
Interleukin-10 Mitigates Doxorubicin-Induced Endoplasmic Reticulum Stress as Well as Cardiomyopathy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cardiomyocyte Isolation and Treatment
2.2. Protein Estimation
2.3. Western Blot Analysis
2.4. Immunofluorescence
2.5. RT-qPCR
2.6. Flow Cytometry
2.7. Statistical Analysis
3. Results
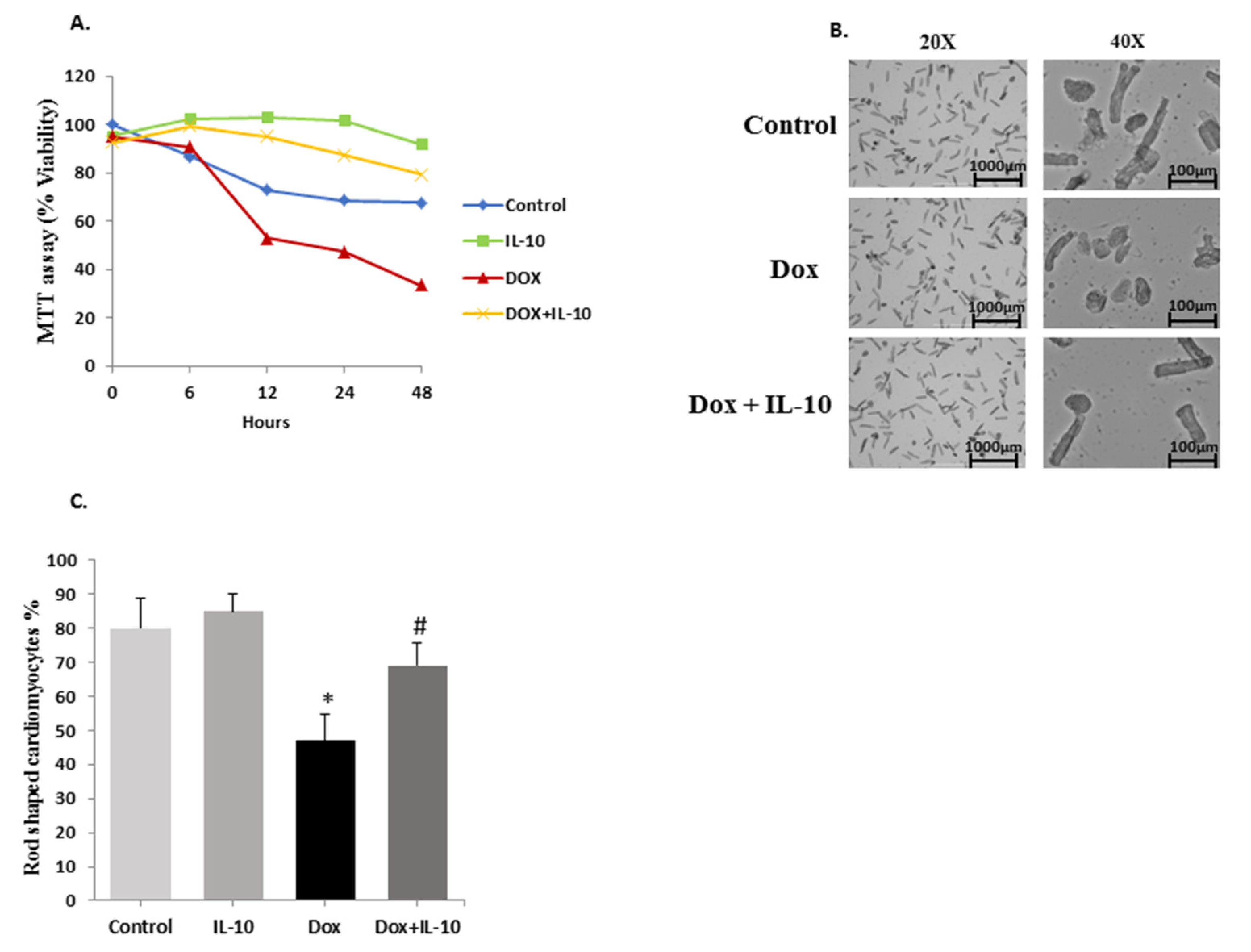
3.1. IL-10 Improves Cardiomyocyte Viability
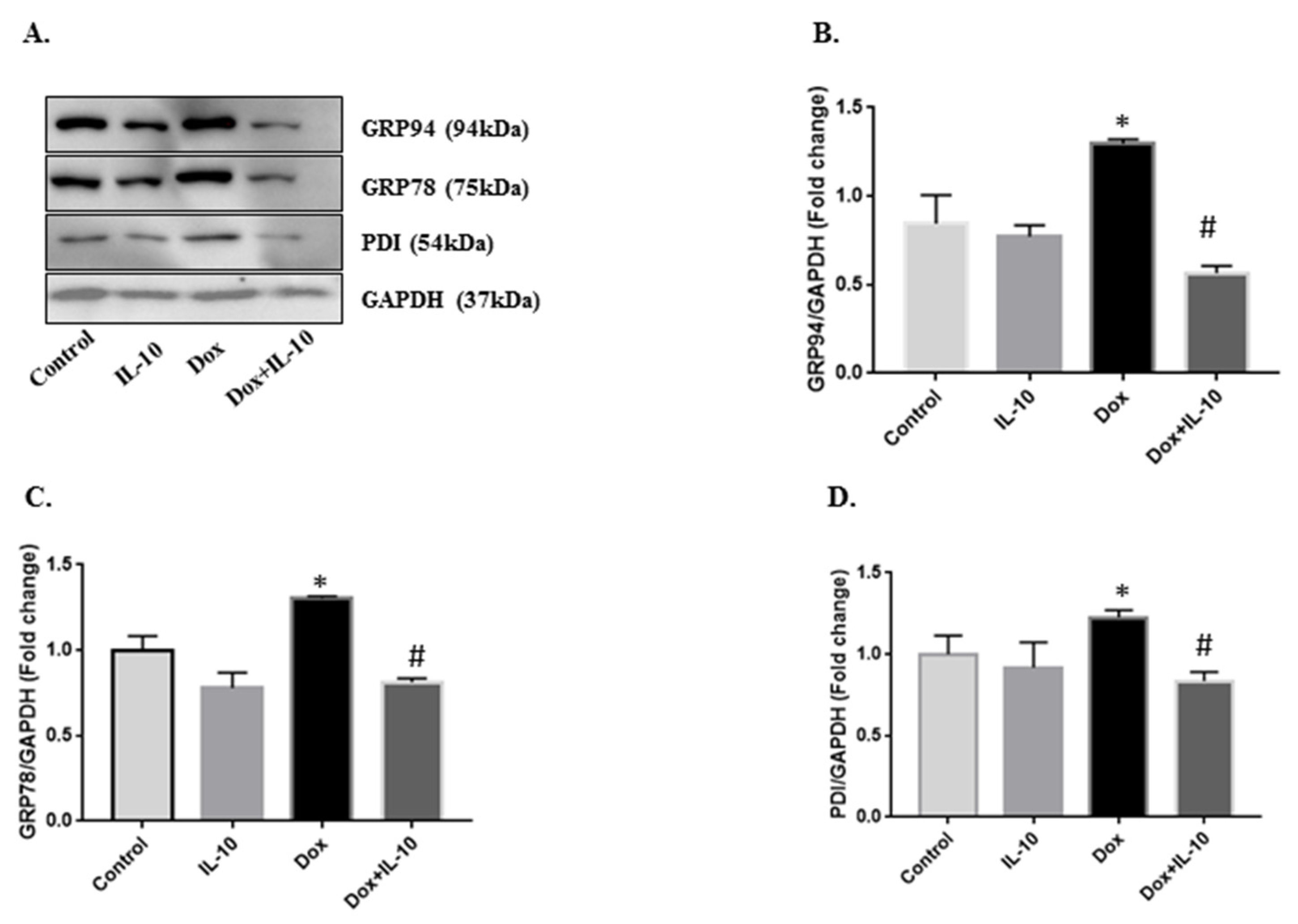
3.2. IL-10 Effects on Dox-Induced Changes in ER-Stress Markers
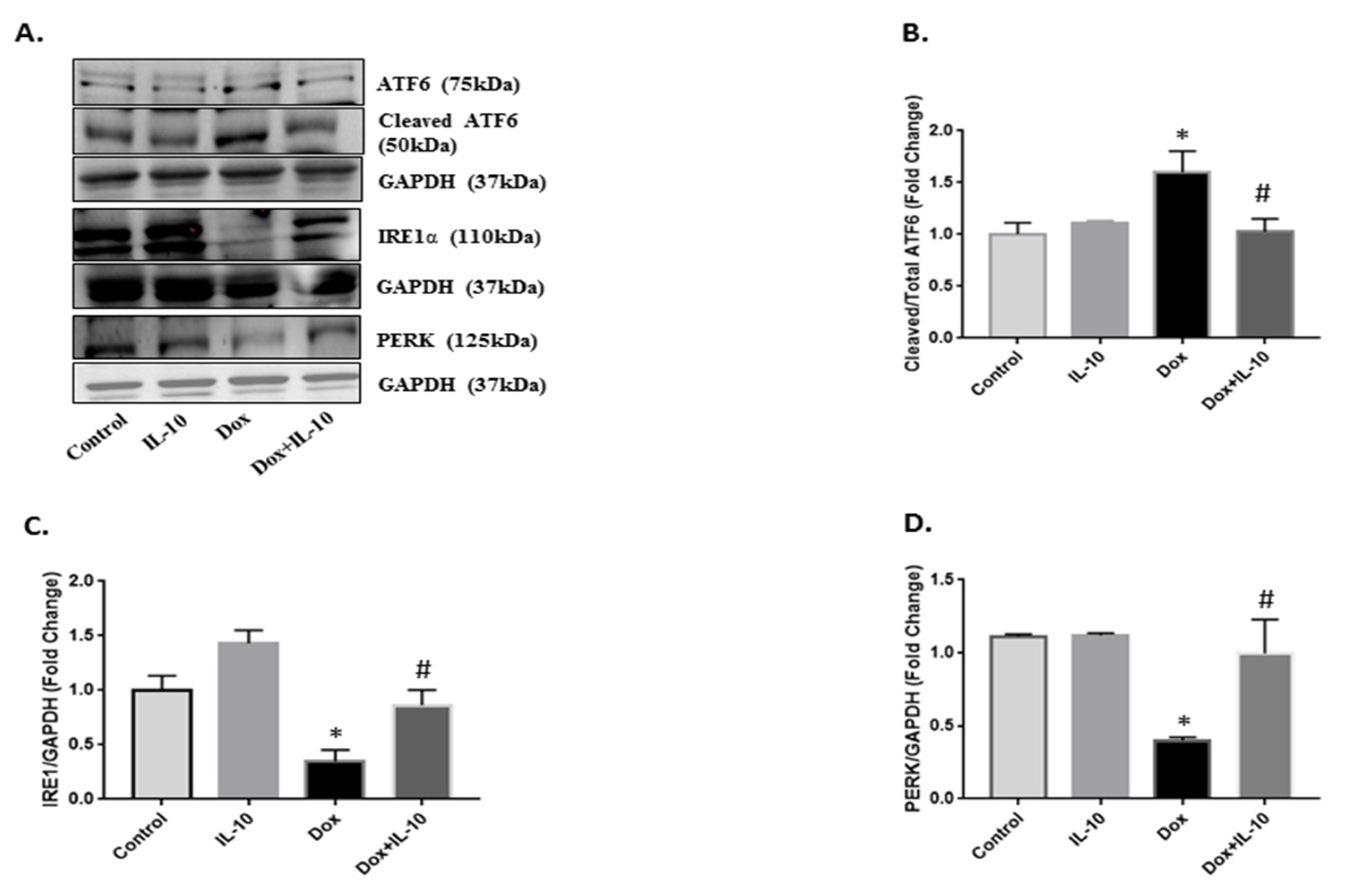
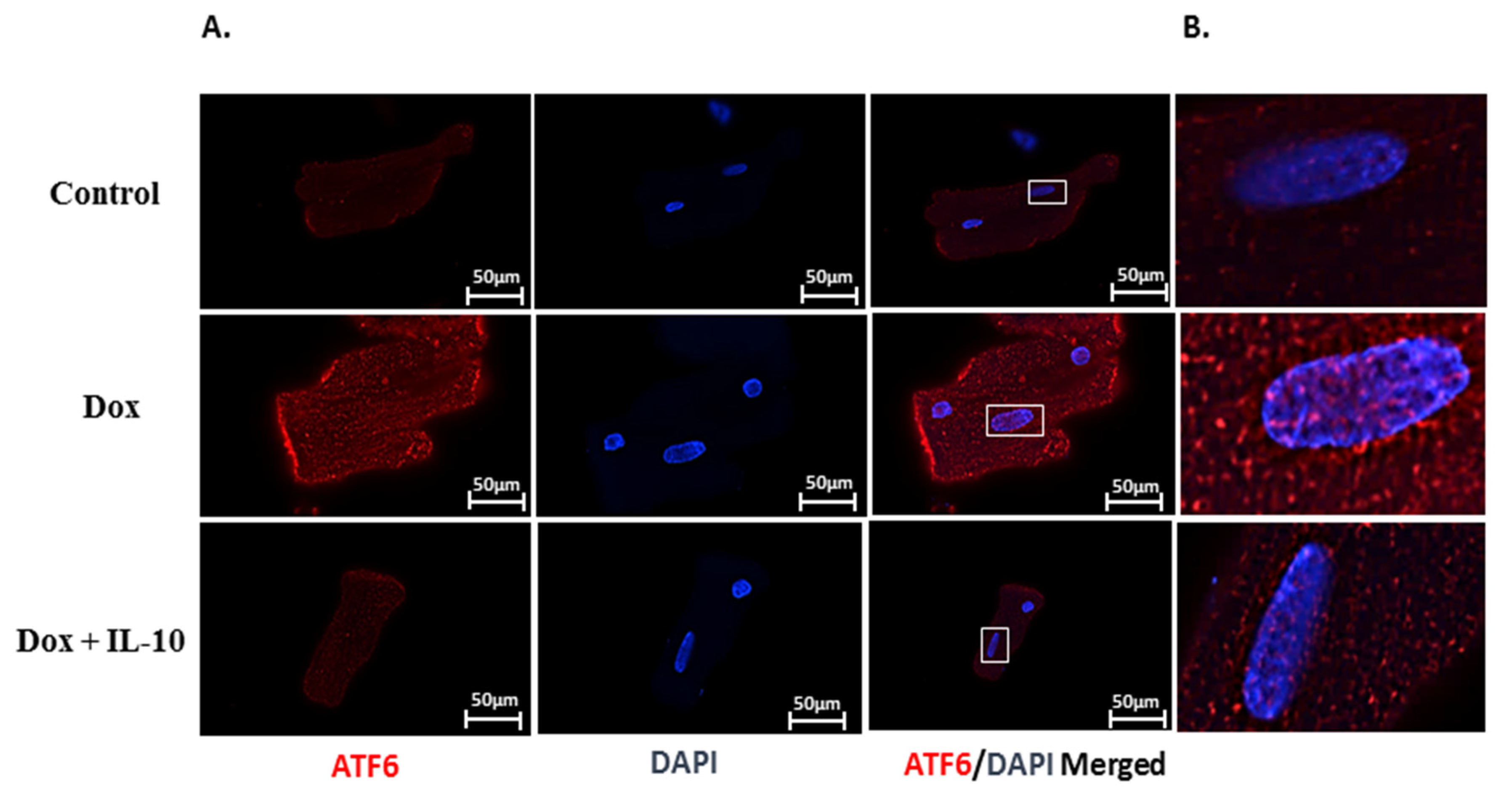
3.3. IL-10 Effect on Dox-Induced Changes in UPR-Signaling Transducers
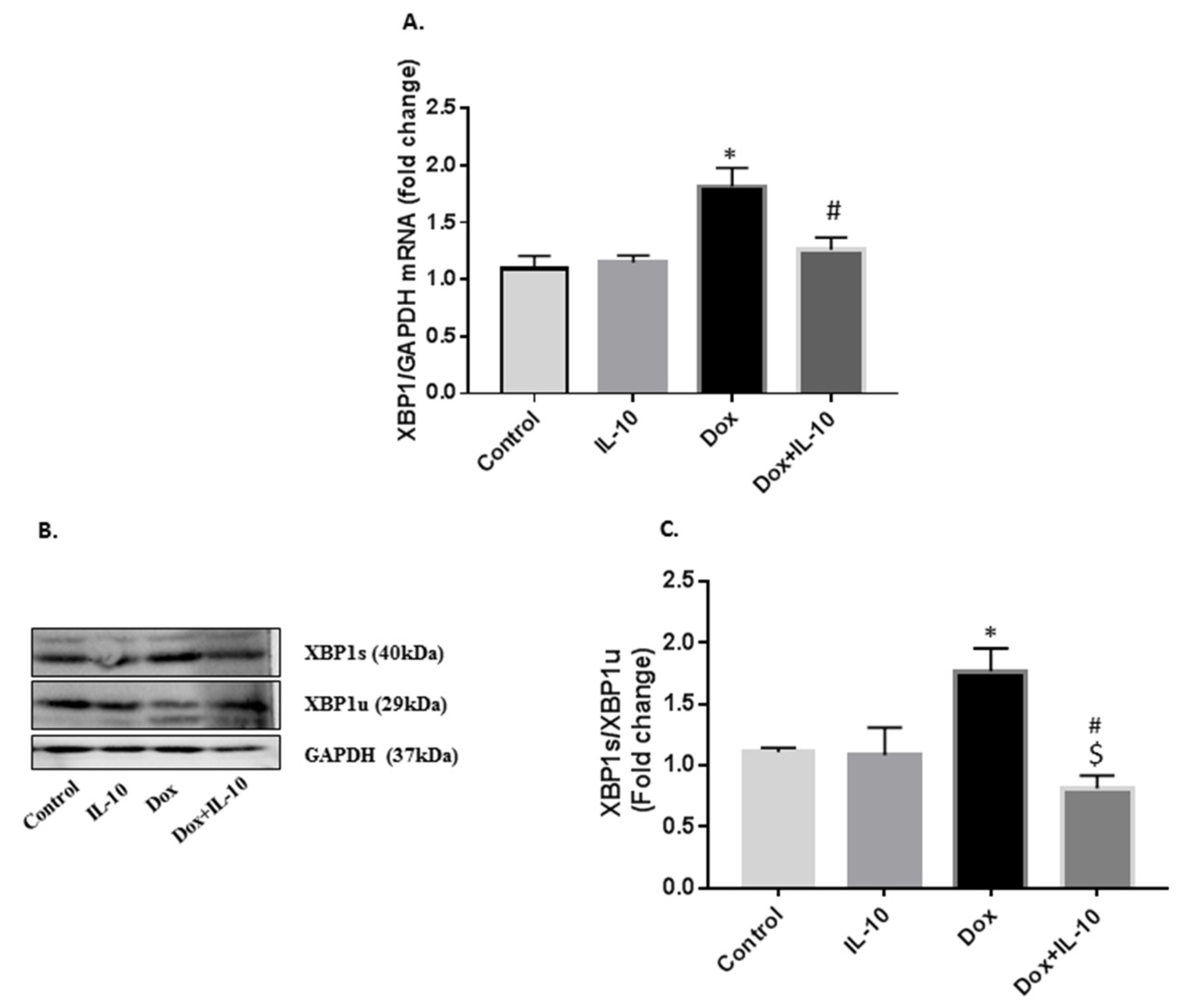
3.4. IL-10 Effects on Dox-Induced Changes in XBP1 mRNA and XBP1s/XBP1u Protein Levels
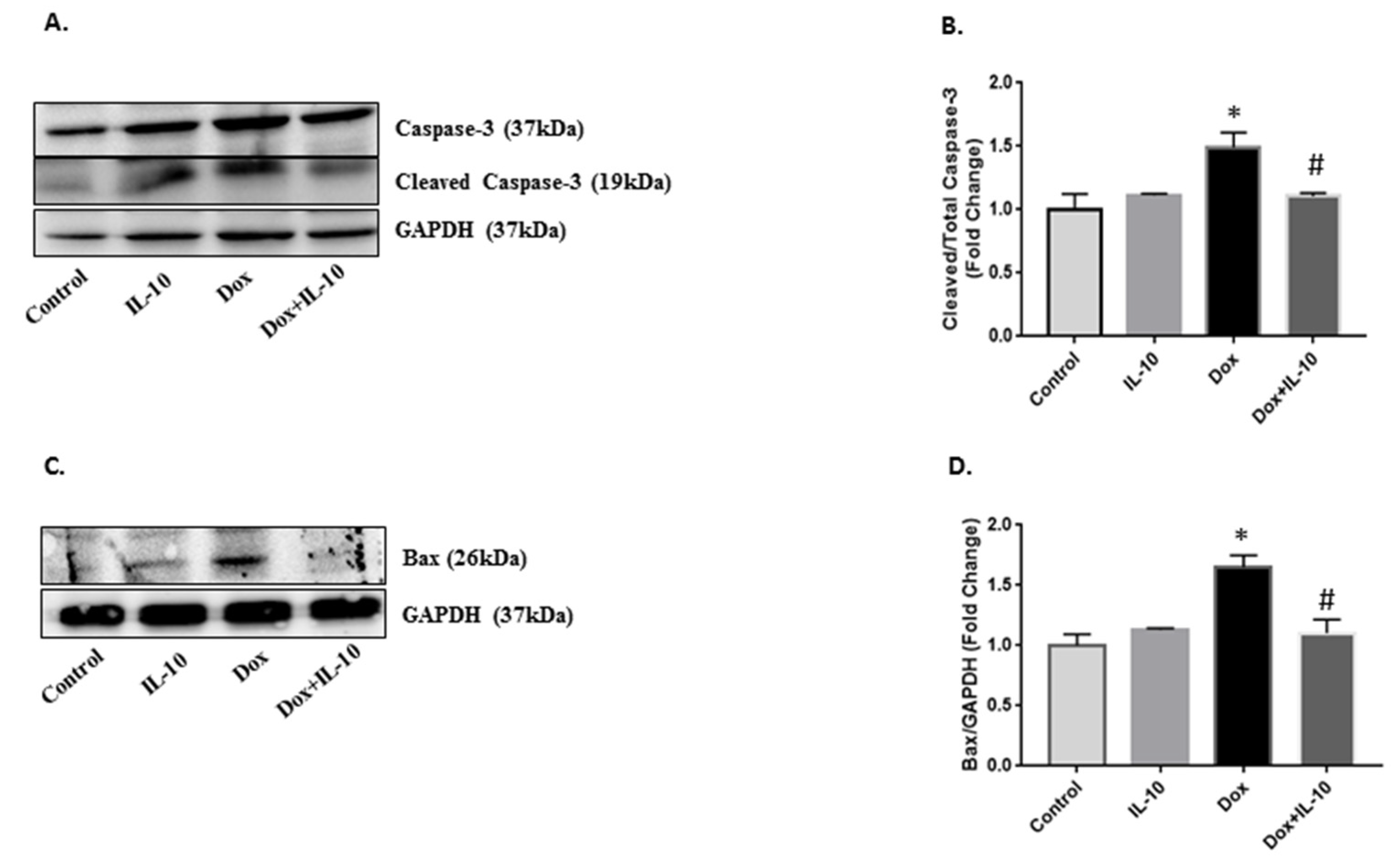
3.5. Effect of IL-10 on Dox-Induced Apoptotic Pathways
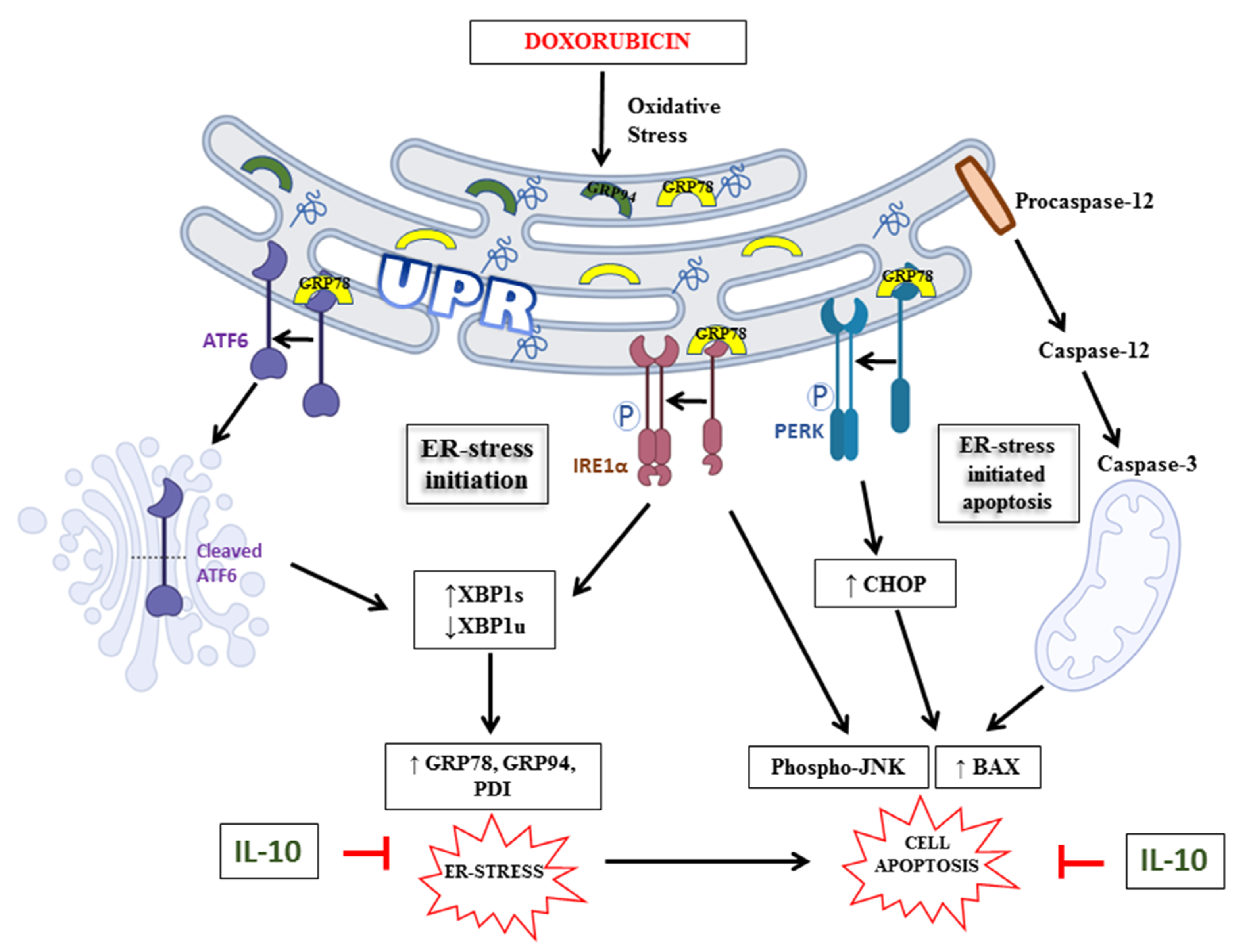
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cassinelli, G. The Roots of Modern Oncology: From Discovery of New Antitumor Anthracyclines to Their Clinical Use. Tumori J. 2016, 102, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Johnson-Arbor, K.; Dubey, R. Doxorubicin. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Lefrak, E.A.; Pitha, J.; Rosenheim, S.; Gottlieb, J.A. A Clinicopathologic Analysis of Adriamycin Cardiotoxicity. Cancer 1973, 32, 302–314. [Google Scholar] [CrossRef]
- Singal, P.K.; Iliskovic, N. Doxorubicin-Induced Cardiomyopathy. N. Engl. J. Med. 1998, 339, 900–905. [Google Scholar] [CrossRef] [PubMed]
- Ludke, A.R.; Sharma, A.K.; Akolkar, G.; Bajpai, G.; Singal, P.K. Downregulation of Vitamin C Transporter SVCT-2 in Doxorubicin-Induced Cardiomyocyte Injury. Am. J. Physiol.-Cell Physiol. 2012, 303, C645–C653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singal, P.; Li, T.; Kumar, D.; Danelisen, I.; Iliskovic, N. Adriamycin-Induced Heart Failure: Mechanisms and Modulation. Mol. Cell. Biochem. 2000, 207, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Akolkar, G.; Bagchi, A.K.; Ayyappan, P.; Jassal, D.S.; Singal, P.K. Doxorubicin-Induced Nitrosative Stress Is Mitigated by Vitamin C via the Modulation of Nitric Oxide Synthases. Am. J. Physiol.-Cell Physiol. 2017, 312, C418–C427. [Google Scholar] [CrossRef] [Green Version]
- Bagchi, A.K.; Malik, A.; Akolkar, G.; Zimmer, A.; Belló-Klein, A.; De Angelis, K.; Jassal, D.S.; Fini, M.A.; Stenmark, K.R.; Singal, P.K. Study of ER Stress and Apoptotic Proteins in the Heart and Tumor Exposed to Doxorubicin. Biochim. Biophys. Acta BBA Mol. Cell Res. 2021, 1868, 119039. [Google Scholar] [CrossRef]
- Gaut, J.R.; Hendershot, L.M. The Modification and Assembly of Proteins in the Endoplasmic Reticulum. Curr. Opin. Cell Biol. 1993, 5, 589–595. [Google Scholar] [CrossRef]
- Krebs, J.; Agellon, L.B.; Michalak, M. Ca(2 +) Homeostasis and Endoplasmic Reticulum (ER) Stress: An Integrated View of Calcium Signaling. Biochem. Biophys. Res. Commun. 2015, 460, 114–121. [Google Scholar] [CrossRef]
- Kaufman, R.J. Orchestrating the Unfolded Protein Response in Health and Disease. J. Clin. Investig. 2002, 110, 1389–1398. [Google Scholar] [CrossRef]
- Hetz, C.; Papa, F.R. The Unfolded Protein Response and Cell Fate Control. Mol. Cell 2018, 69, 169–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walter, P.; Ron, D. The Unfolded Protein Response: From Stress Pathway to Homeostatic Regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [Green Version]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic Interaction of BiP and ER Stress Transducers in the Unfolded-Protein Response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein Translation and Folding Are Coupled by an Endoplasmic-Reticulum-Resident Kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Rozpedek, W.; Pytel, D.; Mucha, B.; Leszczynska, H.; Diehl, J.A.; Majsterek, I. The Role of the PERK/EIF2α/ATF4/CHOP Signaling Pathway in Tumor Progression During Endoplasmic Reticulum Stress. Curr. Mol. Med. 2016, 16, 533–544. [Google Scholar] [CrossRef]
- Wang, X.Z.; Harding, H.P.; Zhang, Y.; Jolicoeur, E.M.; Kuroda, M.; Ron, D. Cloning of Mammalian Ire1 Reveals Diversity in the ER Stress Responses. EMBO J. 1998, 17, 5708–5717. [Google Scholar] [CrossRef] [Green Version]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of Stress in the ER to Activation of JNK Protein Kinases by Transmembrane Protein Kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 MRNA Is Induced by ATF6 and Spliced by IRE1 in Response to ER Stress to Produce a Highly Active Transcription Factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef] [Green Version]
- Glembotski, C.C. Roles for ATF6 and the Sarco/Endoplasmic Reticulum Protein Quality Control System in the Heart. J. Mol. Cell. Cardiol. 2014, 71, 11–15. [Google Scholar] [CrossRef] [Green Version]
- Zinszner, H.; Kuroda, M.; Wang, X.; Batchvarova, N.; Lightfoot, R.T.; Remotti, H.; Stevens, J.L.; Ron, D. CHOP Is Implicated in Programmed Cell Death in Response to Impaired Function of the Endoplasmic Reticulum. Genes Dev. 1998, 12, 982–995. [Google Scholar] [CrossRef]
- Nakagawa, T.; Zhu, H.; Morishima, N.; Li, E.; Xu, J.; Yankner, B.A.; Yuan, J. Caspase-12 Mediates Endoplasmic-Reticulum-Specific Apoptosis and Cytotoxicity by Amyloid-Beta. Nature 2000, 403, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, T.; Imaizumi, K.; Oono, K.; Yui, D.; Gomi, F.; Katayama, T.; Tohyama, M. Activation of Caspase-12, an Endoplastic Reticulum (ER) Resident Caspase, through Tumor Necrosis Factor Receptor-Associated Factor 2-Dependent Mechanism in Response to the ER Stress. J. Biol. Chem. 2001, 276, 13935–13940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szegezdi, E.; Fitzgerald, U.; Samali, A. Caspase-12 and ER-Stress-Mediated Apoptosis: The Story so Far. Ann. N. Y. Acad. Sci. 2003, 1010, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Bagchi, A.K.; Malik, A.; Akolkar, G.; Jassal, D.S.; Singal, P.K. Endoplasmic Reticulum Stress Promotes INOS/NO and Influences Inflammation in the Development of Doxorubicin-Induced Cardiomyopathy. Antioxidants 2021, 10, 1897. [Google Scholar] [CrossRef]
- Yarmohammadi, F.; Rezaee, R.; Haye, A.W.; Karimi, G. Endoplasmic reticulum stress in doxorubicin-induced cardiotoxicity may be therapeutically targeted by natural and chemical compounds: A review. Pharmacol. Res. 2021, 164, 105383. [Google Scholar] [CrossRef]
- Bagchi, A.K.; Akolkar, G.; Mandal, S.; Ayyappan, P.; Yang, X.; Singal, P.K. Toll-like Receptor 2 Dominance over Toll-like Receptor 4 in Stressful Conditions for Its Detrimental Role in the Heart. Am. J. Physiol.-Heart Circ. Physiol. 2017, 312, H1238–H1247. [Google Scholar] [CrossRef] [Green Version]
- Bagchi, A.K.; Surendran, A.; Malik, A.; Jassal, D.S.; Ravandi, A.; Singal, P.K. IL-10 Attenuates OxPCs-Mediated Lipid Metabolic Responses in Ischemia Reperfusion Injury. Sci. Rep. 2020, 10, 12120. [Google Scholar] [CrossRef]
- Verma, S.K.; Krishnamurthy, P.; Barefield, D.; Singh, N.; Gupta, R.; Lambers, E.; Thal, M.; Mackie, A.; Hoxha, E.; Ramirez, V.; et al. Interleukin-10 Treatment Attenuates Pressure Overload–Induced Hypertrophic Remodeling and Improves Heart Function via Signal Transducers and Activators of Transcription 3–Dependent Inhibition of Nuclear Factor-ΚB. Circulation 2012, 126, 418–429. [Google Scholar] [CrossRef] [Green Version]
- O’Garra, A.; Barrat, F.J.; Castro, A.G.; Vicari, A.; Hawrylowicz, C. Strategies for Use of IL-10 or Its Antagonists in Human Disease. Immunol. Rev. 2008, 223, 114–131. [Google Scholar] [CrossRef]
- Krishnamurthy, P.; Rajasingh, J.; Lambers, E.; Qin, G.; Losordo, D.W.; Kishore, R. IL-10 Inhibits Inflammation and Attenuates Left Ventricular Remodeling after Myocardial Infarction via Activation of STAT3 and Suppression of HuR. Circ. Res. 2009, 104, e9–e18. [Google Scholar] [CrossRef]
- Sishi, B.J.N.; Loos, B.; van Rooyen, J.; Engelbrecht, A.-M. Doxorubicin Induces Protein Ubiquitination and Inhibits Proteasome Activity during Cardiotoxicity. Toxicology 2013, 309, 23–29. [Google Scholar] [CrossRef]
- Cappetta, D.; De Angelis, A.; Sapio, L.; Prezioso, L.; Illiano, M.; Quaini, F.; Rossi, F.; Berrino, L.; Naviglio, S.; Urbanek, K. Oxidative Stress and Cellular Response to Doxorubicin: A Common Factor in the Complex Milieu of Anthracycline Cardiotoxicity. Oxid. Med. Cell. Longev. 2017, 2017, e1521020. [Google Scholar] [CrossRef] [PubMed]
- Wenningmann, N.; Knapp, M.; Ande, A.; Vaidya, T.R.; Ait-Oudhia, S. Insights into Doxorubicin-Induced Cardiotoxicity: Molecular Mechanisms, Preventive Strategies, and Early Monitoring. Mol. Pharmacol. 2019, 96, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Kaur, K.; Sharma, A.K.; Singal, P.K. Significance of Changes in TNF-α and IL-10 Levels in the Progression of Heart Failure Subsequent to Myocardial Infarction. Am. J. Physiol.-Heart Circ. Physiol. 2006, 291, H106–H113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhingra, S.; Sharma, A.K.; Arora, R.C.; Slezak, J.; Singal, P.K. IL-10 Attenuates TNF-α-Induced NFκB Pathway Activation and Cardiomyocyte Apoptosis. Cardiovasc. Res. 2009, 82, 59–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagchi, A.K.; Sharma, A.; Dhingra, S.; Lehenbauer Ludke, A.R.; Al-Shudiefat, A.A.-R.; Singal, P.K. Interleukin-10 Activates Toll-like Receptor 4 and Requires MyD88 for Cardiomyocyte Survival. Cytokine 2013, 61, 304–314. [Google Scholar] [CrossRef]
- Steen, E.H.; Wang, X.; Balaji, S.; Butte, M.J.; Bollyky, P.L.; Keswani, S.G. The Role of the Anti-Inflammatory Cytokine Interleukin-10 in Tissue Fibrosis. Adv. Wound Care 2020, 9, 184–198. [Google Scholar] [CrossRef] [Green Version]
- Samanta, A.; Dawn, B. IL-10 for Cardiac Autophagy Modulation: New Direction in the Pursuit of Perfection. J. Mol. Cell. Cardiol. 2016, 91, 204–206. [Google Scholar] [CrossRef] [Green Version]
- Oslowski, C.M.; Urano, F. Measuring ER Stress and the Unfolded Protein Response Using Mammalian Tissue Culture System. Methods Enzymol. 2011, 490, 71–92. [Google Scholar] [CrossRef] [Green Version]
- Tay, K.H.; Luan, Q.; Croft, A.; Jiang, C.C.; Jin, L.; Zhang, X.D.; Tseng, H.-Y. Sustained IRE1 and ATF6 Signaling Is Important for Survival of Melanoma Cells Undergoing ER Stress. Cell. Signal. 2014, 26, 287–294. [Google Scholar] [CrossRef]
- Chen, S.; Henderson, A.; Petriello, M.C.; Romano, K.A.; Gearing, M.; Miao, J.; Schell, M.; Sandoval-Espinola, W.J.; Tao, J.; Sha, B.; et al. Trimethylamine N-Oxide Binds and Activates PERK to Promote Metabolic Dysfunction. Cell Metab. 2019, 30, 1141–1151.e5. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Fu, Y.; Cai, Z.; Yu, F.; Gong, Z.; Dai, R.; Hu, Y.; Zeng, L.; Xu, Q.; Kong, W. Unspliced XBP1 Confers VSMC Homeostasis and Prevents Aortic Aneurysm Formation via FoxO4 Interaction. Circ. Res. 2017, 121, 1331–1345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, H.; Oku, M.; Suzuki, M.; Mori, K. PXBP1(U) Encoded in XBP1 Pre-MRNA Negatively Regulates Unfolded Protein Response Activator PXBP1(S) in Mammalian ER Stress Response. J. Cell Biol. 2006, 172, 565–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tirosh, B.; Iwakoshi, N.N.; Glimcher, L.H.; Ploegh, H.L. Rapid Turnover of Unspliced Xbp-1 as a Factor That Modulates the Unfolded Protein Response. J. Biol. Chem. 2006, 281, 5852–5860. [Google Scholar] [CrossRef] [Green Version]
- Hitomi, J.; Katayama, T.; Taniguchi, M.; Honda, A.; Imaizumi, K.; Tohyama, M. Apoptosis Induced by Endoplasmic Reticulum Stress Depends on Activation of Caspase-3 via Caspase-12. Neurosci. Lett. 2004, 357, 127–130. [Google Scholar] [CrossRef] [Green Version]
- Verma, G.; Datta, M. IL-1beta Induces ER Stress in a JNK Dependent Manner That Determines Cell Death in Human Pancreatic Epithelial MIA PaCa-2 Cells. Apoptosis Int. J. Program. Cell Death 2010, 15, 864–876. [Google Scholar] [CrossRef]
- Zhu, J.; Zhang, J.; Zhang, L.; Du, R.; Xiang, D.; Wu, M.; Zhang, R.; Han, W. Interleukin-1 Signaling Mediates Acute Doxorubicin-Induced Cardiotoxicity. Biomed. Pharmacother. Biomed. Pharmacother. 2011, 65, 481–485. [Google Scholar] [CrossRef]
- Kelly, A.; Lynch, A.; Vereker, E.; Nolan, Y.; Queenan, P.; Whittaker, E.; O’Neill, L.A.; Lynch, M.A. The Anti-Inflammatory Cytokine, Interleukin (IL)-10, Blocks the Inhibitory Effect of IL-1 Beta on Long Term Potentiation. A Role for JNK. J. Biol. Chem. 2001, 276, 45564–45572. [Google Scholar] [CrossRef] [Green Version]
- Bagchi, A.K.; Malik, A.; Akolkar, G.; Bello-Klein, A.; Khaper, N.; Singal, P.K. IL-10: A key molecule in the mitigation of heart failure. In Biomedical Translational Research Volume 2: Disease Diagnosis to Treatment; Sobti, R.C., Krishan, A., Eds.; Springer: Berlin/Heidelberg, Germany, 2022; in press. [Google Scholar]
- Hansen, I.S.; Schoonejans, J.M.; Sritharan, L.; van Burgsteden, J.A.; Ambarus, C.A.; Baeten, D.L.P.; den Dunnen, J. ER stress abrogates the immunosuppressive effect of IL-10 on human macrophages through inhibition of STAT3 activation. Inflamm. Res. 2019, 68, 775–785. [Google Scholar] [CrossRef] [Green Version]
- Marra, L.E.; Zhang, Z.X.; Joe, B.; Campbell, J.; Levy, G.A.; Penninger, J.; Zhang, L. IL-10 Induces Regulatory T Cell Apoptosis by Up-Regulation of the Membrane Form of TNF-α. J. Immunol. 2004, 172, 1028–1035. [Google Scholar] [CrossRef] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malik, A.; Bagchi, A.K.; Jassal, D.S.; Singal, P.K. Interleukin-10 Mitigates Doxorubicin-Induced Endoplasmic Reticulum Stress as Well as Cardiomyopathy. Biomedicines 2022, 10, 890. https://doi.org/10.3390/biomedicines10040890
Malik A, Bagchi AK, Jassal DS, Singal PK. Interleukin-10 Mitigates Doxorubicin-Induced Endoplasmic Reticulum Stress as Well as Cardiomyopathy. Biomedicines. 2022; 10(4):890. https://doi.org/10.3390/biomedicines10040890
Chicago/Turabian StyleMalik, Akshi, Ashim K. Bagchi, Davinder S. Jassal, and Pawan K. Singal. 2022. "Interleukin-10 Mitigates Doxorubicin-Induced Endoplasmic Reticulum Stress as Well as Cardiomyopathy" Biomedicines 10, no. 4: 890. https://doi.org/10.3390/biomedicines10040890