
The Selenoprotein Glutathione Peroxidase 4: From Molecular Mechanisms to Novel Therapeutic Opportunities


Abstract
:1. Introduction
2. Glutathione Peroxidase Family
2.1. The Origins of Glutathione Peroxidase
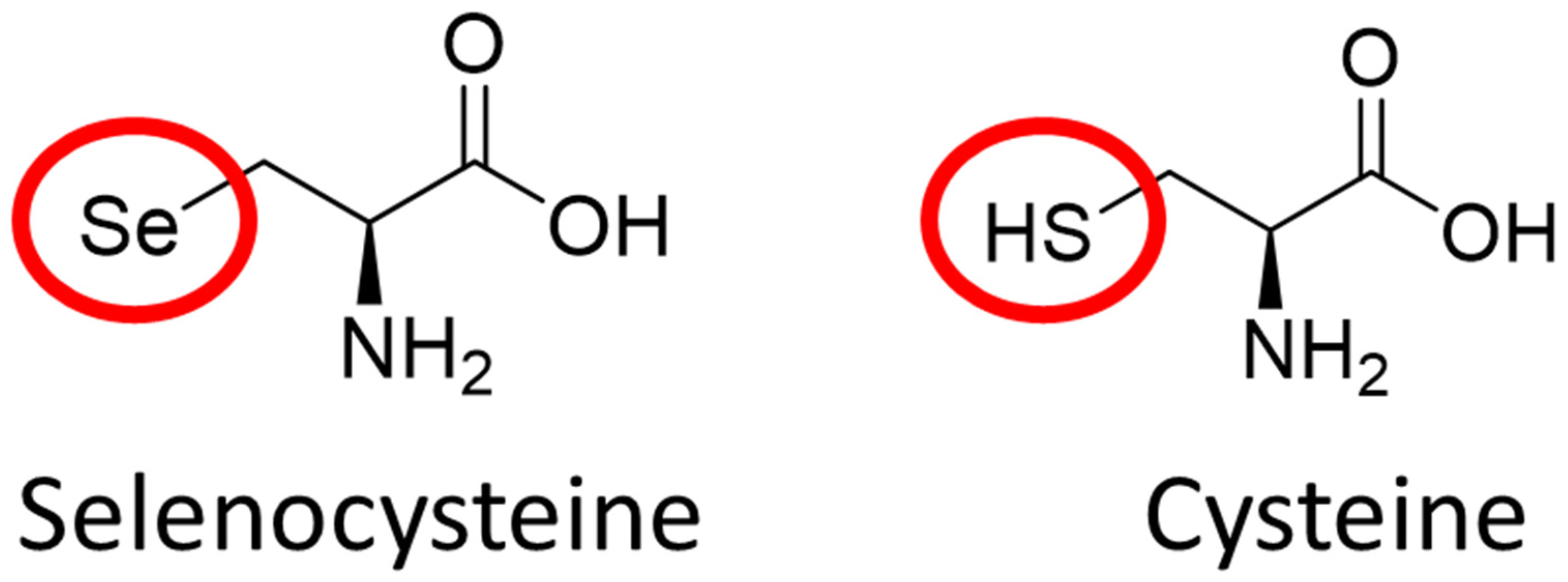
2.2. The Role of Elemental Selenium in Selenoproteins
2.3. The Biochemical Structure of Glutathione Peroxidase
2.4. General Mechanism of Glutathione Peroxidase
2.5. Substrate Specificity
2.6. Role of Oxidative Stress in Biology
2.7. Glutathione Peroxidase in Health and Diseases
3. Glutathione Peroxidase 4 in Biochemistry and Molecular Biology
3.1. The Origins of Glutathione Peroxidase 4
3.2. Clinical Relevance of Glutathione Peroxidase 4
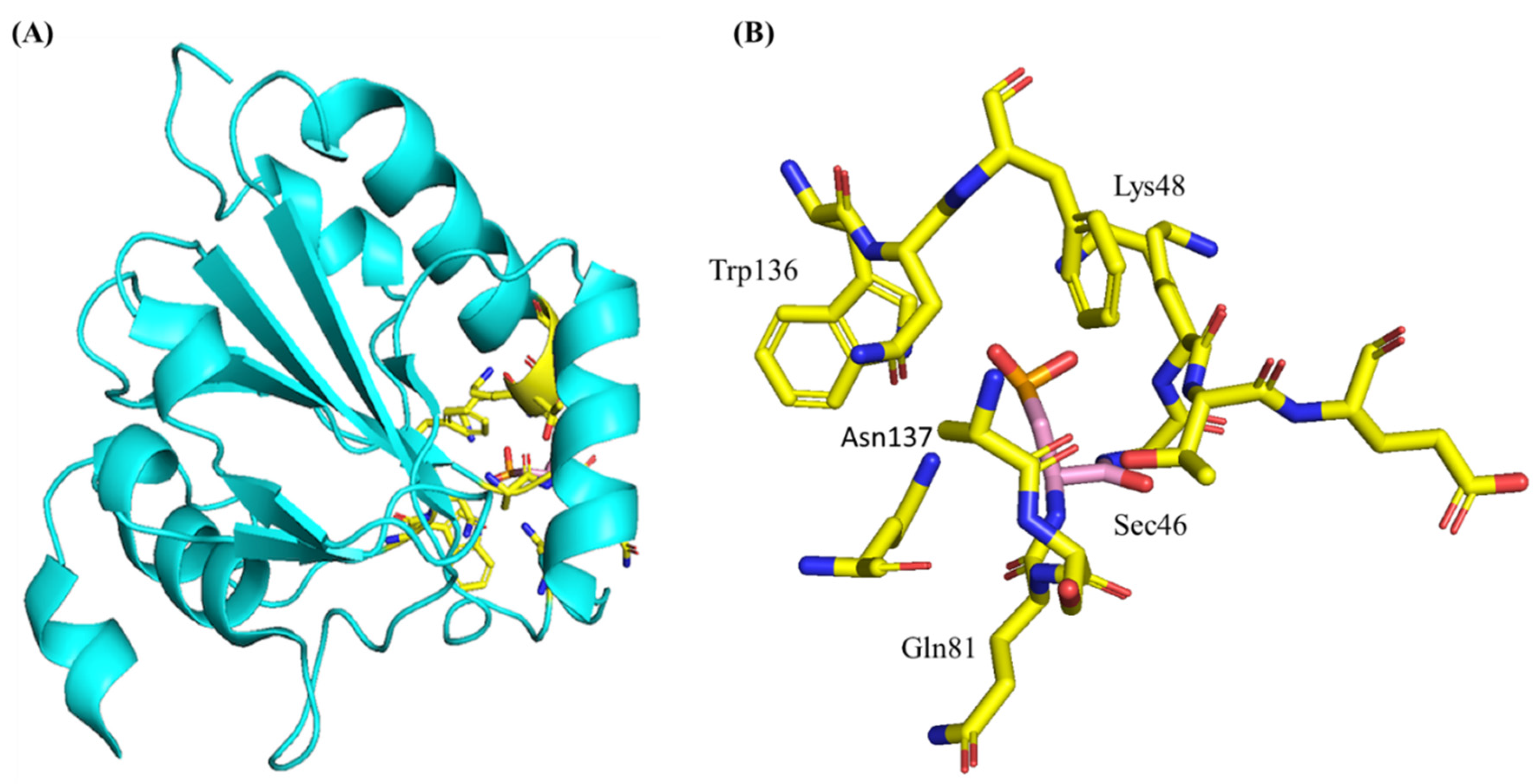
3.3. Structure and Genetics of Glutathione Peroxidase 4
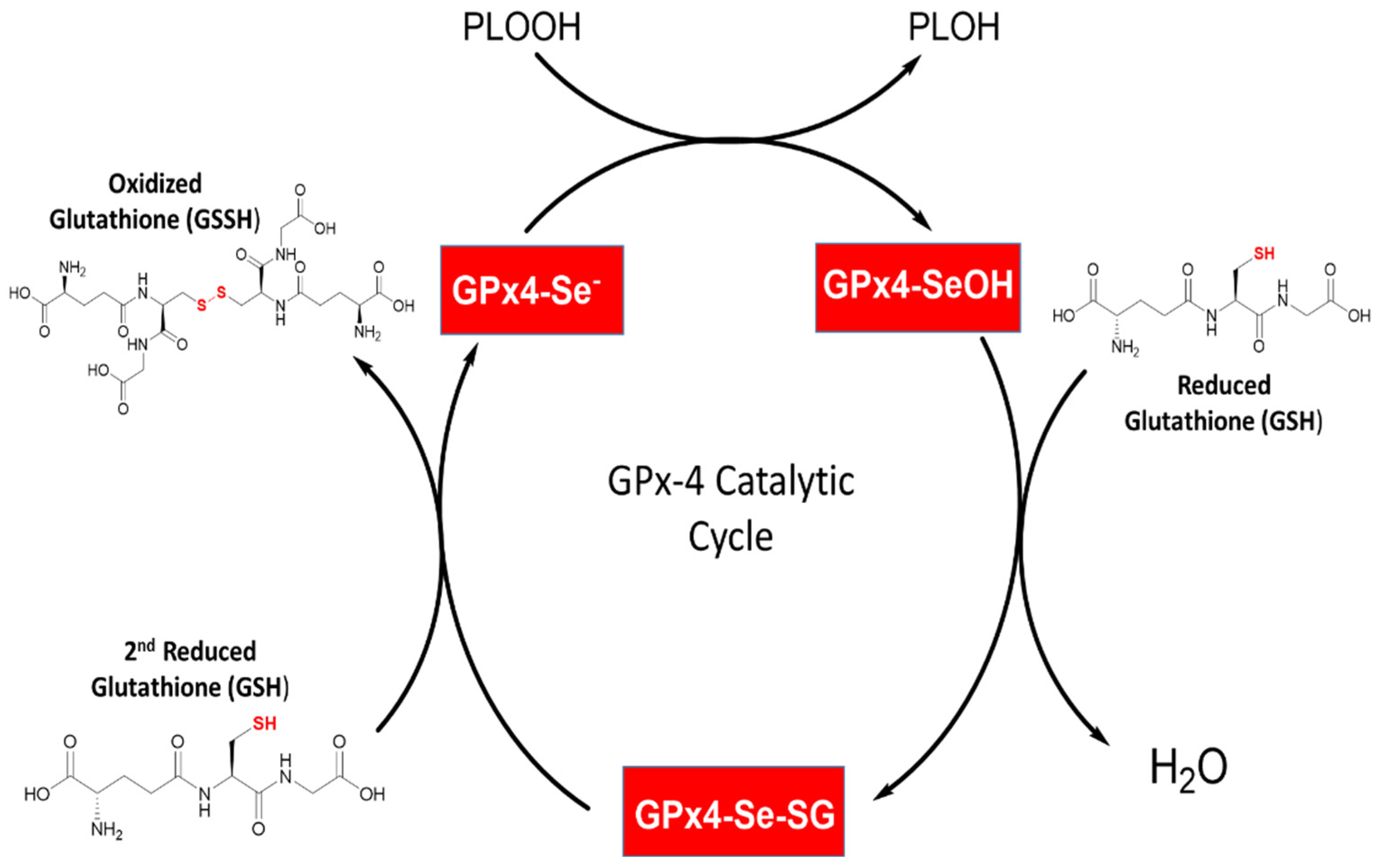
3.4. Enzymology and Kinetics of Glutathione Peroxidase 4 and GSH
3.5. Synthesis, Degradation, and Regulation of Glutathione Peroxidase 4
4. Glutathione Peroxidase 4 as a Chief Regulator of Ferroptosis
4.1. Overview of Ferroptosis
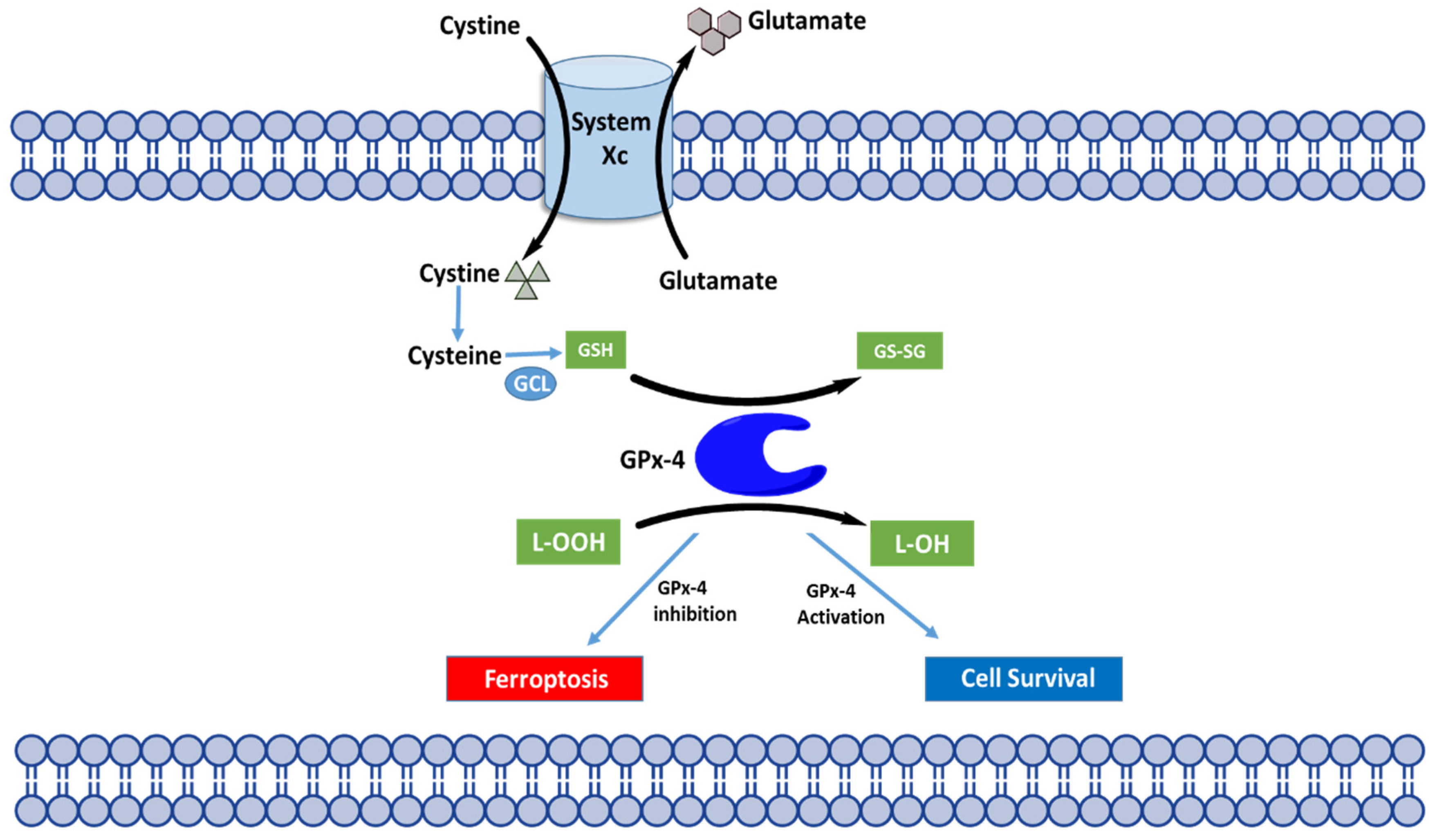
4.2. Molecular Mechanisms of Ferroptosis
4.3. Regulation of Ferroptosis through GPX4
4.4. Modulation of GPX4 to Probe Ferroptosis
4.5. GPX4, Ferroptosis, and Mitochondria
5. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mills, G.C. Hemoglobin Catabolism. I. Glutathione Peroxidase, an Erythrocyte Enzyme Which Protects Hemoglobin from Oxidative Breakdown. J. Biol. Chem. 1957, 229, 189–197. [Google Scholar] [CrossRef]
- Christophersen, B.O. Reduction of Linolenic Acid Hydroperoxide by a Glutathione Peroxidase. Biochim. Biophys. Acta 1969, 176, 463–470. [Google Scholar] [CrossRef]
- Little, C.; Olinescu, R.; Reid, K.G.; O’Brien, P.J. Properties and Regulation of Glutathione Peroxidase. J. Biol. Chem. 1970, 245, 3632–3636. [Google Scholar] [CrossRef]
- Flohe, L.; Günzler, W.A.; Schock, H.H. Glutathione Peroxidase: A Selenoenzyme. FEBS Lett. 1973, 32, 132–134. [Google Scholar] [CrossRef] [Green Version]
- Flohé, L. Glutathione peroxidase: Enzymology and biological aspects. Klin. Wochenschr. 1971, 49, 669–683. [Google Scholar] [CrossRef]
- Forstrom, J.W.; Zakowski, J.J.; Tappel, A.L. Identification of the Catalytic Site of Rat Liver Glutathione Peroxidase as Selenocysteine. Biochemistry 1978, 17, 2639–2644. [Google Scholar] [CrossRef]
- Rotruck, J.T.; Pope, A.L.; Ganther, H.E.; Swanson, A.B.; Hafeman, D.G.; Hoekstra, W.G. Selenium: Biochemical Role as a Component of Glutathione Peroxidase. Science 1973, 179, 588–590. [Google Scholar] [CrossRef]
- Brigelius-Flohé, R.; Maiorino, M. Glutathione Peroxidases. Biochim. Biophys. Acta 2013, 1830, 3289–3303. [Google Scholar] [CrossRef]
- Trenz, T.S.; Delaix, C.L.; Turchetto-Zolet, A.C.; Zamocky, M.; Lazzarotto, F.; Margis-Pinheiro, M. Going Forward and Back: The Complex Evolutionary History of the GPx. Biology 2021, 10, 1165. [Google Scholar] [CrossRef]
- Margis, R.; Dunand, C.; Teixeira, F.K.; Margis-Pinheiro, M. Glutathione Peroxidase Family—An Evolutionary Overview. FEBS J. 2008, 275, 3959–3970. [Google Scholar] [CrossRef]
- Takebe, G.; Yarimizu, J.; Saito, Y.; Hayashi, T.; Nakamura, H.; Yodoi, J.; Nagasawa, S.; Takahashi, K. A Comparative Study on the Hydroperoxide and Thiol Specificity of the Glutathione Peroxidase Family and Selenoprotein P. J. Biol. Chem. 2002, 277, 41254–41258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bersweiler, A.; D’Autréaux, B.; Mazon, H.; Kriznik, A.; Belli, G.; Delaunay-Moisan, A.; Toledano, M.B.; Rahuel-Clermont, S. A Scaffold Protein That Chaperones a Cysteine-Sulfenic Acid in H2O2 Signaling. Nat. Chem. Biol. 2017, 13, 909–915. [Google Scholar] [CrossRef] [Green Version]
- Brigelius-Flohé, R.; Flohé, L. Regulatory Phenomena in the Glutathione Peroxidase Superfamily. Antioxid. Redox Signal. 2020, 33, 498–516. [Google Scholar] [CrossRef] [PubMed]
- McClung, J.P.; Roneker, C.A.; Mu, W.; Lisk, D.J.; Langlais, P.; Liu, F.; Lei, X.G. Development of Insulin Resistance and Obesity in Mice Overexpressing Cellular Glutathione Peroxidase. Proc. Natl. Acad. Sci. USA 2004, 101, 8852–8857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brigelius-Flohé, R.; Kipp, A. Glutathione Peroxidases in Different Stages of Carcinogenesis. Biochim. Biophys. Acta 2009, 1790, 1555–1568. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohé, R.; Kipp, A.P. Physiological Functions of GPx2 and Its Role in Inflammation-Triggered Carcinogenesis. Ann. N. Y. Acad. Sci. 2012, 1259, 19–25. [Google Scholar] [CrossRef]
- Florian, S.; Krehl, S.; Loewinger, M.; Kipp, A.; Banning, A.; Esworthy, S.; Chu, F.-F.; Brigelius-Flohé, R. Loss of GPx2 Increases Apoptosis, Mitosis, and GPx1 Expression in the Intestine of Mice. Free Radic. Biol. Med. 2010, 49, 1694. [Google Scholar] [CrossRef] [Green Version]
- Naiki, T.; Naiki-Ito, A.; Iida, K.; Etani, T.; Kato, H.; Suzuki, S.; Yamashita, Y.; Kawai, N.; Yasui, T.; Takahashi, S. GPX2 Promotes Development of Bladder Cancer with Squamous Cell Differentiation through the Control of Apoptosis. Oncotarget 2018, 9, 15847. [Google Scholar] [CrossRef] [Green Version]
- An, B.C.; Jung, N.-K.; Park, C.Y.; Oh, I.-J.; Choi, Y.-D.; Park, J.-I.; Lee, S.-W. Epigenetic and Glucocorticoid Receptor-Mediated Regulation of Glutathione Peroxidase 3 in Lung Cancer Cells. Mol. Cells 2016, 39, 631–638. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Luo, K.; Tan, L.-Z.; Ren, B.-G.; Gu, L.-Q.; Michalopoulos, G.; Luo, J.-H.; Yu, Y.P. P53-Induced Gene 3 Mediates Cell Death Induced by Glutathione Peroxidase 3. J. Biol. Chem. 2012, 287, 16890. [Google Scholar] [CrossRef] [Green Version]
- Forcina, G.C.; Dixon, S.J. GPX4 at the Crossroads of Lipid Homeostasis and Ferroptosis. Proteomics 2019, 19, e1800311. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingold, I.; Aichler, M.; Yefremova, E.; Roveri, A.; Buday, K.; Doll, S.; Tasdemir, A.; Hoffard, N.; Wurst, W.; Walch, A.; et al. Expression of a Catalytically Inactive Mutant Form of Glutathione Peroxidase 4 (Gpx4) Confers a Dominant-Negative Effect in Male Fertility. J. Biol. Chem. 2015, 290, 14668–14678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fallahi, S.; Rajaei, M.; Hesam, M.J.; Koolivand, M.; Malekzadeh, K. The Effect of Phoenix Dactylifera Pollen on the Expression of NRF2, SOD2, CAT, and GPX4 Genes, and Sperm Parameters of Fertile and Infertile Men: A Controlled Clinical Trial. Int. J. Reprod. Biomed. 2021, 19, 545–558. [Google Scholar] [CrossRef]
- Liu, H.; Forouhar, F.; Seibt, T.; Saneto, R.; Wigby, K.; Friedman, J.; Xia, X.; Shchepinov, M.S.; Ramesh, S.K.; Conrad, M.; et al. Characterization of a Patient-Derived Variant of GPX4 for Precision Therapy. Nat. Chem. Biol. 2022, 18, 91–100. [Google Scholar] [CrossRef]
- Hangauer, M.J.; Viswanathan, V.S.; Ryan, M.J.; Bole, D.; Eaton, J.K.; Matov, A.; Galeas, J.; Dhruv, H.D.; Berens, M.E.; Schreiber, S.L.; et al. Drug-Tolerant Persister Cancer Cells Are Vulnerable to GPX4 Inhibition. Nature 2017, 551, 247–250. [Google Scholar] [CrossRef] [Green Version]
- Chabory, E.; Damon, C.; Lenoir, A.; Kauselmann, G.; Kern, H.; Zevnik, B.; Garrel, C.; Saez, F.; Cadet, R.; Henry-Berger, J.; et al. Epididymis Seleno-Independent Glutathione Peroxidase 5 Maintains Sperm DNA Integrity in Mice. J. Clin. Investig. 2009, 119, 2074. [Google Scholar] [CrossRef]
- Eshraghi, M.; Karunadharma, P.P.; Blin, J.; Shahani, N.; Ricci, E.P.; Michel, A.; Urban, N.T.; Galli, N.; Sharma, M.; Ramírez-Jarquín, U.N.; et al. Mutant Huntingtin Stalls Ribosomes and Represses Protein Synthesis in a Cellular Model of Huntington Disease. Nat. Commun. 2021, 12, 1461. [Google Scholar] [CrossRef]
- Nguyen, V.D.; Saaranen, M.J.; Karala, A.-R.; Lappi, A.-K.; Wang, L.; Raykhel, I.B.; Alanen, H.I.; Salo, K.E.H.; Wang, C.-C.; Ruddock, L.W. Two Endoplasmic Reticulum PDI Peroxidases Increase the Efficiency of the Use of Peroxide during Disulfide Bond Formation. J. Mol. Biol. 2011, 406, 503–515. [Google Scholar] [CrossRef]
- Ramming, T.; Appenzeller-Herzog, C. Destroy and Exploit: Catalyzed Removal of Hydroperoxides from the Endoplasmic Reticulum. Int. J. Cell Biol. 2013, 2013, 180906. [Google Scholar] [CrossRef] [Green Version]
- Hatfield, D.L.; Tsuji, P.A.; Carlson, B.A.; Gladyshev, V.N. Selenium and Selenocysteine: Roles in Cancer, Health, and Development. Trends Biochem. Sci. 2014, 39, 112–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, J.-J.; Yue, S.-Y.; Fang, Y.-H.; Liu, X.-L.; Wang, C.-H. Mechanisms Affecting the Biosynthesis and Incorporation Rate of Selenocysteine. Mol. Basel Switz. 2021, 26, 7120. [Google Scholar] [CrossRef] [PubMed]
- Friedmann Angeli, J.P.; Conrad, M. Selenium and GPX4, a Vital Symbiosis. Free Radic. Biol. Med. 2018, 127, 153–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lothrop, A.P.; Snider, G.W.; Ruggles, E.L.; Hondal, R.J. Why Is Mammalian Thioredoxin Reductase 1 so Dependent upon the Use of Selenium? Biochemistry 2014, 53, 554–565. [Google Scholar] [CrossRef] [PubMed]
- Areti, S.; Verma, S.K.; Bellare, J.; Rao, C.P. Selenocysteine vs Cysteine: Tuning the Derivatization on Benzenesulfonyl Moiety of a Triazole Linked Dansyl Connected Glycoconjugate for Selective Recognition of Selenocysteine and the Applicability of the Conjugate in Buffer, in Serum, on Silica Gel, and in HepG2 Cells. Anal. Chem. 2016, 88, 7259–7267. [Google Scholar] [CrossRef]
- Arnér, E.S.J. Selenoproteins-What Unique Properties Can Arise with Selenocysteine in Place of Cysteine? Exp. Cell Res. 2010, 316, 1296–1303. [Google Scholar] [CrossRef]
- Reich, H.J.; Hondal, R.J. Why Nature Chose Selenium. ACS Chem. Biol. 2016, 11, 821–841. [Google Scholar] [CrossRef]
- Maiorino, M.; Aumann, K.D.; Brigelius-Flohé, R.; Doria, D.; van den Heuvel, J.; McCarthy, J.; Roveri, A.; Ursini, F.; Flohé, L. Probing the Presumed Catalytic Triad of a Selenium-Containing Peroxidase by Mutational Analysis. Z. Ernahrungswiss. 1998, 37, 118–121. [Google Scholar]
- Ingold, I.; Berndt, C.; Schmitt, S.; Doll, S.; Poschmann, G.; Buday, K.; Roveri, A.; Peng, X.; Porto Freitas, F.; Seibt, T.; et al. Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell 2018, 172, 409–422.e21. [Google Scholar] [CrossRef] [Green Version]
- Hariharan, S.; Dharmaraj, S. Selenium and Selenoproteins: It’s Role in Regulation of Inflammation. Inflammopharmacology 2020, 28, 667–695. [Google Scholar] [CrossRef]
- Labunskyy, V.M.; Hatfield, D.L.; Gladyshev, V.N. Selenoproteins: Molecular Pathways and Physiological Roles. Physiol. Rev. 2014, 94, 739–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalimuthu, K.; Keerthana, C.K.; Mohan, M.; Arivalagan, J.; Christyraj, J.R.S.S.; Firer, M.A.; Choudry, M.H.A.; Anto, R.J.; Lee, Y.J. The Emerging Role of Selenium Metabolic Pathways in Cancer: New Therapeutic Targets for Cancer. J. Cell. Biochem. 2021, 123, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Shreenath, A.P.; Ameer, M.A.; Dooley, J. Selenium Deficiency. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Rayman, M.P. Selenium Intake, Status, and Health: A Complex Relationship. Horm. Athens Greece 2020, 19, 9–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Berry, M.J. Selenium and Selenoproteins in the Brain and Brain Diseases. J. Neurochem. 2003, 86, 1–12. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhou, Y.; Schweizer, U.; Savaskan, N.E.; Hua, D.; Kipnis, J.; Hatfield, D.L.; Gladyshev, V.N. Comparative Analysis of Selenocysteine Machinery and Selenoproteome Gene Expression in Mouse Brain Identifies Neurons as Key Functional Sites of Selenium in Mammals. J. Biol. Chem. 2008, 283, 2427–2438. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.-H.; Song, G.-L. Roles of Selenoproteins in Brain Function and the Potential Mechanism of Selenium in Alzheimer’s Disease. Front. Neurosci. 2021, 15, 646518. [Google Scholar] [CrossRef]
- Janowski, R.; Scanu, S.; Niessing, D.; Madl, T. Crystal and Solution Structural Studies of Mouse Phospholipid Hydroperoxide Glutathione Peroxidase 4. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2016, 72, 743–749. [Google Scholar] [CrossRef] [Green Version]
- Labrecque, C.L.; Fuglestad, B. Electrostatic Drivers of GPx4 Interactions with Membrane, Lipids, and DNA. Biochemistry 2021, 60, 2761–2772. [Google Scholar] [CrossRef]
- Borchert, A.; Kalms, J.; Roth, S.R.; Rademacher, M.; Schmidt, A.; Holzhutter, H.-G.; Kuhn, H.; Scheerer, P. Crystal Structure and Functional Characterization of Selenocysteine-Containing Glutathione Peroxidase 4 Suggests an Alternative Mechanism of Peroxide Reduction. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 1095–1107. [Google Scholar] [CrossRef]
- Ursini, F.; Maiorino, M. Lipid Peroxidation and Ferroptosis: The Role of GSH and GPx4. Free Radic. Biol. Med. 2020, 152, 175–185. [Google Scholar] [CrossRef]
- Brieger, K.; Schiavone, S.; Miller, F.J.; Krause, K.-H. Reactive Oxygen Species: From Health to Disease. Swiss Med. Wkly. 2012, 142, w13659. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhong, C. Oxidative Stress in Alzheimer’s Disease. Neurosci. Bull. 2014, 30, 271–281. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Zhu, G.; Wang, G.; Zhang, F. Oxidative Stress and Neuroinflammation Potentiate Each Other to Promote Progression of Dopamine Neurodegeneration. Oxid. Med. Cell. Longev. 2020, 2020, 6137521. [Google Scholar] [CrossRef] [PubMed]
- Kirtonia, A.; Sethi, G.; Garg, M. The Multifaceted Role of Reactive Oxygen Species in Tumorigenesis. Cell. Mol. Life Sci. CMLS 2020, 77, 4459–4483. [Google Scholar] [CrossRef] [PubMed]
- Klaunig, J.E. Oxidative Stress and Cancer. Curr. Pharm. Des. 2018, 24, 4771–4778. [Google Scholar] [CrossRef]
- Zhang, P.; Li, T.; Wu, X.; Nice, E.C.; Huang, C.; Zhang, Y. Oxidative Stress and Diabetes: Antioxidative Strategies. Front. Med. 2020, 14, 583–600. [Google Scholar] [CrossRef]
- Sinha, N.; Dabla, P.K. Oxidative Stress and Antioxidants in Hypertension-a Current Review. Curr. Hypertens. Rev. 2015, 11, 132–142. [Google Scholar] [CrossRef]
- Zhang, Y.; Xin, L.; Xiang, M.; Shang, C.; Wang, Y.; Wang, Y.; Cui, X.; Lu, Y. The Molecular Mechanisms of Ferroptosis and Its Role in Cardiovascular Disease. Biomed. Pharmacother. Biomed. Pharmacother. 2022, 145, 112423. [Google Scholar] [CrossRef]
- Ursini, F.; Maiorino, M.; Valente, M.; Ferri, L.; Gregolin, C. Purification from Pig Liver of a Protein Which Protects Liposomes and Biomembranes from Peroxidative Degradation and Exhibits Glutathione Peroxidase Activity on Phosphatidylcholine Hydroperoxides. Biochim. Biophys. Acta 1982, 710, 197–211. [Google Scholar] [CrossRef]
- Ursini, F.; Maiorino, M.; Gregolin, C. The Selenoenzyme Phospholipid Hydroperoxide Glutathione Peroxidase. Biochim. Biophys. Acta 1985, 839, 62–70. [Google Scholar] [CrossRef]
- Schuckelt, R.; Brigelius-Flohé, R.; Maiorino, M.; Roveri, A.; Reumkens, J.; Strabburger, W.; Ursini, F.; Wolf, B.; Flohé, L. Phospholipid Hydroperoxide Glutathione Peroxidase Is a Seleno-Enzyme Distinct from the Classical Glutathione Peroxidase as Evident from Cdna and Amino Acid Sequencing. Free Radic. Res. Commun. 1991, 14, 343–361. [Google Scholar] [CrossRef] [PubMed]
- Conrad, M.; Schneider, M.; Seiler, A.; Bornkamm, G.W. Physiological Role of Phospholipid Hydroperoxide Glutathione Peroxidase in Mammals. Biol. Chem. 2007, 388, 1019–1025. [Google Scholar] [CrossRef] [PubMed]
- Godeas, C.; Tramer, F.; Micali, F.; Roveri, A.; Maiorino, M.; Nisii, C.; Sandri, G.; Panfili, E. Phospholipid Hydroperoxide Glutathione Peroxidase (PHGPx) in Rat Testis Nuclei Is Bound to Chromatin. Biochem. Mol. Med. 1996, 59, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Maiorino, M.; Roveri, A.; Benazzi, L.; Bosello, V.; Mauri, P.; Toppo, S.; Tosatto, S.C.E.; Ursini, F. Functional Interaction of Phospholipid Hydroperoxide Glutathione Peroxidase with Sperm Mitochondrion-Associated Cysteine-Rich Protein Discloses the Adjacent Cysteine Motif as a New Substrate of the Selenoperoxidase. J. Biol. Chem. 2005, 280, 38395–38402. [Google Scholar] [CrossRef] [Green Version]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the Ferroptosis Regulator Gpx4 Triggers Acute Renal Failure in Mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yant, L.J.; Ran, Q.; Rao, L.; Van Remmen, H.; Shibatani, T.; Belter, J.G.; Motta, L.; Richardson, A.; Prolla, T.A. The Selenoprotein GPX4 Is Essential for Mouse Development and Protects from Radiation and Oxidative Damage Insults. Free Radic. Biol. Med. 2003, 34, 496–502. [Google Scholar] [CrossRef]
- Song, X.; Wang, X.; Liu, Z.; Yu, Z. Role of GPX4-Mediated Ferroptosis in the Sensitivity of Triple Negative Breast Cancer Cells to Gefitinib. Front. Oncol. 2020, 10, 597434. [Google Scholar] [CrossRef]
- da Rocha, T.J.; Silva Alves, M.; Guisso, C.C.; de Andrade, F.M.; Camozzato, A.; de Oliveira, A.A.; Fiegenbaum, M. Association of GPX1 and GPX4 Polymorphisms with Episodic Memory and Alzheimer’s Disease. Neurosci. Lett. 2018, 666, 32–37. [Google Scholar] [CrossRef]
- Hambright, W.S.; Fonseca, R.S.; Chen, L.; Na, R.; Ran, Q. Ablation of Ferroptosis Regulator Glutathione Peroxidase 4 in Forebrain Neurons Promotes Cognitive Impairment and Neurodegeneration. Redox Biol. 2017, 12, 8–17. [Google Scholar] [CrossRef]
- Jakaria, M.; Belaidi, A.A.; Bush, A.I.; Ayton, S. Ferroptosis as a Mechanism of Neurodegeneration in Alzheimer’s Disease. J. Neurochem. 2021, 159, 804–825. [Google Scholar] [CrossRef]
- Tosatto, S.C.E.; Bosello, V.; Fogolari, F.; Mauri, P.; Roveri, A.; Toppo, S.; Flohé, L.; Ursini, F.; Maiorino, M. The Catalytic Site of Glutathione Peroxidases. Antioxid. Redox Signal. 2008, 10, 1515–1526. [Google Scholar] [CrossRef] [PubMed]
- Peeler, J.C.; Weerapana, E. Chemical Biology Approaches to Interrogate the Selenoproteome. Acc. Chem. Res. 2019, 52, 2832–2840. [Google Scholar] [CrossRef] [PubMed]
- Scheerer, P.; Borchert, A.; Krauss, N.; Wessner, H.; Gerth, C.; Höhne, W.; Kuhn, H. Structural Basis for Catalytic Activity and Enzyme Polymerization of Phospholipid Hydroperoxide Glutathione Peroxidase-4 (GPx4). Biochemistry 2007, 46, 9041–9049. [Google Scholar] [CrossRef]
- Eaton, J.K.; Furst, L.; Ruberto, R.A.; Moosmayer, D.; Hilpmann, A.; Ryan, M.J.; Zimmermann, K.; Cai, L.L.; Niehues, M.; Badock, V.; et al. Selective Covalent Targeting of GPX4 Using Masked Nitrile-Oxide Electrophiles. Nat. Chem. Biol. 2020, 16, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Bjørklund, G.; Tinkov, A.A.; Hosnedlová, B.; Kizek, R.; Ajsuvakova, O.P.; Chirumbolo, S.; Skalnaya, M.G.; Peana, M.; Dadar, M.; El-Ansary, A.; et al. The Role of Glutathione Redox Imbalance in Autism Spectrum Disorder: A Review. Free Radic. Biol. Med. 2020, 160, 149–162. [Google Scholar] [CrossRef]
- Kennedy, L.; Sandhu, J.K.; Harper, M.-E.; Cuperlovic-Culf, M. Role of Glutathione in Cancer: From Mechanisms to Therapies. Biomolecules 2020, 10, 1429. [Google Scholar] [CrossRef]
- Wrotek, S.; Sobocińska, J.; Kozłowski, H.M.; Pawlikowska, M.; Jędrzejewski, T.; Dzialuk, A. New Insights into the Role of Glutathione in the Mechanism of Fever. Int. J. Mol. Sci. 2020, 21, 1393. [Google Scholar] [CrossRef] [Green Version]
- Teskey, G.; Abrahem, R.; Cao, R.; Gyurjian, K.; Islamoglu, H.; Lucero, M.; Martinez, A.; Paredes, E.; Salaiz, O.; Robinson, B.; et al. Glutathione as a Marker for Human Disease. Adv. Clin. Chem. 2018, 87, 141–159. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: Overview of Its Protective Roles, Measurement, and Biosynthesis. Mol. Asp. Med. 2009, 30, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Zhu, S.; Zhang, Q.; Sun, X.; Zeh, H.J.; Lotze, M.T.; Kang, R.; Tang, D. HSPA5 Regulates Ferroptotic Cell Death in Cancer Cells. Cancer Res. 2017, 77, 2064–2077. [Google Scholar] [CrossRef] [Green Version]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The Molecular Machinery of Regulated Cell Death. Cell Res. 2019, 29, 347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerr, J.F.R.; Wyllie, A.H.; Currie, A.R. Apoptosis: A Basic Biological Phenomenon with Wide-Ranging Implications in Tissue Kinetics. Br. J. Cancer 1972, 26, 239. [Google Scholar] [CrossRef] [Green Version]
- D’Arcy, M.S. Cell Death: A Review of the Major Forms of Apoptosis, Necrosis and Autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Ko, C.-J.; Gao, S.-L.; Lin, T.-K.; Chu, P.-Y.; Lin, H.-Y. Ferroptosis as a Major Factor and Therapeutic Target for Neuroinflammation in Parkinson’s Disease. Biomedicines 2021, 9, 1679. [Google Scholar] [CrossRef] [PubMed]
- Mahoney-Sánchez, L.; Bouchaoui, H.; Ayton, S.; Devos, D.; Duce, J.A.; Devedjian, J.-C. Ferroptosis and Its Potential Role in the Physiopathology of Parkinson’s Disease. Prog. Neurobiol. 2021, 196, 101890. [Google Scholar] [CrossRef] [PubMed]
- Mazhar, M.; Din, A.U.; Ali, H.; Yang, G.; Ren, W.; Wang, L.; Fan, X.; Yang, S. Implication of Ferroptosis in Aging. Cell Death Discov. 2021, 7, 149. [Google Scholar] [CrossRef]
- Peña-Bautista, C.; Vento, M.; Baquero, M.; Cháfer-Pericás, C. Lipid Peroxidation in Neurodegeneration. Clin. Chim. Acta Int. J. Clin. Chem. 2019, 497, 178–188. [Google Scholar] [CrossRef]
- Ghoochani, A.; Hsu, E.-C.; Aslan, M.; Rice, M.A.; Nguyen, H.M.; Brooks, J.D.; Corey, E.; Paulmurugan, R.; Stoyanova, T. Ferroptosis Inducers Are a Novel Therapeutic Approach for Advanced Prostate Cancer. Cancer Res. 2021, 81, 1583–1594. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Zhu, C.; Tang, D.; Dou, Q.P.; Shen, J.; Chen, X. The Role of Ferroptosis in Lung Cancer. Biomark. Res. 2021, 9, 82. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Deng, H.; Hu, S.; Zhang, Y.; Zheng, L.; Liu, M.; Chen, Y.; Wei, J.; Yang, H.; Lv, X. Role of Ferroptosis in Lung Diseases. J. Inflamm. Res. 2021, 14, 2079–2090. [Google Scholar] [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, Biology and Role in Disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R.; Jiang, X.; Gu, W. Emerging Mechanisms and Disease Relevance of Ferroptosis. Trends Cell Biol. 2020, 30, 478–490. [Google Scholar] [CrossRef] [PubMed]
- Eagle, H. Nutrition Needs of Mammalian Cells in Tissue Culture. Science 1955, 122, 501–514. [Google Scholar] [CrossRef] [PubMed]
- Eagle, H.; Piez, K.A.; Oyama, V.I. The Biosynthesis of Cystine in Human Cell Cultures. J. Biol. Chem. 1961, 236, 1425–1428. [Google Scholar] [CrossRef]
- Mitchell, J.R.; Jollow, D.J.; Potter, W.Z.; Gillette, J.R.; Brodie, B.B. Acetaminophen-Induced Hepatic Necrosis. IV. Protective Role of Glutathione. J. Pharmacol. Exp. Ther. 1973, 187, 211–217. [Google Scholar]
- Bannai, S.; Tsukeda, H.; Okumura, H. Effect of Antioxidants on Cultured Human Diploid Fibroblasts Exposed to Cystine-Free Medium. Biochem. Biophys. Res. Commun. 1977, 74, 1582–1588. [Google Scholar] [CrossRef]
- Maher, P.; Currais, A.; Schubert, D. Using the Oxytosis/Ferroptosis Pathway to Understand and Treat Age-Associated Neurodegenerative Diseases. Cell Chem. Biol. 2020, 27, 1456–1471. [Google Scholar] [CrossRef]
- Murphy, T.H.; Miyamoto, M.; Sastre, A.; Schnaar, R.L.; Coyle, J.T. Glutamate Toxicity in a Neuronal Cell Line Involves Inhibition of Cystine Transport Leading to Oxidative Stress. Neuron 1989, 2, 1547–1558. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Cao, F.; Yin, H.; Huang, Z.; Lin, Z.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, Present and Future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef]
- Kim, S.-W.; Kim, Y.; Kim, S.E.; An, J.-Y. Ferroptosis-Related Genes in Neurodevelopment and Central Nervous System. Biology 2021, 10, 35. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, S.; Chen, Z.; Yang, Y.; Zhang, J.; Tang, J.; Yang, K. Clinical and Biological Significances of a Ferroptosis-Related Gene Signature in Glioma. Front. Oncol. 2020, 10, 590861. [Google Scholar] [CrossRef] [PubMed]
- Mandal, P.K.; Seiler, A.; Perisic, T.; Kölle, P.; Banjac Canak, A.; Förster, H.; Weiss, N.; Kremmer, E.; Lieberman, M.W.; Bannai, S.; et al. System Xc−and Thioredoxin Reductase 1 Cooperatively Rescue Glutathione Deficiency*. J. Biol. Chem. 2010, 285, 22244–22253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seibt, T.M.; Proneth, B.; Conrad, M. Role of GPX4 in Ferroptosis and Its Pharmacological Implication. Free Radic. Biol. Med. 2019, 133, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Zhu, S.; Song, X.; Sun, X.; Fan, Y.; Liu, J.; Zhong, M.; Yuan, H.; Zhang, L.; Billiar, T.R.; et al. The Tumor Suppressor P53 Limits Ferroptosis by Blocking DPP4 Activity. Cell Rep. 2017, 20, 1692–1704. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Shi, J.; Liu, X.; Feng, L.; Gong, Z.; Koppula, P.; Sirohi, K.; Li, X.; Wei, Y.; Lee, H.; et al. BAP1 Links Metabolic Regulation of Ferroptosis to Tumour Suppression. Nat. Cell Biol. 2018, 20, 1181–1192. [Google Scholar] [CrossRef]
- Zheng, J.; Conrad, M. The Metabolic Underpinnings of Ferroptosis. Cell Metab. 2020, 32, 920–937. [Google Scholar] [CrossRef]
- Shah, R.; Shchepinov, M.S.; Pratt, D.A. Resolving the Role of Lipoxygenases in the Initiation and Execution of Ferroptosis. ACS Cent. Sci. 2018, 4, 387–396. [Google Scholar] [CrossRef]
- Wenzel, S.E.; Tyurina, Y.Y.; Zhao, J.; St Croix, C.M.; Dar, H.H.; Mao, G.; Tyurin, V.A.; Anthonymuthu, T.S.; Kapralov, A.A.; Amoscato, A.A.; et al. PEBP1 Wardens Ferroptosis by Enabling Lipoxygenase Generation of Lipid Death Signals. Cell 2017, 171, 628–641.e26. [Google Scholar] [CrossRef] [Green Version]
- Maiorino, M.; Conrad, M.; Ursini, F. GPx4, Lipid Peroxidation, and Cell Death: Discoveries, Rediscoveries, and Open Issues. Antioxid. Redox Signal. 2018, 29, 61–74. [Google Scholar] [CrossRef]
- Seiler, A.; Schneider, M.; Förster, H.; Roth, S.; Wirth, E.K.; Culmsee, C.; Plesnila, N.; Kremmer, E.; Rådmark, O.; Wurst, W.; et al. Glutathione Peroxidase 4 Senses and Translates Oxidative Stress into 12/15-Lipoxygenase Dependent-and AIF-Mediated Cell Death. Cell Metab. 2008, 8, 237–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsushita, M.; Freigang, S.; Schneider, C.; Conrad, M.; Bornkamm, G.W.; Kopf, M. T Cell Lipid Peroxidation Induces Ferroptosis and Prevents Immunity to Infection. J. Exp. Med. 2015, 212, 555–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imai, H.; Matsuoka, M.; Kumagai, T.; Sakamoto, T.; Koumura, T. Lipid Peroxidation-Dependent Cell Death Regulated by GPx4 and Ferroptosis. Curr. Top. Microbiol. Immunol. 2017, 403, 143–170. [Google Scholar] [CrossRef]
- Pennington, L.D.; Muegge, I. Holistic Drug Design for Multiparameter Optimization in Modern Small Molecule Drug Discovery. Bioorg. Med. Chem. Lett. 2021, 41, 128003. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R. Chemical Genetics: Ligand-Based Discovery of Gene Function. Nat. Rev. Genet. 2000, 1, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R.; Jiang, X. The Chemistry and Biology of Ferroptosis. Cell Chem. Biol. 2020, 27, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, V.S.; Ryan, M.J.; Dhruv, H.D.; Gill, S.; Eichhoff, O.M.; Seashore-Ludlow, B.; Kaffenberger, S.D.; Eaton, J.K.; Shimada, K.; Aguirre, A.J.; et al. Dependency of a Therapy-Resistant State of Cancer Cells on a Lipid Peroxidase Pathway. Nature 2017, 547, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Zhao, B.; Zhou, L.; Zhang, Z.; Shen, Y.; Lv, H.; AlQudsy, L.H.H.; Shang, P. Ferroptosis, a Novel Pharmacological Mechanism of Anti-Cancer Drugs. Cancer Lett. 2020, 483, 127–136. [Google Scholar] [CrossRef]
- Dolma, S.; Lessnick, S.L.; Hahn, W.C.; Stockwell, B.R. Identification of Genotype-Selective Antitumor Agents Using Synthetic Lethal Chemical Screening in Engineered Human Tumor Cells. Cancer Cell 2003, 3, 285–296. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Patel, D.N.; Welsch, M.; Skouta, R.; Lee, E.D.; Hayano, M.; Thomas, A.G.; Gleason, C.E.; Tatonetti, N.P.; Slusher, B.S.; et al. Pharmacological Inhibition of Cystine-Glutamate Exchange Induces Endoplasmic Reticulum Stress and Ferroptosis. eLife 2014, 3, e02523. [Google Scholar] [CrossRef]
- Wei, Y.; Lv, H.; Shaikh, A.B.; Han, W.; Hou, H.; Zhang, Z.; Wang, S.; Shang, P. Directly Targeting Glutathione Peroxidase 4 May Be More Effective than Disrupting Glutathione on Ferroptosis-Based Cancer Therapy. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129539. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Stockwell, B.R. Synthetic Lethal Screening Identifies Compounds Activating Iron-Dependent, Nonapoptotic Cell Death in Oncogenic-RAS-Harboring Cancer Cells. Chem. Biol. 2008, 15, 234–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armenta, D.A.; Dixon, S.J. Investigating Nonapoptotic Cell Death Using Chemical Biology approaches. Cell Chem. Biol. 2020, 27, 376–386. [Google Scholar] [CrossRef]
- Gao, M.; Yi, J.; Zhu, J.; Minikes, A.M.; Monian, P.; Thompson, C.B.; Jiang, X. Role of Mitochondria in Ferroptosis. Mol. Cell 2019, 73, 354–363.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cole-Ezea, P.; Swan, D.; Shanley, D.; Hesketh, J. Glutathione Peroxidase 4 Has a Major Role in Protecting Mitochondria from Oxidative Damage and Maintaining Oxidative Phosphorylation Complexes in Gut Epithelial Cells. Free Radic. Biol. Med. 2012, 53, 488–497. [Google Scholar] [CrossRef] [PubMed]
- Gan, B. Mitochondrial Regulation of Ferroptosis. J. Cell Biol. 2021, 220, e202105043. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mammalian GPX Type | Tissue Distribution | Cellular Localization | Primary Function | Biological Relevance/References |
|---|---|---|---|---|
| GPX-1 | Most abundant and ubiquitously expressed GPx. Highly distributed in the lungs, kidney, red blood cells, and liver. | Cytosol and mitochondria. | Reduces hydrogen peroxides in the cytoplasm at the expense of GSH. | Dampens phosphorylation of phosphatases [13], modulator of the insulin signaling pathway [14], acts in an antiapoptotic manner which can support tumor cell survival [15] |
| GPX-2 | Gastrointestinal tract, endothelial cells (particularly malignant tissues and pluripotent stem cells). | Cytosol | Reduces hydrogen peroxide. | Inhibits inflammation-induced carcinogenesis in the gut [13], but also promotes the growth of some cancers including bladder cancer [16,17,18] |
| GPX-3 | Kidney, lung, heart, muscle. | Plasma | Reduces hydrogen peroxide using GSH, Trx, or Grx. | Deficiency facilitates platelet aggregation and is a risk factor for stroke [13]. Acts as a tumor suppressor in many cancers including lung cancer [19,20] |
| GPX-4 | Widespread. Especially testis and spermatozoa kidney, followed by the liver, spleen, pancreas, heart, and brain. | Cytosol, Mitochondria, Plasma. | Reduces hydroperoxides from phospholipids and cholesterol. | Key regulator of ferroptosis [21,22]. Deficiency facilitates male infertility [23,24], Modulator of a rare genetic disorder called SSMD [25]. Implicated in several cancers including CCC and TNBC [26] |
| GPX-5 | Testis, spermatozoa, liver, kidney. | Epididymis | Protects the membranes of spermatozoa from lipid peroxidation. | Deficiency, together with GPX4, decreases male fertility [27] |
| GPX-6 | Embryos and adult olfactory epithelium. | n.d. | n.d. | Reduces the motor defects found in Huntington’s disease [28] |
| GPX-7 | Endoplasmic reticulum | n.d. | Mild glutathione peroxidase activity. Senses ROS levels and transmits redox signals to other thiols. | Contributes to oxidative protein folding in the ER. [29,30] |
| GPX-8 | Endoplasmic reticulum | n.d. | Mild glutathione peroxidase activity. Prevents endoplasmic reticulum oxidation and stress. | Contributes to oxidative protein folding in the ER. [29,30] |
| Mammalian GPX Type | Peroxidic Residue | Uniprot Molecular Weight (kDa) | Structure Type | Human Wild-Type Crystal Structure (PDB Code) | Human Mutant Crystal Structure (PDB Code) | Reference (Uniprot Code) |
|---|---|---|---|---|---|---|
| GPX-1 | Selenocysteine | 22 | Homotetramer | n.d. | U46G (2F8A) | P07203 |
| GPX-2 | Selenocysteine | 21.9 | Homotetramer | n.d. | U46C (2HE3) | P18283 |
| GPX-3 | Selenocystine | 22.5 | Homotetramer | n.d. | U46G (2R37) | P22352 |
| GPX-4 | Selenocysteine | 22 | Monomer | 6ElW | Many mutants (e.g., 7L81, 6HN3, 7L8K, etc.) | P36969 |
| GPX-5 | Cysteine | 25.2 | Homotetramer | 213Y | n.d. | O75715 |
| GPX-6 | Selenocysteine in Humans. Cysteine in rodents | 24.9 | Homotetramer | n.d. | n.d. | P59796 |
| GPX-7 | Cysteine | 20.9 | Monomer | 2P31 | n.d. | Q96SL4 |
| GPX-8 | Cysteine | 23.8 | Monomer | 3CYN | n.d. | Q8TED1 |
| Compound | Mode of Action | PubChem CID |
|---|---|---|
| (1S,3R)-RSL3 | Covalently and irreversibly inhibits GPX4. RSL3 is potent but has poor ADME properties [123] | 1750826 |
| DP12--DP19 | Not well characterized. Exhibits potency and ferroptosis hallmarks [22] | 5728915 |
| Altretamine | GPX4 inhibitor [22] | 2123 26186195 |
| DPI10 & ML210 | Nitroisoxazole moiety generates a nitrile oxide electrophile that may react with GPX4 [117] | 15945537 |
| ML162 | Shares the same chloroacetamide moiety as RSL3 but is otherwise very structurally different. Likely to have different off-target effects [22] | 3689413 |
| DPI17 & DPI18 | Exhibits potency and ferroptosis hallmarks. Likely to be a covalent GPX4 inhibitor [22] | 932617 |
| JKE-1674, JKE-1716 & BSC144988 | Identical function as DPI10. Nitroisozazole moiety leads to a nitrile oxide electrophilic reaction with GPX4 [117] | 145865941 |
| Withaferin A | Acts as a GPX4 inhibitor likely through its electrophilic groups [117] | 265237 |
| Compound | Possible Mode of Action | PubChem CID |
|---|---|---|
| Erastin | Directly inhibits system Xc causing depletion of intracellular GSH, which normally works alongside GPX4 to suppress phospholipid hydroperoxide accumulation [120] | 11214940 |
| Erastin Derivatives (Piperazine & Imidazole Ketone Erastin) | Same proposed mode of action as Erasin. These derivatives have improved ADME properties [22] | 72710858 & 91824786 |
| RSL5 | Displays similar effects as Erastin and may have identical mechanisms, but this has not been experimentally verified [123] | 2863472 |
| Sulfasalazine (FDA-approved drug) | Inhibits system Xc, which causes GSH depletion. Low potency and metabolically unstable in vivo [117] | 5339 |
| Glutamate | Inhibits system Xc likely by inhibiting one of its kinase targets. May induce necrotic cell death at high concentrations [99] | 23672308 |
| Diaryl-isoxazole | Non-competitive System Xc-inhibitor [117,124] | n.a |
| Engineered human cyst(e)inase | Systemic Depletion of Cysteine [117,124] | n.a |
| Tac-beclin1 | System Xc-inhibitor [117,124] | n.a |
| Lanperisone (FDA-approved drug) | Inhibits cystine uptake, Causes GSH depletion [117,124] | 198707 |
| Sorafenib | Inhibits system Xc, Causes GSH depletion. It also activates NRF2 against ferroptosis [22,121] | 216239 |
| FINO2 and FIN56 | Does not directly target GPX4, system Xc, or CoQ10. Rather, it oxidizes iron which leads to the subsequent inactivation of GPX4 activity [124] | n.a |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weaver, K.; Skouta, R. The Selenoprotein Glutathione Peroxidase 4: From Molecular Mechanisms to Novel Therapeutic Opportunities. Biomedicines 2022, 10, 891. https://doi.org/10.3390/biomedicines10040891
Weaver K, Skouta R. The Selenoprotein Glutathione Peroxidase 4: From Molecular Mechanisms to Novel Therapeutic Opportunities. Biomedicines. 2022; 10(4):891. https://doi.org/10.3390/biomedicines10040891
Chicago/Turabian StyleWeaver, Kamari, and Rachid Skouta. 2022. "The Selenoprotein Glutathione Peroxidase 4: From Molecular Mechanisms to Novel Therapeutic Opportunities" Biomedicines 10, no. 4: 891. https://doi.org/10.3390/biomedicines10040891