
Physiological Associations between Vitamin B Deficiency and Diabetic Kidney Disease

Abstract
:1. Introduction
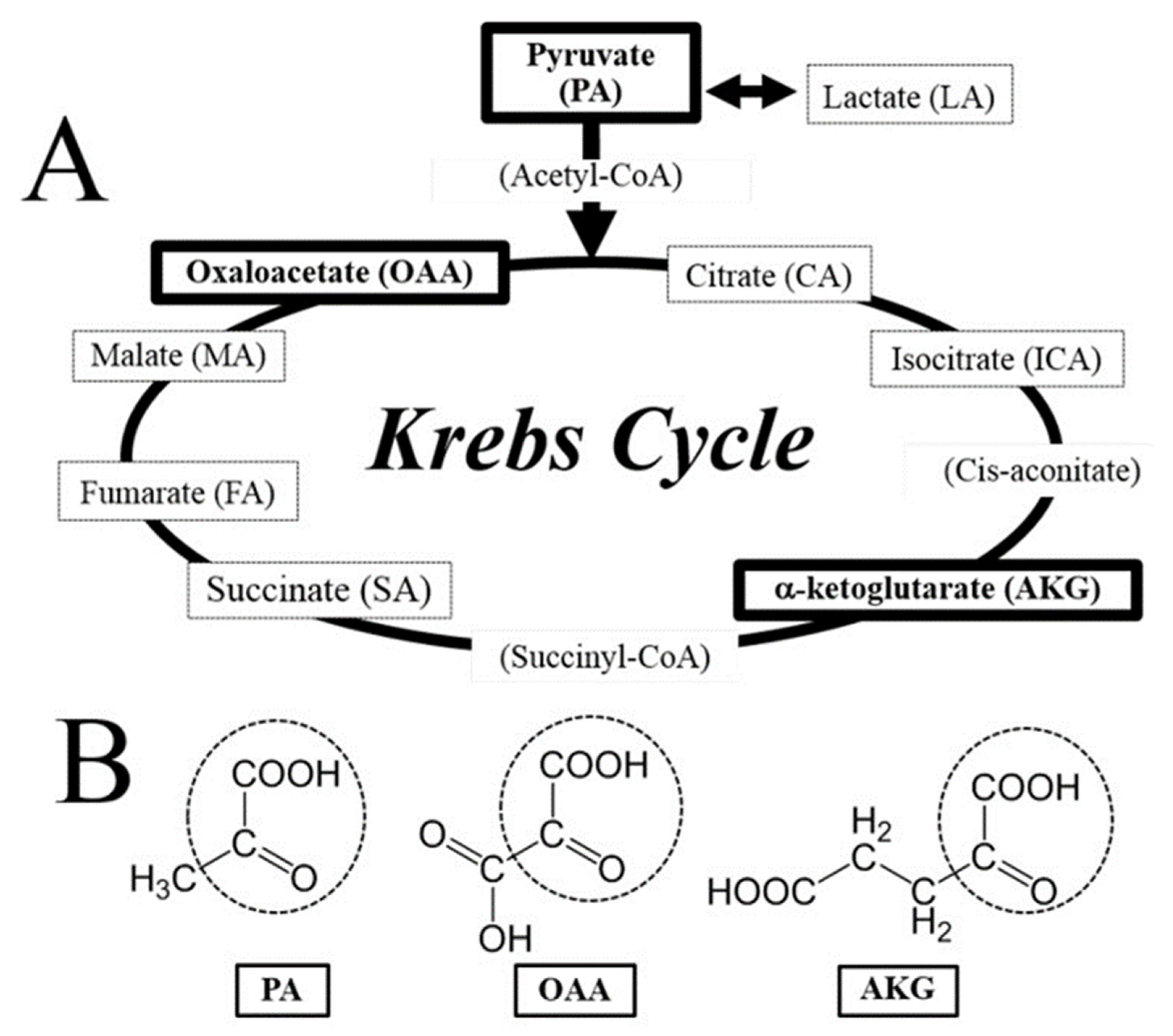
2. Vitamin B1 (Thiamine)
3. Vitamin B2 (Riboflavin)
4. Vitamin B3 (Niacin)
5. Vitamin B5 (Pantothenic Acid)
6. Vitamin B6 (Pyridoxine)
7. Vitamin B8 (Biotin)
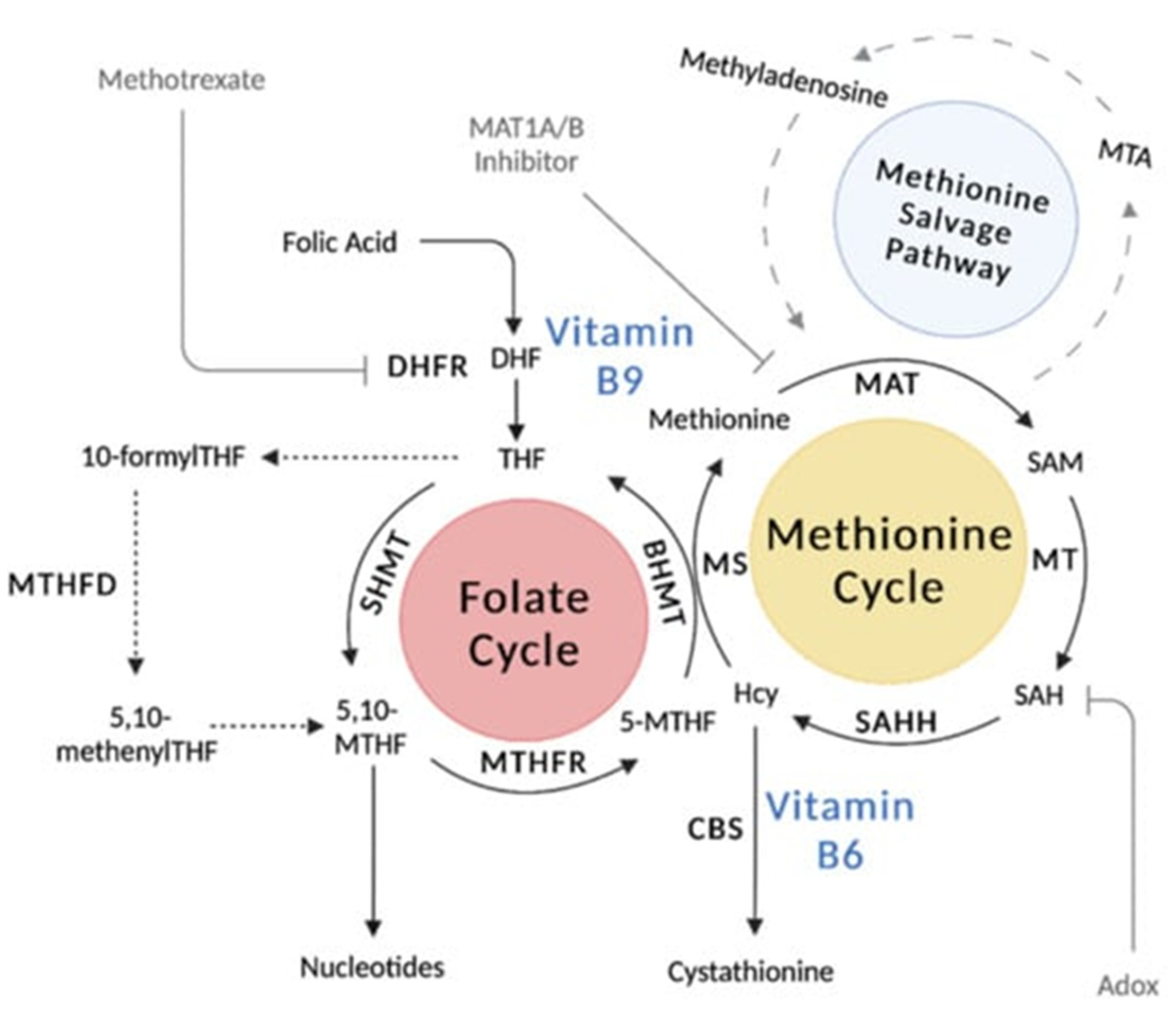
8. Vitamin B9 (Folate)
9. Vitamin B12 (Cobalamin)
10. Summary and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Levin, A.; Stevens, P.E.; Bilous, R.W.; Coresh, J.; De Francisco, A.L.; De Jong, P.E.; Griffith, K.E.; Hemmelgarn, B.R.; Iseki, K.; Lamb, E.J.; et al. Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO 2012 clinical practice guideline for the evaluation and management of chronic kidney disease. Kidney Int. Suppl. 2013, 3, 1–50. [Google Scholar]
- Murphy, D.; McCulloch, C.E.; Lin, F.; Banerjee, T.; Bragg-Gresham, J.L.; Eberhardt, M.S.; Morgenstern, H.; Pavkov, M.E.; Saran, R.; Powe, N.R.; et al. Trends in prevalence of chronic kidney disease in the United States. Ann. Intern. Med. 2016, 165, 473–481. [Google Scholar] [CrossRef] [Green Version]
- Wallace, A.S.; Chang, A.R.; Shin, J.I.; Reider, J.; Echouffo-Tcheugui, J.B.; Grams, M.E.; Selvin, E. Obesity and chronic kidney disease in US adults with type 1 and type 2 diabetes mellitus. J. Endocrinol. Metab. 2022, 107, 1247–1256. [Google Scholar] [CrossRef] [PubMed]
- Altemtam, N.; Russell, J.; El Nahas, M. A study of the natural history of diabetic kidney disease (DKD). Nephrol. Dial. Transplant. 2012, 27, 1847–1854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liyanage, T.; Toyama, T.; Hockham, C.; Ninomiya, T.; Perkovic, V.; Woodward, M.; Fukagawa, M.; Matsushita, K.; Praditpornsilpa, K.; Hooi, L.S.; et al. Prevalence of chronic kidney disease in Asia: A systematic review and analysis. BMJ Glob. Health 2022, 7, e007525. [Google Scholar] [CrossRef]
- Kovesdy, C.P. Epidemiology of chronic kidney disease: An update 2022. Kidney Int. Suppl. 2022, 12, 7–11. [Google Scholar] [CrossRef]
- US Renal Data System. USRDS 2021 Annual Data Report: Chronic Kidney Disease; National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases: Bethesda, MD, USA, 2021; Volume 2021. [Google Scholar]
- US Renal Data System. USRDS 2022 Annual Data Report: Chronic Kidney Disease; National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases: Bethesda, MD, USA, 2022; Volume 2022. [Google Scholar]
- Persson, F.; Rossing, P. Diagnosis of diabetic kidney disease: State of the art and future perspective. Kidney Int. Suppl. 2018, 8, 2–7. [Google Scholar] [CrossRef] [Green Version]
- Gheith, O.; Farouk, N.; Nampoory, N.; Halim, M.A.; Al-Otaibi, T. Diabetic kidney disease: World wide difference of prevalence and risk factors. J. Nephrolpharmacol. 2016, 5, 49–56. [Google Scholar] [CrossRef]
- Mehdi, U.; Toto, R.D. Anemia, diabetes, and chronic kidney disease. Diabetes Care 2009, 32, 1320–1326. [Google Scholar] [CrossRef] [Green Version]
- Sasso, F.C.; De Nicola, L.; Carbonara, O.; Nasti, R.; Minutolo, R.; Salvatore, T.; Conte, G.; Torella, R.; NID-2 (Nephropathy in Diabetes-Type 2) Study Group. Cardiovascular risk factors and disease management in type 2 diabetic patients with diabetic nephropathy. Diabetes Care 2006, 29, 498–503. [Google Scholar] [CrossRef] [Green Version]
- Tang, S.C.; Yiu, W.H. Innate immunity in diabetic kidney disease. Nat. Rev. Nephrol. 2020, 16, 206–222. [Google Scholar] [CrossRef] [PubMed]
- Etgen, T.; Chonchol, M.; Förstl, H.; Sander, D. Chronic kidney disease and cognitive impairment: A systematic review and meta-analysis. Am. J. Nephrol. 2012, 35, 474–482. [Google Scholar] [CrossRef] [PubMed]
- Formiga, F.; Moreno-González, R.; Corsonello, A.; Carlsson, A.; Ärnlöv, J.; Mattace-Raso, F.; Kostka, T.; Weingart, C.; Roller-Wirnsberger, R.; Tap, L.; et al. Diabetes, sarcopenia and chronic kidney disease; the Screening for CKD among Older People across Europe (SCOPE) study. BMC Geriatr. 2022, 22, 254. [Google Scholar] [CrossRef] [PubMed]
- Bellows, L.; Moore, R.; Anderson, J.; Young, L. Water-Soluble Vitamins: B-Complex and Vitamin C; Food and Nutrition Series Health, No. 9.312; Colorado State University: Fort Collins, CO, USA, 2012. [Google Scholar]
- Lyon, P.; Strippoli, V.; Fang, B.; Cimmino, L. B vitamins and one-carbon metabolism: Implications in human health and disease. Nutrients 2020, 12, 2867. [Google Scholar] [CrossRef]
- Chazot, C.; Steiber, A.L.; Kopple, J.D. Vitamin metabolism and requirements in chronic kidney disease and kidney failure. In Nutritional Management of Renal Disease; Academic Press: New York, NY, USA, 2022; pp. 413–465. [Google Scholar]
- Ford, A.H.; Almeida, O.P. Effect of vitamin B supplementation on cognitive function in the elderly: A systematic review and meta-analysis. Drugs Aging 2019, 36, 419–434. [Google Scholar] [CrossRef]
- Almeida, M.R.; Venâncio, V.P.; Aissa, A.F.; Darin, J.D.; Bianchi, M.L.; Antunes, L.M. Effects of maternal vitamin B6 deficiency and over-supplementation on DNA damage and oxidative stress in rat dams and their offspring. Food Chem. Toxicol. 2015, 80, 201–205. [Google Scholar] [CrossRef]
- Fattal-Valevski, A. Thiamine (vitamin B1). J. Evid. Based Complement. Altern. Med. 2011, 16, 12–20. [Google Scholar] [CrossRef]
- Vuilleumier, J.P.; Keller, H.E.; Gysel, D.; Hunziker, F. Clinical chemical methods for the routine assessment of the vitamin status in human populations. Part I: The fat-soluble vitamins A and E.; and beta-carotene. International Journal for Vitamin and Nutrition research. Internationale Zeitschrift fur Vitamin-und Ernahrungsforschung. Int. J. Vitam. Nutr. Res. 1983, 53, 265–272. [Google Scholar]
- Luft, F.C. Lactic acidosis update for critical care clinicians. J. Am. Soc. Nephrol. 2001, 12, S15–S19. [Google Scholar] [CrossRef]
- Frank, R.A.; Kay, C.W.; Hirst, J.; Luisi, B.F. Off-pathway, oxygen-dependent thiamine radical in the Krebs cycle. J. Am. Chem. Soc. 2008, 130, 1662–1668. [Google Scholar] [CrossRef]
- Sawa, K.; Uematsu, T.; Korenaga, Y.; Hirasawa, R.; Kikuchi, M.; Murata, K.; Zhang, J.; Gai, X.; Sakamoto, K.; Koyama, T.; et al. Krebs cycle intermediates protective against oxidative stress by modulating the level of reactive oxygen species in neuronal HT22 cells. Antioxidants 2017, 6, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sterzel, R.B.; Semar, M.; Lonergan, E.T.; Lange, K. Effect of hemodialysis on the inhibition of nervous tissue transketolase and on uremic neuropathy. ASAIO J. 1971, 17, 77–80. [Google Scholar]
- Marcé-Grau, A.; Martí-Sánchez, L.; Baide-Mairena, H.; Ortigoza-Escobar, J.D.; Pérez-Dueñas, B. Genetic defects of thiamine transport and metabolism: A review of clinical phenotypes, genetics, and functional studies. J. Inherit. Metab. Dis. 2019, 42, 581–597. [Google Scholar] [CrossRef] [PubMed]
- Thornalley, P.J. The potential role of thiamine (vitamin B1) in diabetic complications. Curr. Diabetes Rev. 2005, 1, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Thornalley, P.J.; Babaei-Jadidi, R.; Al Ali, H.; Rabbani, N.; Antonysunil, A.; Larkin, J.; Ahmed, A.; Rayman, G.; Bodmer, C.W. High prevalence of low plasma thiamine concentration in diabetes linked to a marker of vascular disease. Diabetologia 2007, 50, 2164–2170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Page, G.L.; Laight, D.; Cummings, M.H. Thiamine deficiency in diabetes mellitus and the impact of thiamine replacement on glucose metabolism and vascular disease. Int. J. Clin. Pract. 2011, 65, 684–690. [Google Scholar] [CrossRef] [Green Version]
- Rabbani, N.; Thornalley, P.J. Emerging role of thiamine therapy for prevention and treatment of early-stage diabetic nephropathy. Diabetes Obes. Metab. 2011, 13, 577–583. [Google Scholar] [CrossRef]
- Antonysunil, A.; Babaei-Jadidi, R.; Rabbani, N.; Ahmed, A.; Rayman, G.; Bodmer, C.W.; Thornalley, P.J. Increased thiamine transporter contents of red blood cells and peripheral blood mononuclear leukocytes in type 1 and type 2 diabetic patients. Diabetes 2007, 56, A609. [Google Scholar]
- Mazzeo, A.; Barutta, F.; Bellucci, L.; Trento, M.; Gruden, G.; Porta, M.; Beltramo, E. Reduced thiamine availability and hyperglycemia impair thiamine transport in renal glomerular cells through modulation of thiamine transporter 2. Biomedicines 2021, 9, 385. [Google Scholar] [CrossRef]
- Larkin, J.R.; Zhang, F.; Godfrey, L.; Molostvov, G.; Zehnder, D.; Rabbani, N.; Thornalley, P.J. Glucose-induced down regulation of thiamine transporters in the kidney proximal tubular epithelium produces thiamine insufficiency in diabetes. PLoS ONE 2012, 7, e53175. [Google Scholar] [CrossRef]
- Bukhari, F.J.; Moradi, H.; Gollapudi, P.; Ju Kim, H.; Vaziri, N.D.; Said, H.M. Effect of chronic kidney disease on the expression of thiamin and folic acid transporters. Nephrol. Dial. Transplant. 2011, 26, 2137–2144. [Google Scholar] [CrossRef] [Green Version]
- Babaei-Jadidi, R.; Karachalias, N.; Kupich, C.; Ahmed, N.; Thornalley, P.J. High-dose thiamine therapy counters dyslipidaemia in streptozotocin-induced diabetic rats. Diabetologia 2004, 47, 2235–2246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babaei-Jadidi, R.; Karachalias, N.; Ahmed, N.; Battah, S.; Thornalley, P.J. Prevention of incipient diabetic nephropathy by high-dose thiamine and benfotiamine. Diabetes 2003, 52, 2110–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patrini, C.; Griziotti, A.; Ricciardi, L. Obese individuals as thiamine storers. Int. J. Obes. 2004, 28, 920–924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tasevska, N.; Runswick, S.A.; McTaggart, A.; Bingham, S.A. Twenty-four-hour urinary thiamine as a biomarker for the assessment of thiamine intake. Eur. J. Clin. Nutr. 2008, 62, 1139–1147. [Google Scholar] [CrossRef] [Green Version]
- Adaikalakoteswari, A.; Perkins, B.; Krolewski, A.S.; Rabbani, N.; Thornalley, P.J. Thiamine status and risk of early renal function decline in type 1 diabetic patients. Diabetologia 2008, 51, S98. [Google Scholar]
- Rabbani, N.; Alam, S.S.; Riaz, S.; Larkin, J.R.; Akhtar, M.W.; Shafi, T.; Thornalley, P.J. High-dose thiamine therapy for patients with type 2 diabetes and microalbuminuria: A randomised, double-blind placebo-controlled pilot study. Diabetologia 2009, 52, 208–212. [Google Scholar] [CrossRef] [Green Version]
- Satchell, S.C.; Tooke, J.E. What is the mechanism of microalbuminuria in diabetes: A role for the glomerular endothelium? Diabetologia 2008, 51, 714–725. [Google Scholar] [CrossRef] [Green Version]
- Hammes, H.P.; Du, X.; Edelstein, D.; Taguchi, T.; Matsumura, T.; Ju, Q.; Lin, J.; Bierhaus, A.; Nawroth, P.; Hannak, D.; et al. Benfotiamine blocks three major pathways of hyperglycemic damage and prevents experimental diabetic retinopathy. Nat. Med. 2003, 9, 294–299. [Google Scholar] [CrossRef]
- Karachalias, N.; Babaei-Jadidi, R.; Rabbani, N.; Thornalley, P.J. Increased protein damage in renal glomeruli, retina, nerve, plasma and urine and its prevention by thiamine and benfotiamine therapy in a rat model of diabetes. Diabetologia 2010, 53, 1506–1516. [Google Scholar] [CrossRef] [Green Version]
- Xue, M.; Qian, Q.; Adaikalakoteswari, A.; Rabbani, N.; Babaei-Jadidi, R.; Thornalley, P.J. Activation of NF-E2–related factor-2 reverses biochemical dysfunction of endothelial cells induced by hyperglycemia linked to vascular disease. Diabetes 2008, 57, 2809–2817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Pinto, J.T.; Zempleni, J. Riboflavin. Adv. Nutr. 2016, 7, 973–975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCormick, D.B. Riboflavin. In Present Knowledge in Nutrition; Academic Press: New York, NY, USA, 2012; pp. 280–292. [Google Scholar]
- Joosten, V.; van Berkel, W.J. Flavoenzymes. Curr. Opin. Chem. Biol. 2007, 11, 195–202. [Google Scholar] [CrossRef]
- Van Berkel, W.J. Flavoenzymes. Molecules 2018, 23, 1957. [Google Scholar] [CrossRef] [Green Version]
- Jankowska, M.; Rutkowski, B.; Dębska-Ślizień, A. Vitamins and microelement bioavailability in different stages of chronic kidney disease. Nutrients 2017, 9, 282. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.E.; Kim, H.E.; Park, J.I.; Cho, H.; Kwak, M.J.; Kim, B.Y.; Yang, S.H.; Lee, J.P.; Kim, D.K.; Joo, K.W.; et al. The association between gut microbiota and uremia of chronic kidney disease. Microorganisms 2020, 8, 907. [Google Scholar] [CrossRef]
- Alam, M.M.; Iqbal, S.; Naseem, I. Ameliorative effect of riboflavin on hyperglycemia, oxidative stress and DNA damage in type-2 diabetic mice: Mechanistic and therapeutic strategies. Arch. Biochem. Biophys. 2015, 584, 10–19. [Google Scholar] [CrossRef]
- Aguilar, A.; Alvarez-Vijande, R.; Capdevila, S.; Alcoberro, J.; Alcaraz, A. Antioxidant patterns (superoxide dismutase, glutathione reductase, and glutathione peroxidase) in kidneys from non–heart-beating-donors: Experimental study. Transplant. Proc. 2007, 39, 249–252. [Google Scholar] [CrossRef]
- Can, B.; Kulaksiz Erkmen, G.; Dalmizrak, O.; Ogus, I.H.; Ozer, N. Purification and characterisation of rat kidney glutathione reductase. Protein J. 2010, 29, 250–256. [Google Scholar] [CrossRef]
- Kirkland, J.B.; Meyer-Ficca, M.L. Niacin. Adv. Food Nutr. Res. 2018, 83, 83–149. [Google Scholar] [PubMed]
- Kamanna, V.S.; Kashyap, M.L. Mechanism of action of niacin. Am. J. Cardiol. 2008, 101, S20–S26. [Google Scholar] [CrossRef] [PubMed]
- Comai, S.; Bertazzo, A.; Brughera, M.; Crotti, S. Tryptophan in health and disease. Adv. Clin. Chem. 2020, 95, 165–218. [Google Scholar] [PubMed]
- Yaman, M.; Çatak, J.; Uğur, H.; Gürbüz, M.; Belli, İ.; Tanyıldız, S.N.; Yıldırım, H.; Cengiz, S.; Yavuz, B.B.; Kişmiroğlu, C.; et al. The bioaccessibility of water-soluble vitamins: A review. Trends Food Sci. Technol. 2021, 109, 552–563. [Google Scholar] [CrossRef]
- Pollak, N.; Dölle, C.; Ziegler, M. The power to reduce: Pyridine nucleotides–small molecules with a multitude of functions. Biochem. J. 2007, 402, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Oka, S.I.; Hsu, C.P.; Sadoshima, J. Regulation of cell survival and death by pyridine nucleotides. Circ. Res. 2012, 111, 611–627. [Google Scholar] [CrossRef] [Green Version]
- Depeint, F.; Bruce, W.R.; Shangari, N.; Mehta, R.; O’Brien, P.J. Mitochondrial function and toxicity: Role of the B vitamin family on mitochondrial energy metabolism. Chem. Biol. Interact. 2006, 163, 94–112. [Google Scholar] [CrossRef]
- Wu, Y.; Li, S.; Wang, W.; Zhang, D. Associations of dietary vitamin B1, vitamin B2, niacin, vitamin B6, vitamin B12 and folate equivalent intakes with metabolic syndrome. Int. J. Food Sci. Nutr. 2020, 71, 738–749. [Google Scholar] [CrossRef]
- Goel, H.; Dunbar, R.L. Niacin alternatives for dyslipidemia: Fool’s gold or gold mine? Part II: Novel niacinS mimetics. Curr. Atheroscler. Rep. 2016, 18, 17. [Google Scholar] [CrossRef] [Green Version]
- Xiang, D.; Zhang, Q.; Wang, Y.T. Effectiveness of niacin supplementation for patients with type 2 diabetes: A meta-analysis of randomized controlled trials. Medicine 2020, 99, e21235. [Google Scholar] [CrossRef]
- Ke, P.; Jiang, H.; Dowling, R.; Zhong, L.; Ke, L.; Xu, M.; Wang, C.; Tian, Q.; He, Y.; Lu, K.; et al. Relationship between dietary niacin intake and diabetes mellitus in the National Health and Nutrition Examination Survey (NHANES) 2003–2018. Eat. Weight. Disord. EWD 2022, 27, 2425–2434. [Google Scholar] [CrossRef] [PubMed]
- Olson, E.J.; Mahar, K.M.; Haws, T.F.; Fossler, M.J.; Gao, F.; de Gouville, A.C.; Sprecher, D.L.; Lepore, J.J. A Randomized, Placebo-Controlled Trial to Assess the Effects of 8 Weeks of Administration of GSK256073, a Selective GPR109A Agonist, on High-Density Lipoprotein Cholesterol in Subjects With Dyslipidemia. Clin. Pharmacol. Drug Dev. 2019, 8, 871–883. [Google Scholar] [CrossRef] [PubMed]
- Berns, J.S. Niacin and related compounds for treating hyperphosphatemia in dialysis patients. Semin Dial. 2008, 21, 203–205. [Google Scholar] [CrossRef] [PubMed]
- Lenglet, A.; Liabeuf, S.; El Esper, N.; Brisset, S.; Mansour, J.; Lemaire-Hurtel, A.S.; Mary, A.; Brazier, M.; Kamel, S.; Mentaverri, R.; et al. Efficacy and safety of nicotinamide in haemodialysis patients: The NICOREN study. Nephrol. Dial. Transplant. 2017, 32, 870–879. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Ma, T.; Zhang, P. Efficacy and safety of nicotinamide on phosphorus metabolism in hemodialysis patients: A systematic review and meta-analysis. Medicine 2018, 97, e12731. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.W.; Rucker, R.B. Pantothenic acid. In Present Knowledge in Nutrition; Academic Press: New York, NY, USA, 2020; pp. 273–287. [Google Scholar]
- Fry, P.C.; Fox, H.M.; Tao, H.G. Metabolic response to a pantothenic acid deficient diet in Humans. J. Nutr. Sci. Vitaminol. 1976, 22, 339–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velazquez-Arellano, A.; Hernandez-Vazquez, A.D. Vitamins as cofactors for energy homeostasis and their genomic control, with special reference to biotin, thiamine, and pantothenic acid. In Principles of Nutrigenetics and Nutrigenomics; Academic Press: New York, NY, USA, 2020; pp. 271–277. [Google Scholar]
- Tutun, B.; Elbe, H.; Vardi, N.; Parlakpinar, H.A.; Polat, A.; Gunaltili, M.; Guclu, M.M.; Yasar, E.N. Dexpanthenol reduces diabetic nephropathy and renal oxidative stress in rats. Biotech. Histochem. 2019, 94, 84–91. [Google Scholar] [CrossRef]
- Poyan Mehr, A.; Tran, M.T.; Ralto, K.M.; Leaf, D.E.; Washco, V.; Messmer, J.; Lerner, A.; Kher, A.; Kim, S.H.; Khoury, C.C.; et al. De novo NAD+ biosynthetic impairment in acute kidney injury in humans. Nat. Med. 2018, 24, 1351–1359. [Google Scholar] [CrossRef]
- Mackenzie, J.C.; Ford, J.E.; Waters, A.H.; Harding, N.; Cattell, W.R.; Anderson, B.B. Erythropoiesis in patients undergoing regular dialysis treatment (RDT) without transfusion. Proc. Eur. Dial. Transplant. Assoc. 1968, 5, 172–178. [Google Scholar]
- Machado, A.D.; Gómez, L.M.; Marchioni, D.M.; Dos Anjos, F.S.; Molina, M.D.; Lotufo, P.A.; Benseñor, I.J.; Titan, S.M. Association between dietary intake and coronary artery calcification in non-dialysis chronic kidney disease: The PROGREDIR study. Nutrients 2018, 10, 372. [Google Scholar] [CrossRef] [Green Version]
- Spinneker, A.; Sola, R.; Lemmen, V.; Castillo, M.J.; Pietrzik, K.; Gonzalez-Gross, M. Vitamin B6 status, deficiency and its consequences-an overview. Nutr. Hosp. 2007, 22, 7–24. [Google Scholar] [PubMed]
- Ueland, P.M.; Ulvik, A.; Rios-Avila, L.; Midttun, Ø.; Gregory, J.F. Direct and functional biomarkers of vitamin B6 status. Ann. Rev. Nutr. 2015, 35, 33–70. [Google Scholar] [CrossRef] [PubMed]
- Stach, K.; Stach, W.; Augoff, K. Vitamin B6 in health and disease. Nutrients 2021, 13, 3229. [Google Scholar] [CrossRef] [PubMed]
- Roth-Maier, D.A.; Kettler, S.I.; Kirchgessner, M. Availability of vitamin B6 from different food sources. Int. J. Food Sci. Nutr. 2002, 53, 171–179. [Google Scholar] [CrossRef]
- Echevarría, G.R.; Santos, J.G.; Basagoitia, A.; Blanco, F.G. Kinetic and thermodynamic study of the reaction of pyridoxal 5′-phosphate with l-tryptophan. J. Phys. Org. Chem. 2005, 18, 546–551. [Google Scholar] [CrossRef]
- McCormick, D.B. Bioorganic mechanisms important to coenzyme functions. In Handbook of Vitamins, 4th ed.; Academic Press: New York, NY, USA, 2007; pp. 175–192. [Google Scholar]
- Kiran, S.G.; Dorisetty, R.K.; Umrani, M.R.; Boindala, S.; Bhonde, R.R.; Chalsani, M.; Singh, H.; Venkatesan, V. Pyridoxal 5′ phosphate protects islets against streptozotocin-induced beta-cell dysfunction–in vitro and in vivo. Exp. Biol. Med. 2011, 236, 456–465. [Google Scholar] [CrossRef]
- Endo, N.; Nishiyama, K.; Otsuka, A.; Kanouchi, H.; Taga, M.; Oka, T. Antioxidant activity of vitamin B6 delays homocysteine-induced atherosclerosis in rats. Br. J. Nutr. 2006, 95, 1088–1093. [Google Scholar] [CrossRef] [Green Version]
- Salvà, A.; Donoso, J.; Frau, J.; Muñoz, F. DFT studies on Schiff base formation of vitamin B6 analogues. J. Phys. Chem. 2003, 107, 9409–9414. [Google Scholar] [CrossRef]
- Di Salvo, M.L.; Contestabile, R.; Safo, M.K. Vitamin B6 salvage enzymes: Mechanism, structure and regulation. Biochim. Biophys. Acta 2011, 1814, 1597–1608. [Google Scholar] [CrossRef]
- Calderón-Ospina, C.A.; Nava-Mesa, M.O. B Vitamins in the nervous system: Current knowledge of the biochemical modes of action and synergies of thiamine, pyridoxine, and cobalamin. CNS Neurosci. Ther. 2020, 26, 5–13. [Google Scholar] [CrossRef] [Green Version]
- Tully, D.B.; Allgood, V.E.; Cidlowski, J.A. Modulation of steroid receptor-mediated gene expression by vitamin B6. FASEB J. 1994, 8, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Talwar, D.; Catchpole, A.; Wadsworth, J.M.; Toole, B.J.; McMillan, D.C. The relationship between plasma albumin, alkaline phosphatase and pyridoxal phosphate concentrations in plasma and red cells: Implications for assessing vitamin B6 status. Clin. Nutr. 2020, 39, 2824–2831. [Google Scholar] [CrossRef] [PubMed]
- Vasilaki, A.T.; McMillan, D.C.; Kinsella, J.; Duncan, A.; O’Reilly, D.S.; Talwar, D. Relation between pyridoxal and pyridoxal phosphate concentrations in plasma, red cells, and white cells in patients with critical illness. Am. J. Clin. Nutr. 2008, 88, 140–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theofylaktopoulou, D.; Ulvik, A.; Midttun, Ø.; Ueland, P.M.; Vollset, S.E.; Nygård, O.; Hustad, S.; Tell, G.S.; Eussen, S.J. Vitamins B2 and B6 as determinants of kynurenines and related markers of interferon-γ-mediated immune activation in the community-based Hordaland Health Study. Br. J. Nutr. 2014, 112, 1065–1072. [Google Scholar] [CrossRef] [Green Version]
- Leon, A.S.; Spiegel, H.E.; Thomas, G.; Abrams, W.B. Pyridoxine antagonism of levodopa in parkinsonism. JAMA 1971, 218, 1924–1927. [Google Scholar] [CrossRef]
- Nix, W.A.; Zirwes, R.; Bangert, V.; Kaiser, R.P.; Schilling, M.; Hostalek, U.; Obeid, R. Vitamin B status in patients with type 2 diabetes mellitus with and without incipient nephropathy. Diab. Res. Clin. Pract. 2015, 107, 157–165. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Lai, C.Q.; Mattei, J.; Ordovas, J.M.; Tucker, K.L. Association of vitamin B-6 status with inflammation, oxidative stress, and chronic inflammatory conditions: The Boston Puerto Rican Health Study. Am. J. Clin. Nutr. 2010, 91, 337–342. [Google Scholar] [CrossRef] [Green Version]
- Friso, S.; Jacques, P.F.; Wilson, P.W.; Rosenberg, I.H.; Selhub, J. Low circulating vitamin B6 is associated with elevation of the inflammation marker C-reactive protein independently of plasma homocysteine levels. Circulation 2001, 103, 2788–2791. [Google Scholar] [CrossRef] [Green Version]
- Mascolo, E.; Vernì, F. Vitamin B6 and diabetes: Relationship and molecular mechanisms. Int. J. Mol. Sci. 2020, 21, 3669. [Google Scholar] [CrossRef]
- Oxenkrug, G. Insulin resistance and dysregulation of tryptophan–kynurenine and kynurenine–nicotinamide adenine dinucleotide metabolic pathways. Mol. Neurobiol. 2013, 48, 294–301. [Google Scholar] [CrossRef] [Green Version]
- Fields, A.M.; Welle, K.; Ho, E.S.; Mesaros, C.; Susiarjo, M. Vitamin B6 deficiency disrupts serotonin signaling in pancreatic islets and induces gestational diabetes in mice. Commun. Biol. 2021, 4, 421. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.C.; Cheng, C.H.; Hsu, C.L.; Lee, W.J.; Huang, S.C.; Huang, Y.C. Role of vitamin B6 status on antioxidant defenses, glutathione, and related enzyme activities in mice with homocysteine-induced oxidative stress. Food Nutr. Res. 2015, 59, 25702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pusceddu, I.; Herrmann, W.; Kleber, M.E.; Scharnagl, H.; Hoffmann, M.M.; Winklhofer-Roob, B.M.; März, W.; Herrmann, M. Subclinical inflammation, telomere shortening, homocysteine, vitamin B6, and mortality: The Ludwigshafen Risk and Cardiovascular Health Study. Eur. J. Nutr. 2020, 59, 1399–1411. [Google Scholar] [CrossRef] [Green Version]
- Wotherspoon, F.; Laight, D.W.; Shaw, K.M.; Cummings, M.H. Homocysteine, endothelial dysfunction and oxidative stress in type 1 diabetes mellitus. Br. J. Diab. Vasc. Dis. 2003, 3, 334–340. [Google Scholar] [CrossRef]
- Chen, C.H.; Yang, W.C.; Hsiao, Y.H.; Huang, S.C.; Huang, Y.C. High homocysteine, low vitamin B-6, and increased oxidative stress are independently associated with the risk of chronic kidney disease. Nutrition 2016, 32, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Mikkelsen, K.; Apostolopoulos, V. Vitamin B1, B2, B3, B5, and B6 and the Immune System. In Nutrition and Immunity; Springer: Berlin/Heidelberg, Germany, 2019; pp. 115–125. [Google Scholar]
- Dobbelstein, H.; Körner, W.F.; Mempel, W.; Grosse-Wilde, H.; Edel, H.H. Vitamin B6 deficiency in uremia and its implications for the depression of immune responses. Kidney Int. 1974, 5, 233–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.H.; Yeh, E.L.; Chen, C.C.; Huang, S.C.; Huang, Y.C. Vitamin B-6, independent of homocysteine, is a significant factor in relation to inflammatory responses for chronic kidney disease and hemodialysis patients. BioMed. Res. Int. 2017, 2017, 7367831. [Google Scholar] [CrossRef] [Green Version]
- Elmadfa, I.; Meyer, A.L. The role of the status of selected micronutrients in shaping the immune function. Endocr. Metab. Immune Disord. 2019, 19, 1100–1115. [Google Scholar] [CrossRef]
- Mydlík, M.; Derzsiová, K. Vitamin B6 and oxalic acid in clinical nephrology. J. Renal. Nutr. 2010, 20, S95–S102. [Google Scholar] [CrossRef] [PubMed]
- Morgan, S.H.; Maher, E.R.; Purkiss, P.; Watts, R.W.; Curtis, J.R. Oxalate metabolism in end-stage renal disease: The effect of ascorbic acid and pyridoxine. Nephrol. Dial. Transplant. 1988, 3, 28–32. [Google Scholar]
- Vanholder, R.; De Smet, R.; Glorieux, G.; Argilés, A.; Baurmeister, U.; Brunet, P.; Clark, W.; Cohen, G.; De Deyn, P.P.; Deppisch, R.; et al. Review on uremic toxins: Classification, concentration, and interindividual variability. Kidney Int. 2003, 63, 1934–1943. [Google Scholar] [CrossRef] [Green Version]
- Fargue, S.; Knight, J.; Holmes, R.P.; Rumsby, G.; Danpure, C.J. Effects of alanine: Glyoxylate aminotransferase variants and pyridoxine sensitivity on oxalate metabolism in a cell-based cytotoxicity assay. Biochim. Biophys. Acta 2016, 1862, 1055–1062. [Google Scholar] [CrossRef]
- Zempleni, J.; Mock, D. Biotin biochemistry and human requirements. J. Nutr. Biochem. 1999, 10, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Zempleni, J.; Wijeratne, S.S.; Hassan, Y.I. Biotin. Biofactors 2009, 35, 36–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leon-Del-Rio, A. Biotin in metabolism, gene expression, and human disease. J. Inherit. Metab. Dis. 2019, 42, 647–654. [Google Scholar] [CrossRef]
- Said, H.M.; Redha, R.; Nylander, W. Biotin transport in the human intestine: Inhibition by anticonvulsant drugs. Am. J. Clin. Nutr. 1989, 49, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Jungert, A.; Ellinger, S.; Watzl, B.; Richter, M.; German Nutrition Society (DGE). Revised DA-CH reference values for the intake of biotin. Eur. J. Nutr. 2022, 61, 1779–1787. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ding, Y.; Fan, Y.; Xu, Y.; Lu, Y.; Zhai, L.; Wang, L. Influence of biotin intervention on glycemic control and lipid profile in patients with type 2 diabetes mellitus: A systematic review and meta-analysis. Front. Nutr. 2022, 9, 2682. [Google Scholar] [CrossRef] [PubMed]
- De La Vega-Monroy, M.L.; Larrieta, E.; German, M.S.; Baez-Saldana, A.; Fernandez-Mejia, C. Effects of biotin supplementation in the diet on insulin secretion, islet gene expression, glucose homeostasis and beta-cell proportion. J. Nutr. Biochem. 2013, 24, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Larrieta, E.; Velasco, F.; Vital, P.; López-Aceves, T.; Lazo-de-la-Vega, M.L.; Rojas, A.; Fernandez-Mejia, C. Pharmacological concentrations of biotin reduce serum triglycerides and the expression of lipogenic genes. Eur. J. Pharmacol. 2010, 644, 263–268. [Google Scholar] [CrossRef]
- McCarty, M.F. In type 1 diabetics, high-dose biotin may compensate for low hepatic insulin exposure, promoting a more normal expression of glycolytic and gluconeogenic enyzymes and thereby aiding glycemic control. Med. Hypotheses 2016, 95, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Jung, U.; Helbich-Endermann, M.; Bitsch, R.; Schneider, S.; Stein, G. Are patients with chronic renal failure (CRF) deficient in biotin and is regular biotin supplementation required? Z. Ernahr. 1998, 37, 363–367. [Google Scholar] [CrossRef]
- Descombes, E.; Hanck, A.B.; Fellay, G. Water soluble vitamins in chronic hemodialysis patients and need for supplementation. Kidney Int. 1993, 43, 1319–1328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujiwara, M.; Ando, I.; Yagi, S.; Nishizawa, M.; Oguma, S.; Satoh, K.; Sato, H.; Imai, Y. Plasma levels of biotin metabolites are elevated in hemodialysis patients with cramps. Tohoku J. Exp. Med. 2016, 239, 263–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aldahmash, B.A.; El-Nagar, D.M.; Ibrahim, K.E. Attenuation of hepatotoxicity and oxidative stress in diabetes STZ-induced type 1 by biotin in Swiss albino mice. Saudi J. Biol. Sci. 2016, 23, 311–317. [Google Scholar] [CrossRef] [Green Version]
- Donnelly, J.G. Folic acid. Crit. Rev. Clin. Lab. Sci. 2001, 38, 183–223. [Google Scholar] [CrossRef]
- Shulpekova, Y.; Nechaev, V.; Kardasheva, S.; Sedova, A.; Kurbatova, A.; Bueverova, E.; Kopylov, A.; Malsagova, K.; Dlamini, J.C.; Ivashkin, V. The concept of folic acid in health and disease. Molecules 2021, 26, 3731. [Google Scholar] [CrossRef]
- Crider, K.S.; Bailey, L.B.; Berry, R.J. Folic acid food fortification—Its history, effect, concerns, and future directions. Nutrients 2011, 3, 370–384. [Google Scholar] [CrossRef] [Green Version]
- Devlin, A.M.; Ling, E.H.; Peerson, J.M.; Fernando, S.; Clarke, R.; Smith, A.D.; Halsted, C.H. Glutamate carboxypeptidase II: A polymorphism associated with lower levels of serum folate and hyperhomocysteinemia. Hum. Mol. Genet. 2000, 9, 2837–2844. [Google Scholar] [CrossRef] [Green Version]
- Bernstein, L.H.; Gutstein, S.; Weiner, S.; Efron, G. The absorption and malabsorption of folic acid and its polyglutamates. Am. J. Med. 1970, 48, 570–579. [Google Scholar] [CrossRef]
- Farrell, C.J.; Kirsch, S.H.; Herrmann, M. Red cell or serum folate: What to do in clinical practice? Clin. Chem. Lab. Med. 2013, 51, 555–569. [Google Scholar] [CrossRef]
- Steinberg, S.E.; Campbell, C.L.; Hillman, R.S. Kinetics of the normal folate enterohepatic cycle. J. Clin. Investig. 1979, 64, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Franco, C.N.; Seabrook, L.J.; Nguyen, S.T.; Leonard, J.T.; Albrecht, L.V. Simplifying the B Complex: How Vitamins B6 and B9 Modulate One Carbon Metabolism in Cancer and Beyond. Metabolites 2022, 12, 961. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.H.L.; Wang, A.Y.M. Vitamin B12 and chronic kidney disease. Vita. Horm. 2022, 119, 325–353. [Google Scholar]
- Waxman, S.; Metz, J.; Herbert, V. Defective DNA synthesis in human megaloblastic bone marrow: Effects of homocysteine and methionine. J. Clin. Investig. 1969, 48, 284–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Froese, D.S.; Fowler, B.; Baumgartner, M.R. Vitamin B12, folate, and the methionine remethylation cycle—Biochemistry, pathways, and regulation. J. Inherit. Metab. Dis. 2019, 42, 673–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, J.T.; Stover, P.J. Folate-mediated one-carbon metabolism. Vita. Horm. 2008, 79, 1–44. [Google Scholar]
- Alqarni, A.M.; Zeidler, M.P. How does methotrexate work? Biochem. Soc. Trans. 2020, 48, 559–567. [Google Scholar] [CrossRef]
- Cristalli, C.P.; Zannini, C.; Comai, G.; Baraldi, O.; Cuna, V.; Cappuccilli, M.; Mantovani, V.; Natali, N.; Cianciolo, G.; La Manna, G. Methylenetetrahydrofolate reductase, MTHFR; polymorphisms and predisposition to different multifactorial disorders. Genes. Genom. 2017, 39, 689–699. [Google Scholar] [CrossRef]
- Poduri, A.; Mukherjee, D.; Sud, K.; Kohli, H.S.; Sakhuja, V.; Khullar, M. MTHFR A1298C polymorphism is associated with cardiovascular risk in end stage renal disease in North Indians. Mol. Cell. Biochem. 2008, 308, 43–50. [Google Scholar] [CrossRef]
- Ramanathan, G.; Harichandana, B.; Kannan, S.; Elumalai, R.; Sfd, P. Association between end-stage diabetic nephropathy and MTHFR (C677T and A1298C) gene polymorphisms. Nephrology 2019, 24, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Yuan, M.M.; Yuan, L.; Huang, L.L.; Liao, J.H.; Yu, X.L.; Su, C.; Chen, Y.H.; Yang, Y.Y.; Yu, H.; et al. Chronic folate deficiency induces glucose and lipid metabolism disorders and subsequent cognitive dysfunction in mice. PLoS ONE 2018, 13, e0202910. [Google Scholar] [CrossRef]
- Hsu, H.C.; Chiou, J.F.; Wang, Y.H.; Chen, C.H.; Mau, S.Y.; Ho, C.T.; Chang, P.J.; Liu, T.Z.; Chen, C.H. Folate deficiency triggers an oxidative-nitrosative stress-mediated apoptotic cell death and impedes insulin biosynthesis in RINm5F pancreatic islet β–cells: Relevant to the pathogenesis of diabetes. PLoS ONE 2013, 8, e77931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Najib, S.; Sanchez-Margalet, V. Homocysteine thiolactone inhibits insulin-stimulated DNA and protein synthesis: Possible role of mitogen-activated protein kinase (MAPK), glycogen synthase kinase-3 (GSK-3) and p70 S6K phosphorylation. J. Mol. Endocrinol. 2005, 34, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.S.; Cao, E.H.; Qin, J.F. Homocysteine induces cell cycle G1 arrest in endothelial cells through the PI3K/Akt/FOXO signaling pathway. Pharmacologys 2005, 74, 57–64. [Google Scholar] [CrossRef]
- Jiang, Q.; Wang, L.; Si, X.; Tian, J.L.; Zhang, Y.; Gui, H.L.; Li, B.; Tan, D.H. Current progress on the mechanisms of hyperhomocysteinemia-induced vascular injury and use of natural polyphenol compounds. Eur. J. Pharmacol. 2021, 905, 174168. [Google Scholar] [CrossRef]
- Li, H.; Lewis, A.; Brodsky, S.; Rieger, R.; Iden, C.; Goligorsky, M.S. Homocysteine induces 3-hydroxy-3-methylglutaryl coenzyme a reductase in vascular endothelial cells: A mechanism for development of atherosclerosis? Circulation 2002, 105, 1037–1043. [Google Scholar] [CrossRef] [Green Version]
- Pecoits-Filho, R.; Lindholm, B.; Stenvinkel, P. The malnutrition, inflammation, and atherosclerosis (MIA) syndrome—The heart of the matter. Nephrol. Dial. Transplant. 2002, 17, 28–31. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Fernandez, N.; Jacobs-Cachá, C.; Mora-Gutiérrez, J.M.; Vergara, A.; Orbe, J.; Soler, M.J. Matrix metalloproteinases in diabetic kidney disease. J. Clin. Med. 2020, 9, 472. [Google Scholar] [CrossRef] [Green Version]
- Achour, O.; Elmtaoua, S.; Zellama, D.; Omezzine, A.; Moussa, A.; Rejeb, J.; Boumaiza, I.; Bouacida, L.; Rejeb, N.B.; Achour, A.; et al. The C677T MTHFR genotypes influence the efficacy of B9 and B12 vitamins supplementation to lowering plasma total homocysteine in hemodialysis. J. Nephrol. 2016, 29, 691–698. [Google Scholar] [CrossRef]
- Brown, K.L. Chemistry and enzymology of vitamin B12. Chem. Rev. 2005, 105, 2075–2150. [Google Scholar] [CrossRef] [PubMed]
- Stabler, S.P. Vitamin B12. In Present Knowledge in Nutrition; Academic Press: New York, NY, USA, 2020; pp. 257–271. [Google Scholar]
- Hodgkin, D.C.; Kamper, J.; Mackay, M.; Pickworth, J.; Trueblood, K.N.; White, J.G. Structure of vitamin B12. Nature 1956, 178, 64–66. [Google Scholar] [CrossRef] [PubMed]
- McMahon, G.M.; Hwang, S.J.; Tanner, R.M.; Jacques, P.F.; Selhub, J.; Muntner, P.; Fox, C.S. The association between vitamin B12, albuminuria and reduced kidney function: An observational cohort study. BMC Nephrol. 2015, 16, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, F.; Bito, T. Vitamin B12 sources and microbial interaction. Exp. Biol. Med. 2018, 243, 148–158. [Google Scholar] [CrossRef]
- Watanabe, F.; Yabuta, Y.; Bito, T.; Teng, F. Vitamin B12-containing plant food sources for vegetarians. Nutrients 2014, 6, 1861–1873. [Google Scholar] [CrossRef] [Green Version]
- Festen, H.P. Intrinsic factor secretion and cobalamin absorption: Physiology and pathophysiology in the gastrointestinal tract. Scand. J. Gastroenterol. 1991, 26, 1–7. [Google Scholar] [CrossRef]
- Alpers, D.H.; Russell-Jones, G. Gastric intrinsic factor: The gastric and small intestinal stages of cobalamin absorption. A personal journey. Biochimie 2013, 95, 989–994. [Google Scholar] [CrossRef]
- Wuerges, J.; Garau, G.; Geremia, S.; Fedosov, S.N.; Petersen, T.E.; Randaccio, L. Structural basis for mammalian vitamin B12 transport by transcobalamin. Proc. Natl. Acad. Sci. USA 2006, 103, 4386–4391. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Geng, T.; Wan, Z.; Lu, Q.; Zhang, X.; Qiu, Z.; Li, L.; Zhu, K.; Liu, L.; Pan, A.; et al. Associations of serum folate and vitamin B12 levels with cardiovascular disease mortality among patients with type 2 diabetes. JAMA Netw. Open 2022, 5, e2146124. [Google Scholar] [CrossRef]
- Martinelli, D.; Deodato, F.; Dionisi-Vici, C. Cobalamin C defect: Natural history, pathophysiology, and treatment. J. Inherit. Metab. Dis. 2011, 34, 127–135. [Google Scholar] [CrossRef]
- Lemoine, M.; Grangé, S.; Guerrot, D. Kidney disease in cobalamin C deficiency. Nephrol. Ther. 2019, 15, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Cervantes-Laurean, D.; McElvaney, N.G.; Moss, J. Niacin. In Modern Nutrition in Health and Disease, 9th ed.; Williams & Wilkins: Baltimore, MD, USA, 1999; pp. 401–411. [Google Scholar]
- Niafar, M.; Hai, F.; Porhomayon, J.; Nader, N.D. The role of metformin on vitamin B12 deficiency: A meta-analysis review. Intern. Emerg. Med. 2015, 10, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Chapman, L.E.; Darling, A.L.; Brown, J.E. Association between metformin and vitamin B12 deficiency in patients with type 2 diabetes: A systematic review and meta-analysis. Diabetes Metab. 2016, 42, 316–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, M.A. Metformin and vitamin B12 deficiency: Where do we stand? J. Pharm. Pharm. Sci. 2016, 19, 382–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Infante, M.; Leoni, M.; Caprio, M.; Fabbri, A. Long-term metformin therapy and vitamin B12 deficiency: An association to bear in mind. World J. Diabetes 2021, 12, 916–931. [Google Scholar] [CrossRef] [PubMed]
- Wooley, A.C.; Kerr, J.L. Monitoring patients on metformin: Recent changes and rationales. J. Pharm. Technol. 2018, 34, 28–36. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.W. Proton pump inhibitors, H2-receptor antagonists, metformin, and vitamin B-12 deficiency: Clinical implications. Adv. Nutr. 2018, 9, 511S–518S. [Google Scholar] [CrossRef] [Green Version]
- Bauman, W.A.; Shaw, S.; Jayatilleke, E.; Spungen, A.M.; Herbert, V. Increased intake of calcium reverses vitamin B12 malabsorption induced by metformin. Diabetes Care 2000, 23, 1227–1231. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Ahn, C.W.; Fang, S.; Lee, H.S.; Park, J.S. Association between metformin dose and vitamin B12 deficiency in patients with type 2 diabetes. Medicine 2019, 98, e17918. [Google Scholar] [CrossRef]
- De Block, C.E.; De Leeuw, I.H.; Van Gaal, L.F. Autoimmune gastritis in type 1 diabetes: A clinically oriented review. J. Clin. Endocrinol. Metab. 2008, 93, 363–371. [Google Scholar] [CrossRef]
- De Block, C.E.; De Leeuw, I.H.; Rooman, R.P.; Winnock, F.; Du Caju, M.V.; Van Gaal, L.F.; Registry, T.B. Gastric parietal cell antibodies are associated with glutamic acid decarboxylase-65 antibodies and the HLA DQA1* 0501-DQB1* 0301 haplotype in Type 1 diabetes mellitus. Diabet. Med. 2000, 17, 618–622. [Google Scholar] [CrossRef] [PubMed]
- Seetharam, B.; Li, N. Transcobalamin II and its cell surface receptor. Vita. Horm. 2000, 59, 337–366. [Google Scholar]
- Park, J.; Ahmadi, S.F.; Streja, E.; Molnar, M.Z.; Flegal, K.M.; Gillen, D.; Kovesdy, C.P.; Kalantar-Zadeh, K. Obesity paradox in end-stage kidney disease patients. Prog. Cardiovasc. Dis. 2014, 56, 415–425. [Google Scholar] [CrossRef] [Green Version]
- Obeid, R.; Shannan, B.; Herrmann, W. Advanced glycation end products overload might explain intracellular cobalamin deficiency in renal dysfunction, diabetes and aging. Med. Hypotheses 2011, 77, 884–888. [Google Scholar] [CrossRef] [PubMed]
- Nowotny, K.; Jung, T.; Höhn, A.; Weber, D.; Grune, T. Advanced glycation end products and oxidative stress in type 2 diabetes mellitus. Biomolecules 2015, 5, 194–222. [Google Scholar] [CrossRef] [Green Version]
- van de Lagemaat, E.E.; de Groot, L.C.; van den Heuvel, E.G. Vitamin B12 in relation to oxidative stress: A systematic review. Nutrients 2019, 11, 482. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Vitamin B Sub-Form | Summary of Evidence |
|---|---|
| Thiamine (Vitamin B1) |
|
| Riboflavin (Vitamin B2) |
|
| Niacin (Vitamin B3) | |
| Pantothenic Acid (Vitamin B5) |
|
| Pyridoxine (Vitamin B6) |
|
| Biotin (Vitamin B8) |
|
| Folic Acid (Vitamin B9) |
|
| Cobalamin (Vitamin B12) |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, H.H.L.; McDonnell, T.; Chinnadurai, R. Physiological Associations between Vitamin B Deficiency and Diabetic Kidney Disease. Biomedicines 2023, 11, 1153. https://doi.org/10.3390/biomedicines11041153
Wu HHL, McDonnell T, Chinnadurai R. Physiological Associations between Vitamin B Deficiency and Diabetic Kidney Disease. Biomedicines. 2023; 11(4):1153. https://doi.org/10.3390/biomedicines11041153
Chicago/Turabian StyleWu, Henry H. L., Thomas McDonnell, and Rajkumar Chinnadurai. 2023. "Physiological Associations between Vitamin B Deficiency and Diabetic Kidney Disease" Biomedicines 11, no. 4: 1153. https://doi.org/10.3390/biomedicines11041153