; Zichner, T.; Weischenfeldt, J.; Jäger, N.; Remke, M.; Shih, D.; Northcott, P.A.; et al. Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell 2012, 148, 59–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gualberto, A.; Aldape, K.; Kozakiewicz, K.; Tlsty, T.D. An oncogenic form of p53 confers a dominant, gain-of-function phenotype that disrupts spindle checkpoint control. Proc. Natl. Acad. Sci. USA 1998, 95, 5166–5171. [Google Scholar] [CrossRef] [Green Version]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 mutations in human cancers: Origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2010, 2, a001008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.-H.; Wu, C.-F.; Rajasekaran, N.; Shin, Y.K. Loss of Tumor Suppressor Gene Function in Human Cancer: An Overview. Cell. Physiol. Biochem. 2018, 51, 2647–2693. [Google Scholar] [CrossRef] [PubMed]
- Aubrey, B.J.; Janic, A.; Chen, Y.; Chang, C.; Lieschke, E.C.; Diepstraten, S.T.; Kueh, A.J.; Bernardini, J.P.; Dewson, G.; O’Reilly, L.A.; et al. Mutant TRP53 exerts a target gene-selective dominant-negative effect to drive tumor development. Genes Dev. 2018, 32, 1420–1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gencel-Augusto, J.; Lozano, G. p53 tetramerization: At the center of the dominant-negative effect of mutant p53. Genes Dev. 2020, 34, 1128–1146. [Google Scholar] [CrossRef]
- Lang, G.A.; Iwakuma, T.; Suh, Y.-A.; Liu, G.; Rao, V.A.; Parant, J.M.; Valentin-Vega, Y.A.; Terzian, T.; Caldwell, L.C.; Strong, L.C.; et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 2004, 119, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Olive, K.P.; Tuveson, D.A.; Ruhe, Z.C.; Yin, B.; Willis, N.A.; Bronson, R.T.; Crowley, D.; Jacks, T. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell 2004, 119, 847–860. [Google Scholar] [CrossRef] [Green Version]
- Doyle, B.; Morton, J.P.; Delaney, D.W.; Ridgway, R.A.; Wilkins, J.A.; Sansom, O.J. p53 mutation and loss have different effects on tumourigenesis in a novel mouse model of pleomorphic rhabdomyosarcoma. J. Pathol. 2010, 222, 129–137. [Google Scholar] [CrossRef]
- Morton, J.P.; Timpson, P.; Karim, S.A.; Ridgway, R.A.; Athineos, D.; Doyle, B.; Jamieson, N.B.; Oien, K.A.; Lowy, A.M.; Brunton, V.G.; et al. Mutant p53 drives metastasis and overcomes growth arrest/senescence in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 246–251. [Google Scholar] [CrossRef] [Green Version]
- Yue, X.; Zhao, Y.; Xu, Y.; Zheng, M.; Feng, Z.; Hu, W. Mutant p53 in Cancer: Accumulation, Gain-of-Function, and Therapy. J. Mol. Biol. 2017, 429, 1595–1606. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Liu, J.; Xu, D.; Zhang, T.; Hu, W.; Feng, Z. Gain-of-function mutant p53 in cancer progression and therapy. J. Mol. Cell Biol. 2020, 12, 674–687. [Google Scholar] [CrossRef] [PubMed]
- Alvarado-Ortiz, E.; de la Cruz-López, K.G.; Becerril-Rico, J.; Sarabia-Sánchez, M.A.; Ortiz-Sánchez, E.; García-Carrancá, A. Mutant p53 Gain-of-Function: Role in Cancer Development, Progression, and Therapeutic Approaches. Front. Cell Dev. Biol. 2021, 8, 607670. [Google Scholar] [CrossRef] [PubMed]
- Boettcher, S.; Miller, P.G.; Sharma, R.; McConkey, M.; Leventhal, M.; Krivtsov, A.V.; Giacomelli, A.O.; Wong, W.; Kim, J.; Chao, S.; et al. A dominant-negative effect drives selection of TP53 missense mutations in myeloid malignancies. Science 2019, 365, 599–604. [Google Scholar] [CrossRef]
- Chen, C.; Liu, Y.; Rappaport, A.R.; Kitzing, T.; Schultz, N.; Zhao, Z.; Shroff, A.S.; Dickins, R.A.; Vakoc, C.R.; Bradner, J.E.; et al. MLL3 is a haploinsufficient 7q tumor suppressor in acute myeloid leukemia. Cancer Cell 2014, 25, 652–665. [Google Scholar] [CrossRef] [Green Version]
- Loizou, E.; Banito, A.; Livshits, G.; Ho, Y.-J.; Koche, R.P.; Sánchez-Rivera, F.J.; Mayle, A.; Chen, C.-C.; Kinalis, S.; Bagger, F.O.; et al. A Gain-of-Function p53-Mutant Oncogene Promotes Cell Fate Plasticity and Myeloid Leukemia through the Pluripotency Factor FOXH1. Cancer Discov. 2019, 9, 962–979. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Chen, C.; Xu, Z.; Scuoppo, C.; Rillahan, C.D.; Gao, J.; Spitzer, B.; Bosbach, B.; Kastenhuber, E.R.; Baslan, T.; et al. Deletions linked to TP53 loss drive cancer through p53-independent mechanisms. Nature 2016, 531, 471–475. [Google Scholar] [CrossRef] [Green Version]
- Göhring, G.; Giagounidis, A.; Büsche, G.; Kreipe, H.H.; Zimmermann, M.; Hellström-Lindberg, E.; Aul, C.; Schlegelberger, B. Patients with del(5q) MDS who fail to achieve sustained erythroid or cytogenetic remission after treatment with lenalidomide have an increased risk for clonal evolution and AML progression. Ann. Hematol. 2010, 89, 365–374. [Google Scholar] [CrossRef] [Green Version]
- Lindsley, R.C.; Gibson, C.J.; Murdock, H.M.; Stone, R.M.; Cortes, J.E.; Uy, G.L.; Lin, T.L.; Ritchie, E.K.; Prebet, T.; Ryan, R.J.; et al. Genetic Characteristics and Outcomes By Mutation Status in a Phase 3 Study of CPX-351 Versus 7+3 in Older Adults with Newly Diagnosed, High-Risk/Secondary Acute Myeloid Leukemia (AML). Blood 2019, 134, 15. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Döhner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N. Engl. J. Med. 2020, 383, 617–629. [Google Scholar] [CrossRef]
- Britt, A.; Mohyuddin, G.R.; McClune, B.; Singh, A.; Lin, T.; Ganguly, S.; Abhyankar, S.; Shune, L.; McGuirk, J.; Skikne, B.; et al. Acute myeloid leukemia or myelodysplastic syndrome with chromosome 17 abnormalities and long-term outcomes with or without hematopoietic stem cell transplantation. Leuk. Res. 2020, 95, 106402. [Google Scholar] [CrossRef]
- Poiré, X.; Labopin, M.; Polge, E.; Forcade, E.; Ganser, A.; Volin, L.; Michallet, M.; Blaise, D.; Yakoub-Agha, I.; Maertens, J.; et al. Allogeneic stem cell transplantation using HLA-matched donors for acute myeloid leukemia with deletion 5q or monosomy 5: A study from the Acute Leukemia Working Party of the EBMT. Haematologica 2020, 105, 414–423. [Google Scholar] [CrossRef] [Green Version]
- Garderet, L.; Ziagkos, D.; van Biezen, A.; Iacobelli, S.; Finke, J.; Maertens, J.; Volin, L.; Ljungman, P.; Chevallier, P.; Passweg, J.; et al. Allogeneic Stem Cell Transplantation for Myelodysplastic Syndrome Patients with a 5q Deletion. Biol. Blood Marrow Transplant. 2018, 24, 507–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Bykov, V.J.N.; Wiman, K.G.; Zawacka-Pankau, J. APR-246 reactivates mutant p53 by targeting cysteines 124 and 277. Cell Death Dis. 2018, 9, 439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, J.M.R.; Gorzov, P.; Veprintsev, D.B.; Söderqvist, M.; Segerbäck, D.; Bergman, J.; Fersht, A.R.; Hainaut, P.; Wiman, K.G.; Bykov, V.J.N. PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell 2009, 15, 376–388. [Google Scholar] [CrossRef] [Green Version]
- Peng, X.; Zhang, M.-Q.-Z.; Conserva, F.; Hosny, G.; Selivanova, G.; Bykov, V.J.N.; Arnér, E.S.J.; Wiman, K.G. APR-246/PRIMA-1MET inhibits thioredoxin reductase 1 and converts the enzyme to a dedicated NADPH oxidase. Cell Death Dis. 2013, 4, e881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tessoulin, B.; Descamps, G.; Moreau, P.; Maïga, S.; Lodé, L.; Godon, C.; Marionneau-Lambot, S.; Oullier, T.; Le Gouill, S.; Amiot, M.; et al. PRIMA-1Met induces myeloma cell death independent of p53 by impairing the GSH/ROS balance. Blood 2014, 124, 1626–1636. [Google Scholar] [CrossRef] [Green Version]
- Gourley, C.; Gabra, H.; Vergote, I.; Basu, B.; Brenton, J.; Von Euler, M.; Björklund, U.; Smith, A.M.; Green, J. EUTROC PiSARRO: A phase Ib study combining APR-246 with standard chemotherapy in platinum sensitive relapsed high grade serous ovarian carcinoma (HGSOC). J. Clin. Oncol. 2015, 33, TPS5605. [Google Scholar] [CrossRef]
- Sallman, D.A.; DeZern, A.E.; Garcia-Manero, G.; Steensma, D.P.; Roboz, G.J.; Sekeres, M.A.; Cluzeau, T.; Sweet, K.L.; McLemore, A.; McGraw, K.L.; et al. Eprenetapopt (APR-246) and Azacitidine in TP53-Mutant Myelodysplastic Syndromes. J. Clin. Oncol. 2021, 39, 1584–1594. [Google Scholar] [CrossRef]
- Cluzeau, T.; Sebert, M.; Rahmé, R.; Cuzzubbo, S.; Lehmann-Che, J.; Madelaine, I.; Peterlin, P.; Bève, B.; Attalah, H.; Chermat, F.; et al. Eprenetapopt Plus Azacitidine in TP53-Mutated Myelodysplastic Syndromes and Acute Myeloid Leukemia: A Phase II Study by the Groupe Francophone des Myélodysplasies (GFM). J. Clin. Oncol. 2021, 39, 1575–1583. [Google Scholar] [CrossRef]
- Oldenborg, P.-A. CD47: A Cell Surface Glycoprotein Which Regulates Multiple Functions of Hematopoietic Cells in Health and Disease. ISRN Hematol. 2013, 2013, 614619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murata, Y.; Kotani, T.; Ohnishi, H.; Matozaki, T. The CD47-SIRPα signalling system: Its physiological roles and therapeutic application. J. Biochem. 2014, 155, 335–344. [Google Scholar] [CrossRef] [Green Version]
- Jaiswal, S.; Jamieson, C.H.M.; Pang, W.W.; Park, C.Y.; Chao, M.P.; Majeti, R.; Traver, D.; van Rooijen, N.; Weissman, I.L. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell 2009, 138, 271–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majeti, R.; Chao, M.P.; Alizadeh, A.A.; Pang, W.W.; Jaiswal, S.; Gibbs, K.D.; van Rooijen, N.; Weissman, I.L. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell 2009, 138, 286–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daver, N.; Senapati, J.; Maiti, A.; Loghavi, S.; Kadia, T.M.; DiNardo, C.D.; Pemmaraju, N.; Jabbour, E.; Montalban-Bravo, G.; Tang, G.; et al. Phase I/II Study of Azacitidine (AZA) with Venetoclax (VEN) and Magrolimab (Magro) in Patients (pts) with Newly Diagnosed (ND) Older/Unfit or High-Risk Acute Myeloid Leukemia (AML) and Relapsed/Refractory (R/R) AML. Blood 2022, 140, 141–144. [Google Scholar] [CrossRef]
- Yamaguchi, R.; Lartigue, L.; Perkins, G. Targeting Mcl-1 and other Bcl-2 family member proteins in cancer therapy. Pharmacol. Ther. 2019, 195, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Thijssen, R.; Diepstraten, S.T.; Moujalled, D.; Chew, E.; Flensburg, C.; Shi, M.X.; Dengler, M.A.; Litalien, V.; MacRaild, S.; Chen, M.; et al. Intact TP-53 function is essential for sustaining durable responses to BH3-mimetic drugs in leukemias. Blood 2021, 137, 2721–2735. [Google Scholar] [CrossRef]
- Ichim, R.; Lopez, J.; Ahmed, S.U.; Muthalagu, N.; Giampazolias, E.; Delgado, E.M.; Haller, M.; Riley, J.S.; Mason, S.M.; Athineos, D.; et al. Limited mitochondrial permeabilization causes DNA damage and genomic instability in the absence of cell death. Mol. Cell 2015, 57, 860–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez, J.; Bessou, M.; Riley, J.S.; Giampazolias, E.; Todt, F.; Rochegüe, T.; Oberst, A.; Green, D.R.; Edlich, F.; Ichim, G.; et al. Mito-priming as a method to engineer Bcl-2 addiction. Nat. Commun. 2016, 7, 10538. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Wu, J.-L.; Liang, Y.; Tang, Y.-G.; Song, H.-X.; Wu, L.-L.; Xing, Y.-F.; Yan, N.; Li, Y.-T.; Wang, Z.-Y.; et al. Arsenic Trioxide Rescues Structural p53 Mutations through a Cryptic Allosteric Site. Cancer Cell 2021, 39, 225–239.e8. [Google Scholar] [CrossRef]
- Weinbach, E.C.; Garbus, J. Mechanism of action of reagents that uncouple oxidative phosphorylation. Nature 1969, 221, 1016–1018. [Google Scholar] [CrossRef] [PubMed]
- Al-Hadiya, B.M.H. Niclosamide: Comprehensive profile. Profiles Drug Subst. Excip. Relat. Methodol. 2005, 32, 67–96. [Google Scholar] [PubMed]
- Maragos, W.F.; Korde, A.S. Mitochondrial uncoupling as a potential therapeutic target in acute central nervous system injury. J. Neurochem. 2004, 91, 257–262. [Google Scholar] [CrossRef]
- Amara, C.E.; Shankland, E.G.; Jubrias, S.A.; Marcinek, D.J.; Kushmerick, M.J.; Conley, K.E. Mild mitochondrial uncoupling impacts cellular aging in human muscles in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 1057–1062. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.; Lu, Z.; Ding, K.; Li, J.; Du, X.; Chen, C.; Sun, X.; Wu, Y.; Zhou, J.; Pan, J. Antineoplastic mechanisms of niclosamide in acute myelogenous leukemia stem cells: Inactivation of the NF-kappaB pathway and generation of reactive oxygen species. Cancer Res. 2010, 70, 2516–2527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helfman, D.M. Niclosamide: An established antihelminthic drug as a potential therapy against S100A4-mediated metastatic colon tumors. J. Natl. Cancer Inst. 2011, 103, 991–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osada, T.; Chen, M.; Yang, X.Y.; Spasojevic, I.; Vandeusen, J.B.; Hsu, D.; Clary, B.M.; Clay, T.M.; Chen, W.; Morse, M.A.; et al. Antihelminth compound niclosamide downregulates Wnt signaling and elicits antitumor responses in tumors with activating APC mutations. Cancer Res. 2011, 71, 4172–4182. [Google Scholar] [CrossRef] [Green Version]
- Fonseca, B.D.; Diering, G.H.; Bidinosti, M.A.; Dalal, K.; Alain, T.; Balgi, A.D.; Forestieri, R.; Nodwell, M.; Rajadurai, C.V.; Gunaratnam, C.; et al. Structure-activity analysis of niclosamide reveals potential role for cytoplasmic pH in control of mammalian target of rapamycin complex 1 (mTORC1) signaling. J. Biol. Chem. 2012, 287, 17530–17545. [Google Scholar] [CrossRef] [Green Version]
- Arend, R.C.; Londoño-Joshi, A.I.; Gangrade, A.; Katre, A.A.; Kurpad, C.; Li, Y.; Samant, R.S.; Li, P.-K.; Landen, C.N.; Yang, E.S.; et al. Niclosamide and its analogs are potent inhibitors of Wnt/β-catenin, mTOR and STAT3 signaling in ovarian cancer. Oncotarget 2016, 7, 86803–86815. [Google Scholar] [CrossRef] [Green Version]
- Jin, B.; Wang, C.; Li, J.; Du, X.; Ding, K.; Pan, J. Anthelmintic Niclosamide Disrupts the Interplay of p65 and FOXM1/β-catenin and Eradicates Leukemia Stem Cells in Chronic Myelogenous Leukemia. Clin. Cancer Res. 2017, 23, 789–803. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.; Coronel, L.; Somalanka, B.; Raju, A.; Aning, O.A.; An, O.; Ho, Y.S.; Chen, S.; Mak, S.Y.; Hor, P.Y.; et al. Mitochondrial uncoupling reveals a novel therapeutic opportunity for p53-defective cancers. Nat. Commun. 2018, 9, 3931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaelin, W.G. The concept of synthetic lethality in the context of anticancer therapy. Nat. Rev. Cancer 2005, 5, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Tahaney, W.M.; Mazumdar, A.; Savage, M.I.; Brown, P.H. Molecularly targeted therapies for p53-mutant cancers. Cell. Mol. Life Sci. 2017, 74, 4171–4187. [Google Scholar] [CrossRef] [PubMed]
- Gurpinar, E.; Vousden, K.H. Hitting cancers’ weak spots: Vulnerabilities imposed by p53 mutation. Trends Cell Biol. 2015, 25, 486–495. [Google Scholar] [CrossRef]
- Kwok, M.; Davies, N.; Agathanggelou, A.; Smith, E.; Oldreive, C.; Petermann, E.; Stewart, G.; Brown, J.; Lau, A.; Pratt, G.; et al. ATR inhibition induces synthetic lethality and overcomes chemoresistance in TP53- or ATM-defective chronic lymphocytic leukemia cells. Blood 2016, 127, 582–595. [Google Scholar] [CrossRef] [Green Version]
- Menendez, D.; Anand, J.R.; Murphy, C.C.; Bell, W.J.; Fu, J.; Slepushkina, N.; Buehler, E.; Martin, S.E.; Lal-Nag, M.; Nitiss, J.L.; et al. Etoposide-induced DNA damage is increased in p53 mutants: Identification of ATR and other genes that influence effects of p53 mutations on Top2-induced cytotoxicity. Oncotarget 2022, 13, 332–346. [Google Scholar] [CrossRef]
- Leijen, S.; van Geel, R.M.J.M.; Pavlick, A.C.; Tibes, R.; Rosen, L.; Razak, A.R.A.; Lam, R.; Demuth, T.; Rose, S.; Lee, M.A.; et al. Phase I Study Evaluating WEE1 Inhibitor AZD1775 As Monotherapy and in Combination With Gemcitabine, Cisplatin, or Carboplatin in Patients With Advanced Solid Tumors. J. Clin. Oncol. 2016, 34, 4371–4380. [Google Scholar] [CrossRef]
- Leijen, S.; van Geel, R.M.J.M.; Sonke, G.S.; de Jong, D.; Rosenberg, E.H.; Marchetti, S.; Pluim, D.; van Werkhoven, E.; Rose, S.; Lee, M.A.; et al. Phase II Study of WEE1 Inhibitor AZD1775 Plus Carboplatin in Patients With TP53-Mutated Ovarian Cancer Refractory or Resistant to First-Line Therapy Within 3 Months. J. Clin. Oncol. 2016, 34, 4354–4361. [Google Scholar] [CrossRef] [Green Version]
- Malekzadeh, P.; Pasetto, A.; Robbins, P.F.; Parkhurst, M.R.; Paria, B.C.; Jia, L.; Gartner, J.J.; Hill, V.; Yu, Z.; Restifo, N.P.; et al. Neoantigen screening identifies broad TP53 mutant immunogenicity in patients with epithelial cancers. J. Clin. Investig. 2019, 129, 1109–1114. [Google Scholar] [CrossRef]
- Lo, W.; Parkhurst, M.; Robbins, P.F.; Tran, E.; Lu, Y.-C.; Jia, L.; Gartner, J.J.; Pasetto, A.; Deniger, D.; Malekzadeh, P.; et al. Immunologic Recognition of a Shared p53 Mutated Neoantigen in a Patient with Metastatic Colorectal Cancer. Cancer Immunol. Res. 2019, 7, 534–543. [Google Scholar] [CrossRef]
- González-Galarza, F.F.; Takeshita, L.Y.C.; Santos, E.J.M.; Kempson, F.; Maia, M.H.T.; da Silva, A.L.S.; Teles e Silva, A.L.; Ghattaoraya, G.S.; Alfirevic, A.; Jones, A.R.; et al. Allele frequency net 2015 update: New features for HLA epitopes, KIR and disease and HLA adverse drug reaction associations. Nucleic Acids Res. 2015, 43, D784–D788. [Google Scholar] [CrossRef] [PubMed]
- Hsiue, E.H.-C.; Wright, K.M.; Douglass, J.; Hwang, M.S.; Mog, B.J.; Pearlman, A.H.; Paul, S.; DiNapoli, S.R.; Konig, M.F.; Wang, Q.; et al. Targeting a neoantigen derived from a common TP53 mutation. Science 2021, 371, eabc8697. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Shapiro, G.I.; Gao, X.; Mahipal, A.; Starr, J.; Furqan, M.; Singh, P.; Ahrorov, A.; Gandhi, L.; Ghosh, A.; et al. Phase Ib study of eprenetapopt (APR-246) in combination with pembrolizumab in patients with advanced or metastatic solid tumors. ESMO Open 2022, 7, 100573. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Tamari, R.; DeZern, A.E.; Byrne, M.T.; Gooptu, M.; Chen, Y.-B.; Deeg, H.J.; Sallman, D.; Gallacher, P.; Wennborg, A.; et al. Eprenetapopt Plus Azacitidine After Allogeneic Hematopoietic Stem-Cell Transplantation for TP53-Mutant Acute Myeloid Leukemia and Myelodysplastic Syndromes. J. Clin. Oncol. 2022, 40, 3985–3993. [Google Scholar] [CrossRef] [PubMed]
- Ray-Coquard, I.; Braicu, I.; Berger, R.; Mahner, S.; Sehouli, J.; Pujade-Lauraine, E.; Cassier, P.A.; Moll, U.M.; Ulmer, H.; Leunen, K.; et al. Part I of GANNET53: A European Multicenter Phase I/II Trial of the Hsp90 Inhibitor Ganetespib Combined With Weekly Paclitaxel in Women With High-Grade, Platinum-Resistant Epithelial Ovarian Cancer-A Study of the GANNET53 Consortium. Front. Oncol. 2019, 9, 832. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Janku, F.; Piha-Paul, S.; Hess, K.; Broaddus, R.; Liu, L.; Shi, N.; Overman, M.; Kopetz, S.; Subbiah, V.; et al. Phase I studies of vorinostat with ixazomib or pazopanib imply a role of antiangiogenesis-based therapy for TP53 mutant malignancies. Sci. Rep. 2020, 10, 3080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.F.; Xiong, N.; Campos, S.M.; Wright, A.A.; Krasner, C.; Schumer, S.; Horowitz, N.; Veneris, J.; Tayob, N.; Morrissey, S.; et al. Phase II Study of the WEE1 Inhibitor Adavosertib in Recurrent Uterine Serous Carcinoma. J. Clin. Oncol. 2021, 39, 1531–1539. [Google Scholar] [CrossRef] [PubMed]
- Bondar, T.; Medzhitov, R. p53-mediated hematopoietic stem and progenitor cell competition. Cell Stem Cell 2010, 6, 309–322. [Google Scholar] [CrossRef] [Green Version]
- Marusyk, A.; Porter, C.C.; Zaberezhnyy, V.; DeGregori, J. Irradiation selects for p53-deficient hematopoietic progenitors. PLoS Biol. 2010, 8, e1000324. [Google Scholar] [CrossRef] [Green Version]
- Durkin, S.G.; Glover, T.W. Chromosome fragile sites. Annu. Rev. Genet. 2007, 41, 169–192. [Google Scholar] [CrossRef]
- Barlow, J.H.; Faryabi, R.B.; Callén, E.; Wong, N.; Malhowski, A.; Chen, H.T.; Gutierrez-Cruz, G.; Sun, H.-W.; McKinnon, P.; Wright, G.; et al. Identification of early replicating fragile sites that contribute to genome instability. Cell 2013, 152, 620–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjerregaard, V.A.; Özer, Ö.; Hickson, I.D.; Liu, Y. The Detection and Analysis of Chromosome Fragile Sites. Methods Mol. Biol. 2018, 1672, 471–482. [Google Scholar] [PubMed]
- Halazonetis, T.D.; Gorgoulis, V.G.; Bartek, J. An oncogene-induced DNA damage model for cancer development. Science 2008, 319, 1352–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, H.; Hollstein, M.; Xu, Y. p53 gain-of-function cancer mutants induce genetic instability by inactivating ATM. Nat. Cell Biol. 2007, 9, 573–580. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahmé, R.; Braun, T.; Manfredi, J.J.; Fenaux, P. TP53 Alterations in Myelodysplastic Syndromes and Acute Myeloid Leukemia. Biomedicines 2023, 11, 1152. https://doi.org/10.3390/biomedicines11041152
Rahmé R, Braun T, Manfredi JJ, Fenaux P. TP53 Alterations in Myelodysplastic Syndromes and Acute Myeloid Leukemia. Biomedicines. 2023; 11(4):1152. https://doi.org/10.3390/biomedicines11041152
Chicago/Turabian StyleRahmé, Ramy, Thorsten Braun, James J. Manfredi, and Pierre Fenaux. 2023. "TP53 Alterations in Myelodysplastic Syndromes and Acute Myeloid Leukemia" Biomedicines 11, no. 4: 1152. https://doi.org/10.3390/biomedicines11041152