
Inhibitory Effects of Astaxanthin on CML-HSA-Induced Inflammatory and RANKL-Induced Osteoclastogenic Gene Expression in RAW 264.7 Cells



Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Chemicals
2.2. Cell Culture
2.3. Cell Cytotoxicity
2.4. TNFα Measurement by ELISA
2.5. In Vitro Osteoclastogenesis
2.6. TRAP Activity
2.7. TRAP Staining
2.8. Isolation of Total RNA and RT-PCR
2.9. F-Actin Ring Formation
2.10. Immunofluorescence Assay
2.11. Statistical Analysis
3. Results
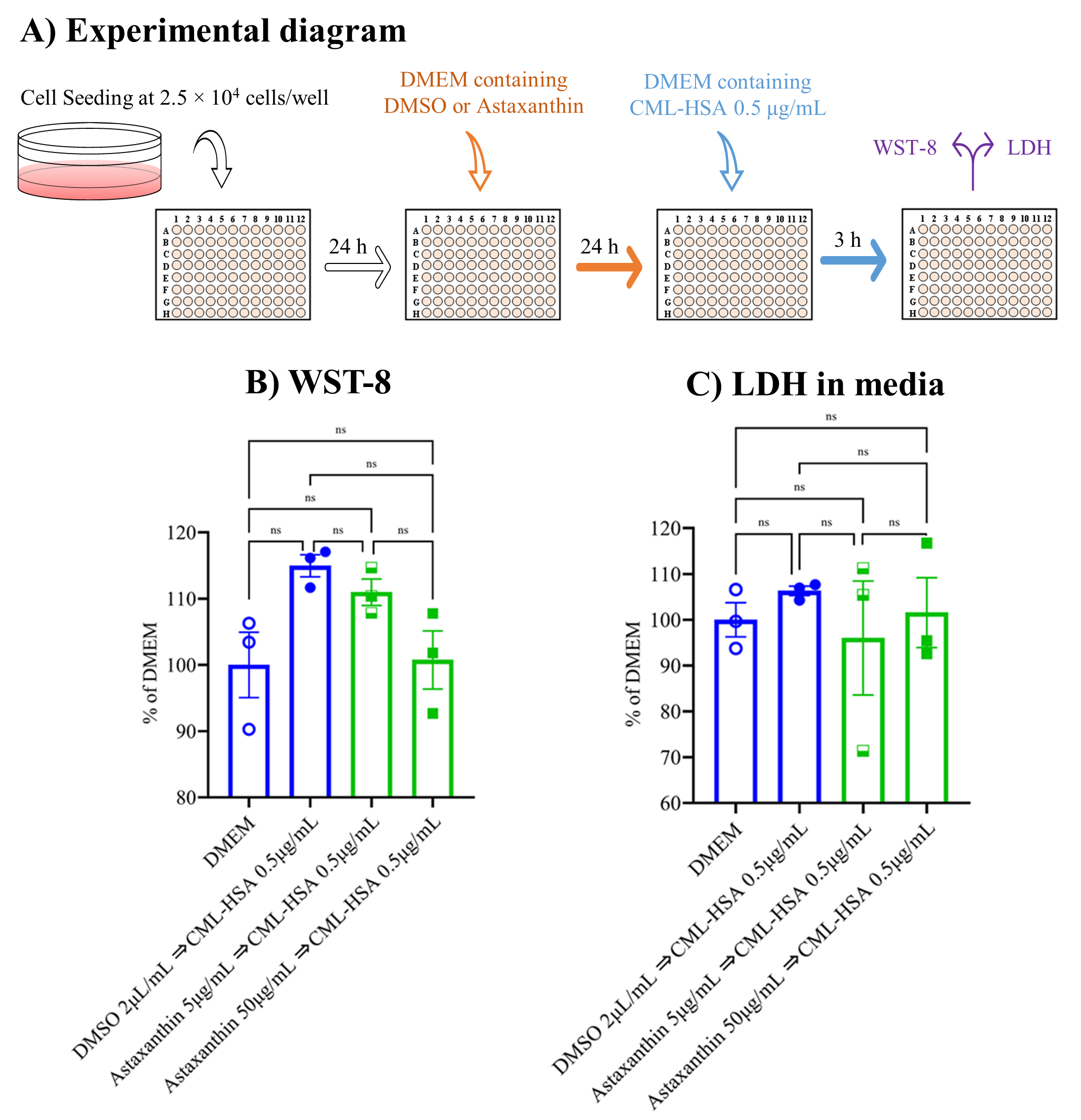
3.1. Neither Astaxanthin nor CML-HSA Produced Any Cytotoxicity
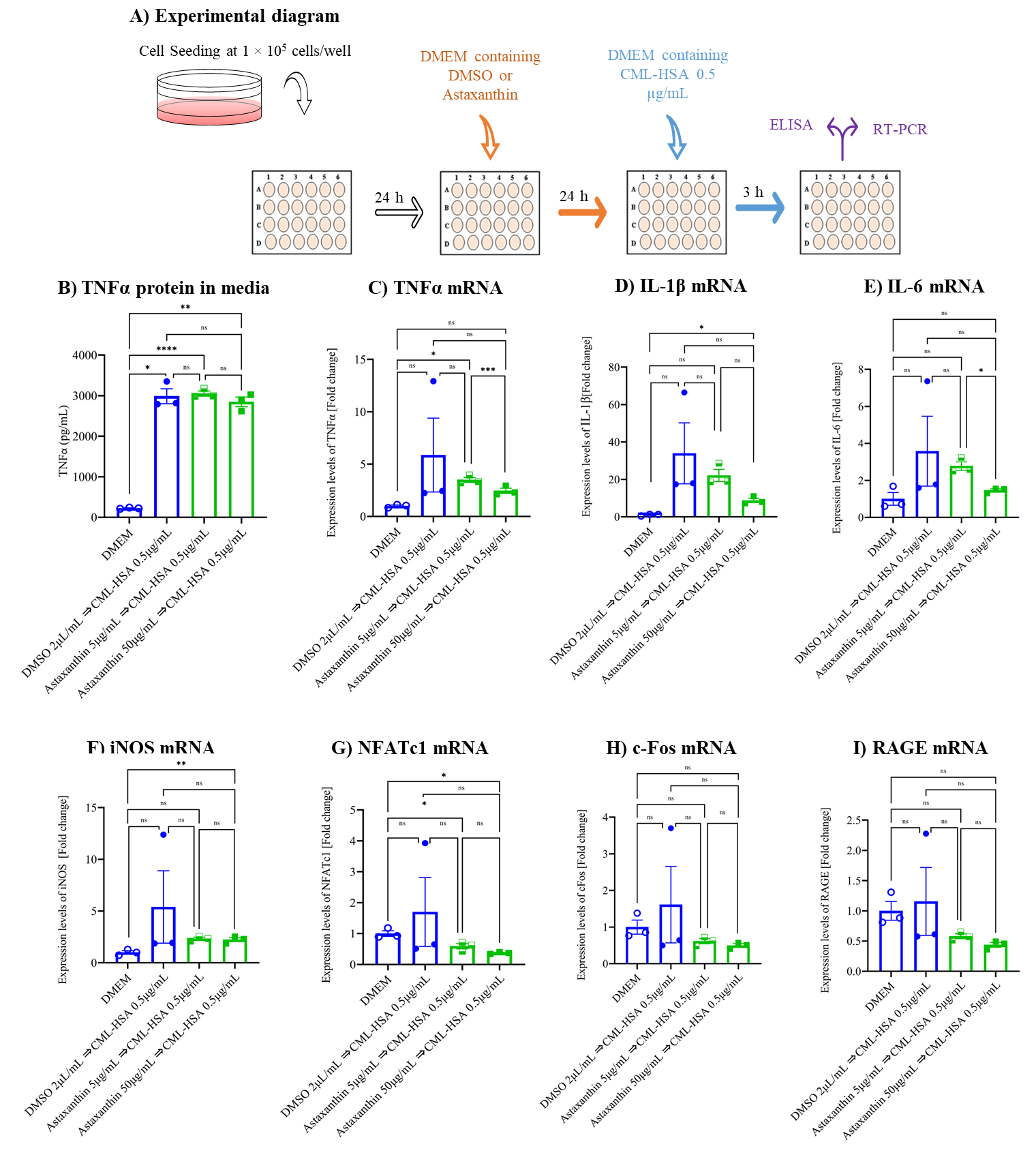
3.2. Astaxanthin Inhibits CML-HSA-Induced Inflammation and Autoinflammation in Cell Culture Model
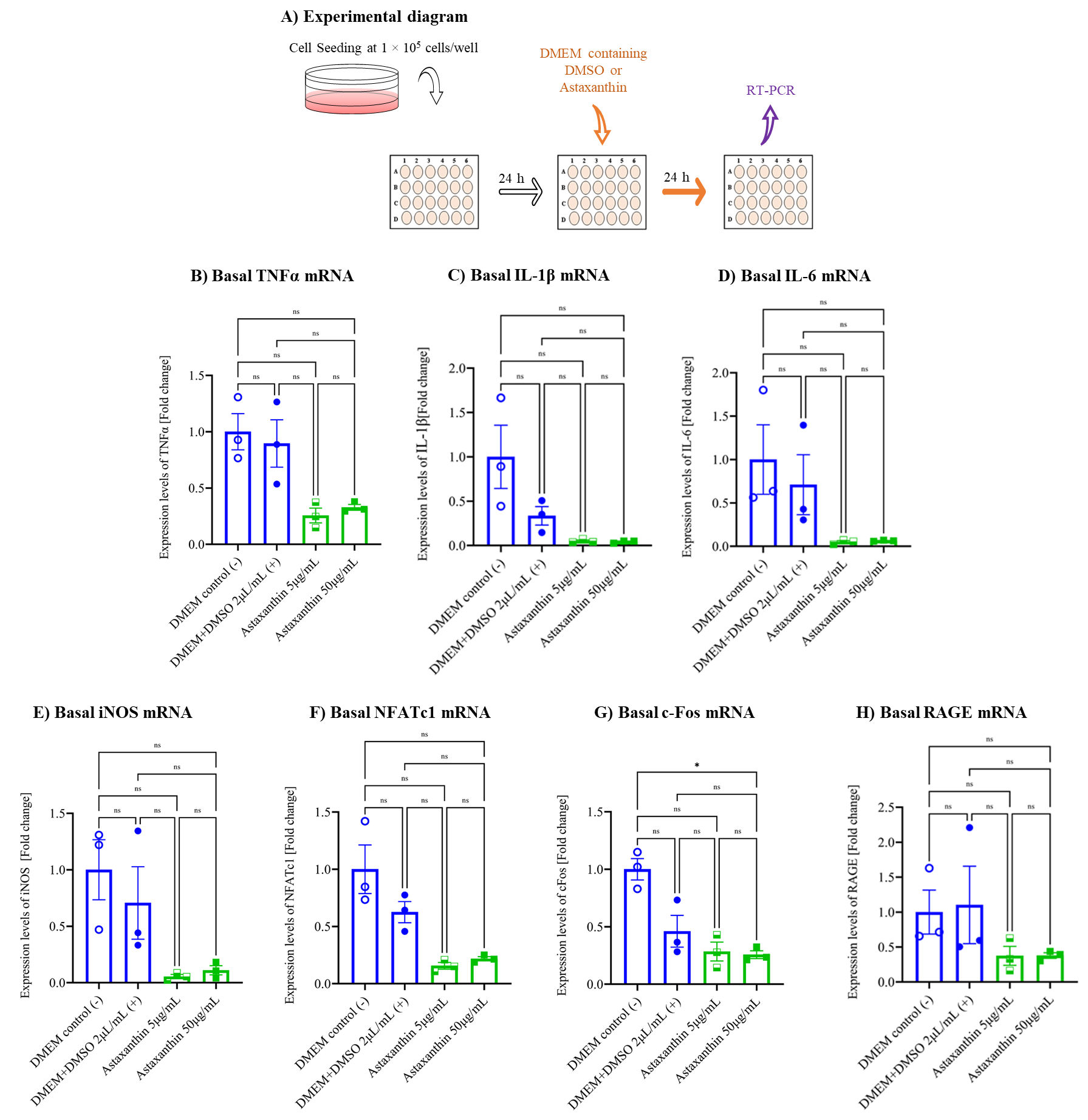
3.3. Astaxanthin Inhibits Autoinflammation in the Cell Culture Model
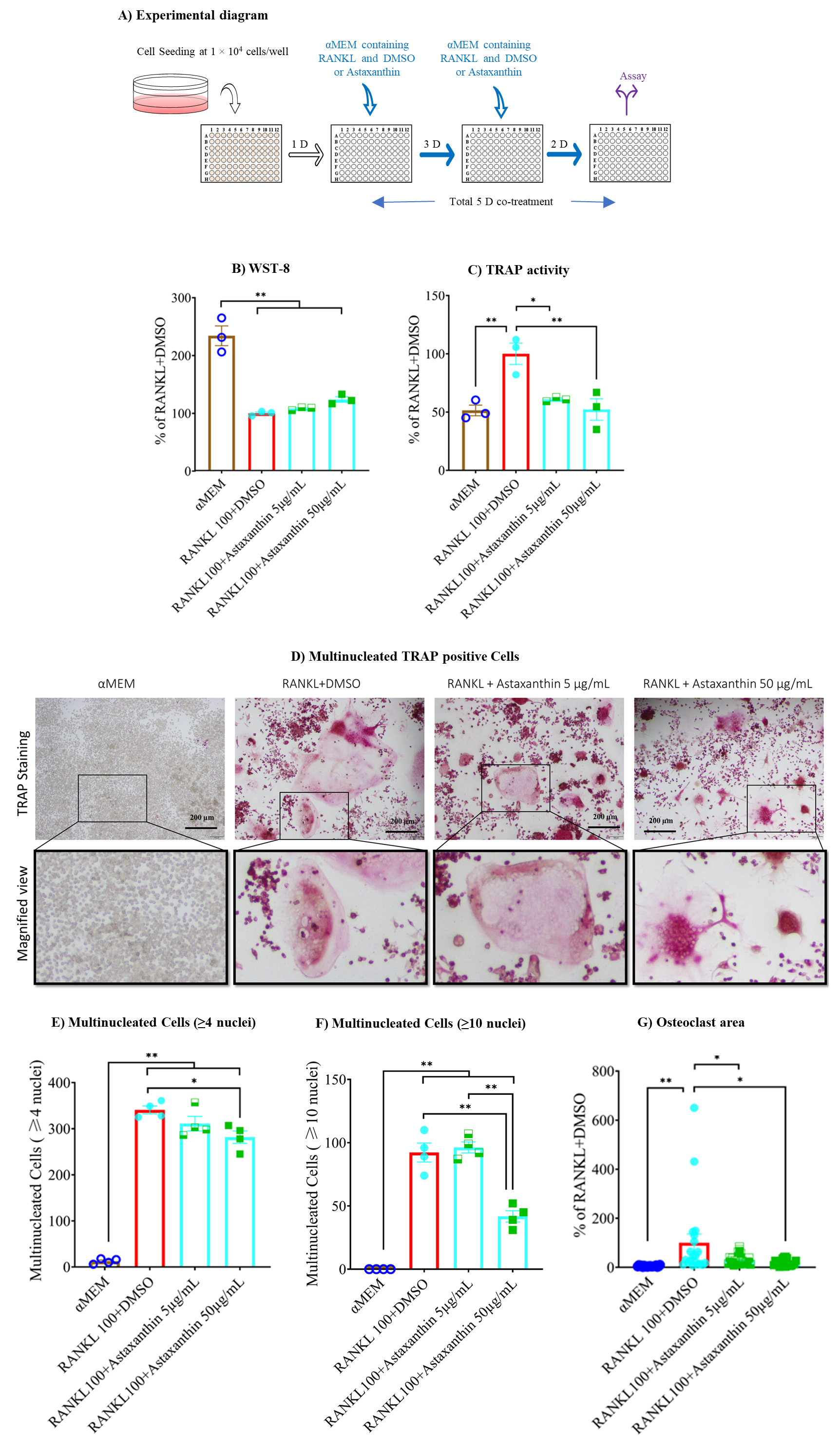
3.4. Astaxanthin Inhibits TRAP Activity without Altering Cell Viability
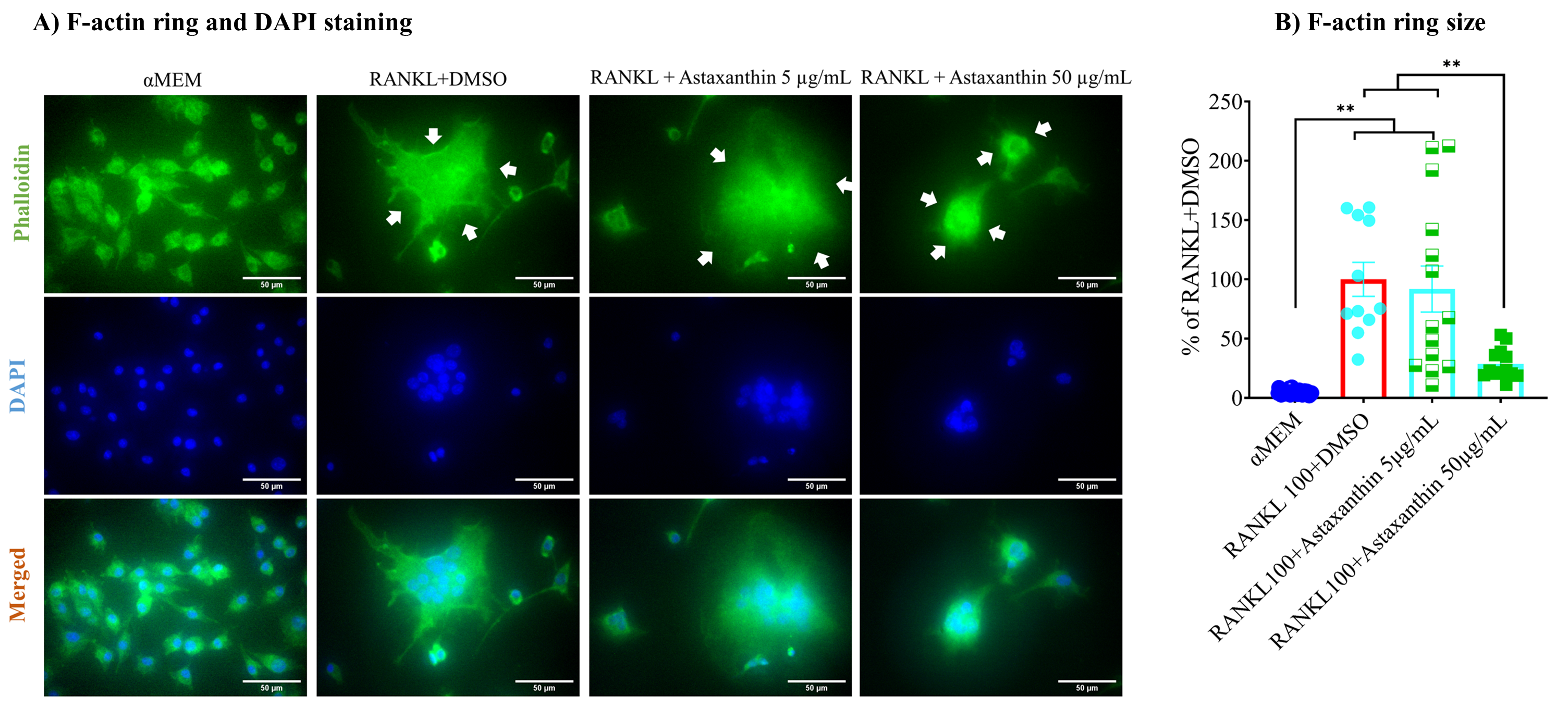
3.5. F-Actin Ring Size Was Reduced by Astaxanthin
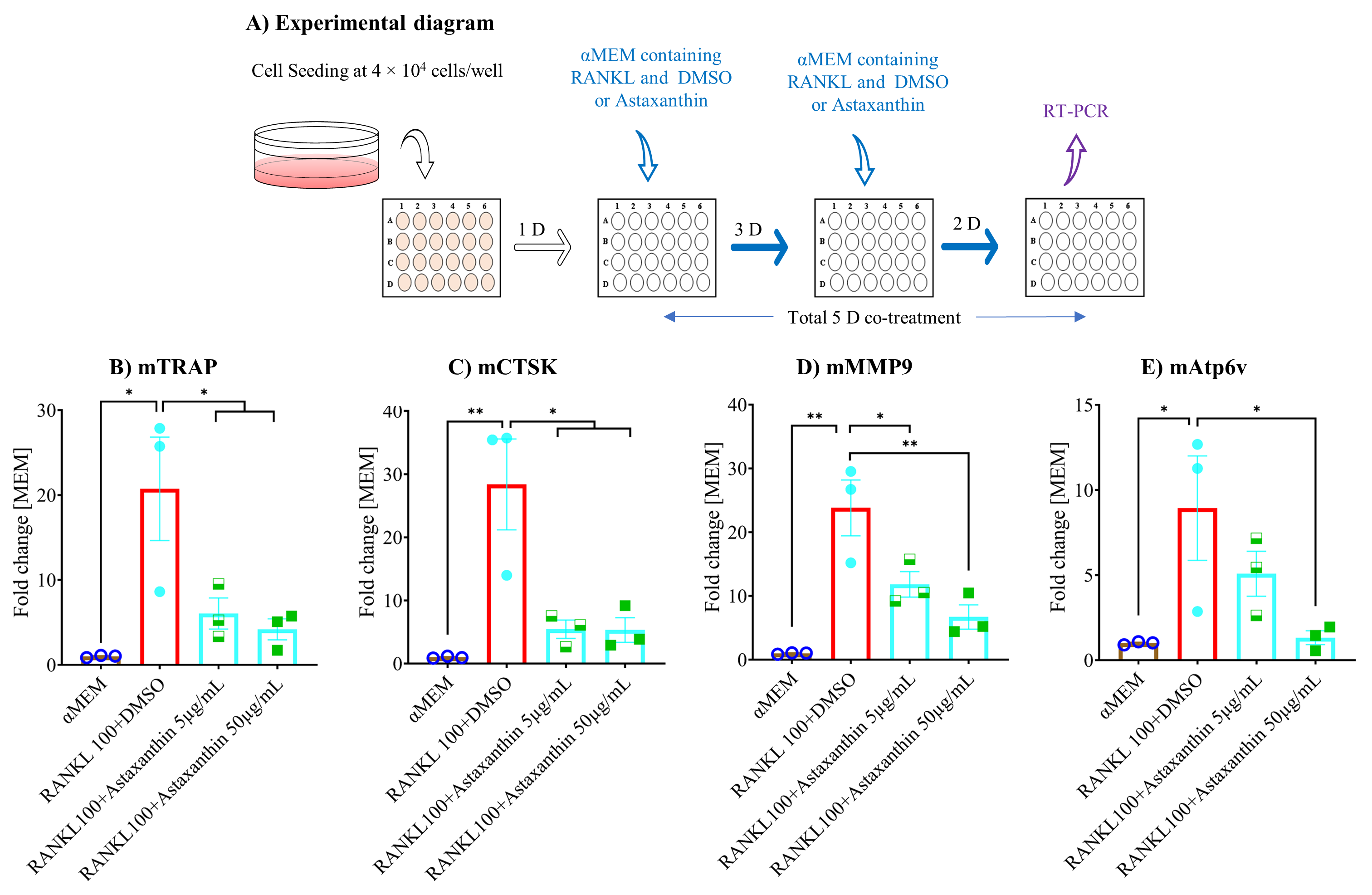
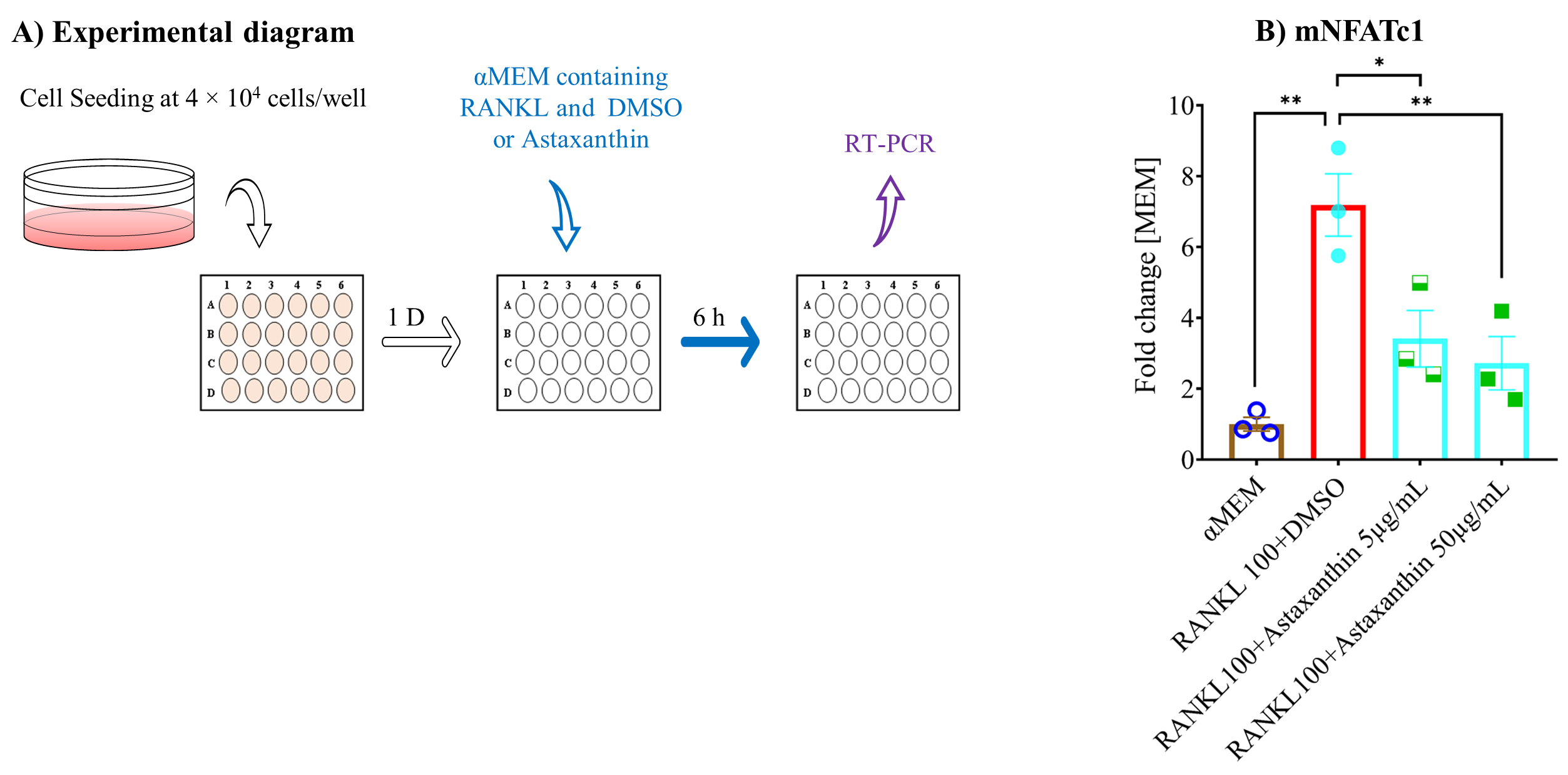
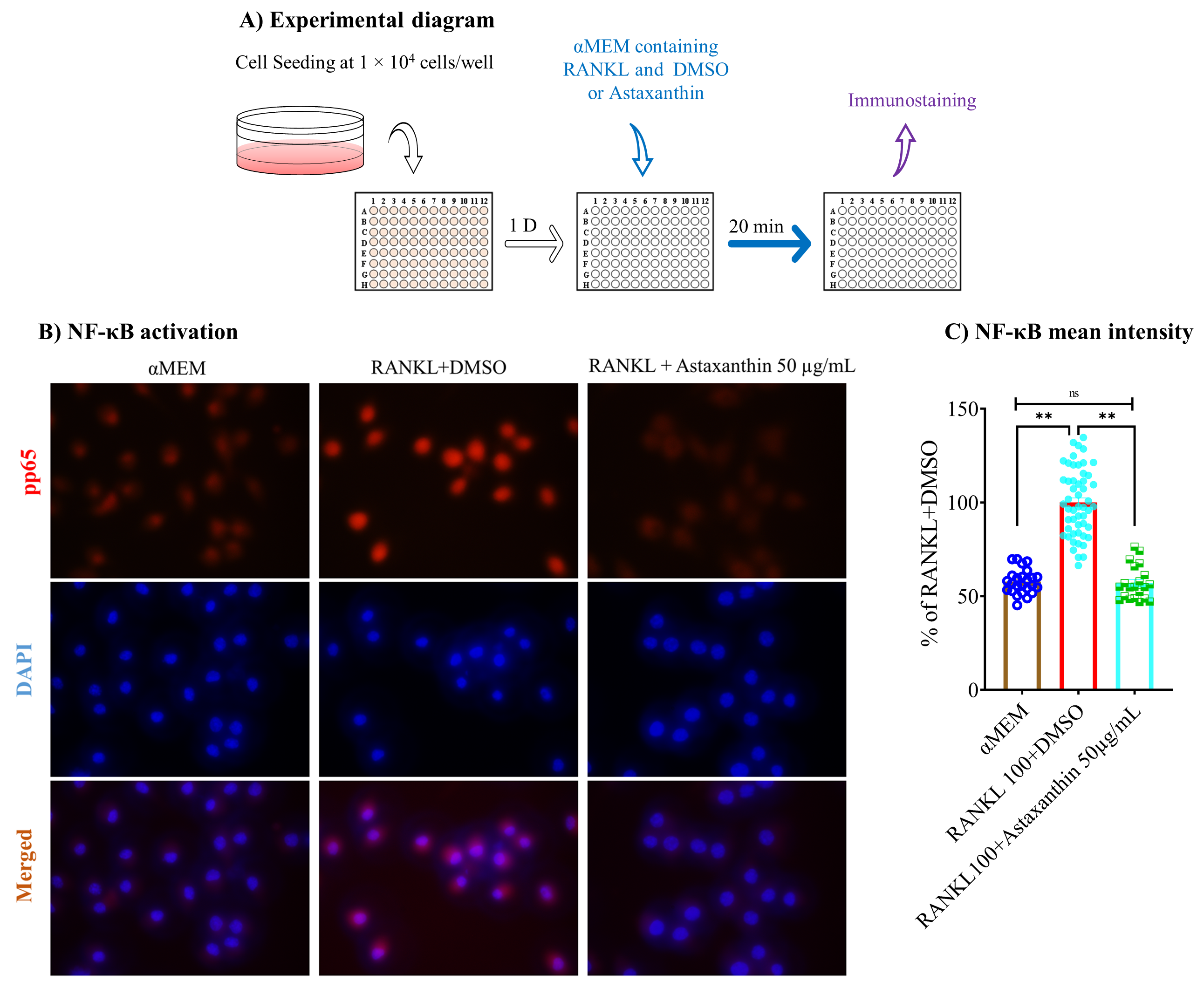
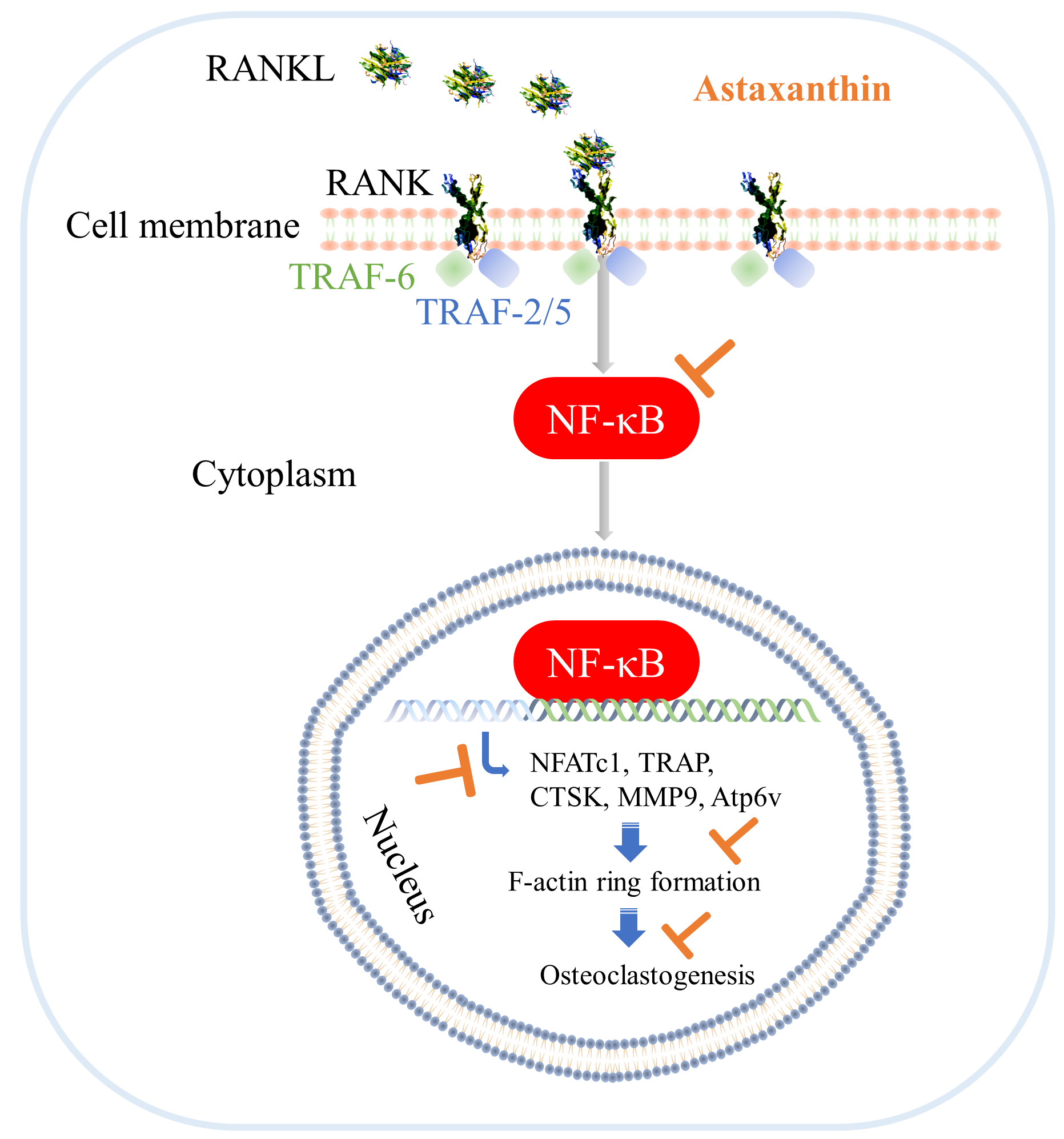
3.6. Astaxanthin Inhibits Osteoclastogenic Gene Expression and NF-κB Pathway Activation
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Montalcini, T.; Romeo, S.; Ferro, Y.; Migliaccio, V.; Gazzaruso, C.; Pujia, A. Osteoporosis in chronic inflammatory disease: The role of malnutrition. Endocrine 2013, 43, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Chabaud, M.; Durand, J.M.; Buchs, N.; Page, G.; Frappart, L.; Miossec, P. Human interleukin-17: A T cell-derived proinflammatory cytokine produced by the rheumatoid synovium. Arthritis Rheumatol. 1999, 42, 963–970. [Google Scholar] [CrossRef]
- Belenska-Todorova, L.; Lambova, S.N.; Stoyanova, S.; Georgieva, E.; Batsalova, T.; Moten, D.; Kolchakova, D.; Dzhambazov, B. Disease-modifying potential of metformin and alendronate in an experimental mouse model of osteoarthritis. Biomedicines 2021, 9, 1017. [Google Scholar] [CrossRef] [PubMed]
- Gemmell, E.; Marshall, R.I.; Seymour, G.J. Cytokines and prostaglandins in immune homeostasis and tissue destruction in periodontal disease. Periodontology 2000 1997, 14, 112–143. [Google Scholar] [CrossRef] [PubMed]
- Alblowi, J.; Kayal, R.A.; Siqueria, M.; McKenzie, E.; Krothapalli, N.; McLean, J.; Conn, J.; Nikolajczyk, B.; Einhorn, T.A.; Gerstenfeld, L.; et al. High levels of tumor necrosis factor-α contribute to accelerated loss of cartilage in diabetic fracture healing. Am. J. Pathol. 2009, 175, 1574–1585. [Google Scholar] [CrossRef] [Green Version]
- Glantschnig, H.; Fisher, J.E.; Wesolowski, G.; Rodan, G.A.; Reszka, A.A. M-CSF, TNFα and RANK ligand promote osteoclast survival by signaling through mTOR/S6 kinase. Cell Death Differ. 2003, 10, 1165–1177. [Google Scholar] [CrossRef] [Green Version]
- Luo, G.; Li, F.; Li, X.; Wang, Z.G.; Zhang, B. TNF-α and RANKL promote osteoclastogenesis by upregulating RANK via the NF-κB pathway. Mol. Med. Rep. 2018, 17, 6605–6611. [Google Scholar] [CrossRef] [Green Version]
- Wei, S.; Kitaura, H.; Zhou, P.; Patrick Ross, F.; Teitelbaum, S.L. IL-1 mediates TNF-induced osteoclastogenesis. J. Clin. Investig. 2005, 115, 282–290. [Google Scholar] [CrossRef] [Green Version]
- Hee Lee, Z.; Eun Lee, S.; Kim, C.-W.; Ho Lee, S.; Kim, S.W.; Kwack, K.; Walsh, K.; Kim, H.-H. IL-lα stimulation of osteoclast survival through the PI 3-Kinase/Akt and ERK Pathways 1. J. Biochem. 2002, 131, 161–166. [Google Scholar] [CrossRef]
- Chen, Z.; Su, L.; Xu, Q.; Katz, J.; Michalek, S.M.; Fan, M.; Feng, X.; Zhang, P. IL-1R/TLR2 through MyD88 divergently modulates osteoclastogenesis through regulation of nuclear factor of activated T cells c1 (NFATc1) and B lymphocyte-induced maturation protein-1 (Blimp1). J. Biol. Chem. 2015, 290, 30163–30174. [Google Scholar] [CrossRef] [Green Version]
- De Martinis, M.; Ginaldi, L.; Sirufo, M.M.; Pioggia, G.; Calapai, G.; Gangemi, S.; Mannucci, C. Alarmins in osteoporosis, RAGE, IL-1, and IL-33 pathways: A literature review. Medicina 2020, 56, 138. [Google Scholar] [CrossRef] [Green Version]
- Kany, S.; Vollrath, J.T.; Relja, B. Cytokines in inflammatory disease. Int. J. Mol. Sci. 2019, 20, 6008. [Google Scholar] [CrossRef] [Green Version]
- Lechner, J.; Rudi, T.; Von Baehr, V. Osteoimmunology of tumor necrosis factor-alpha, IL-6, and RANTES/CCL5: A review of known and poorly understood inflammatory patterns in osteonecrosis. Clin. Cosmet. Investig. Dent. 2018, 10, 251–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokota, K.; Sato, K.; Miyazaki, T.; Aizaki, Y.; Tanaka, S.; Sekikawa, M.; Kozu, N.; Kadono, Y.; Oda, H.; Mimura, T. Characterization and function of tumor necrosis factor and Interleukin-6–induced osteoclasts in rheumatoid arthritis. Arthritis Rheumatol. 2021, 73, 1145–1154. [Google Scholar] [CrossRef]
- Ascone, G.; Cao, Y.; Jansen, I.D.C.; Di Ceglie, I.; Van Den Bosch, M.H.J.; Blom, A.B.; Van Lent, P.L.E.M.; Everts, V.; De Vries, T.J. Increase in the number of bone marrow osteoclast precursors at different skeletal sites, particularly in long bone and Jaw Marrow in mice lacking IL-1RA. Int. J. Mol. Sci. 2020, 21, 3774. [Google Scholar] [CrossRef] [PubMed]
- Gravallese, E.M.; Goldring, S.R. Cellular mechanisms and the role of cytokines in bone erosions in rheumatoid arthritis. Arthritis Rheum. 2000, 43, 2143–2151. [Google Scholar] [CrossRef]
- Marahleh, A.; Kitaura, H.; Ohori, F.; Kishikawa, A.; Ogawa, S.; Shen, W.R.; Qi, J.; Noguchi, T.; Nara, Y.; Mizoguchi, I. TNF-α directly enhances osteocyte rankl expression and promotes osteoclast formation. Front. Immunol. 2019, 10, 2925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Y.; Jansen, I.D.C.; Sprangers, S.; De Vries, T.J.; Everts, V. TNF-α has both stimulatory and inhibitory effects on mouse monocyte-derived osteoclastogenesis. J. Cell. Physiol. 2017, 232, 3273–3285. [Google Scholar] [CrossRef] [Green Version]
- O’Gradaigh, D.; Ireland, D.; Bord, S.; Compston, J.E. Joint erosion in rheumatoid arthritis: Interactions between tumour necrosis factor α, interleukin 1, and receptor activator of nuclear factor κB ligand (RANKL) regulate osteoclasts. Ann. Rheum. Dis. 2004, 63, 354–359. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, K.; Takahashi, N.; Jimi, E.; Udagawa, N.; Takami, M.; Kotake, S.; Nakagawa, N.; Kinosaki, M.; Yamaguchi, K.; Shima, N.; et al. Tumor necrosis factor alpha stimulates osteoclast differentiation by a mechanism independent of the ODF/RANKL-RANK interaction. J. Exp. Med. 2000, 191, 275–286. [Google Scholar] [CrossRef]
- AlQranei, M.S.; Senbanjo, L.T.; Aljohani, H.; Hamza, T.; Chellaiah, M.A. Lipopolysaccharide- TLR-4 Axis regulates Osteoclastogenesis independent of RANKL/RANK signaling. BMC Immunol. 2021, 22, 23. [Google Scholar] [CrossRef]
- Tanaka, U.; Kajioka, S.; Finoti, L.S.; Palioto, D.B.; Kinane, D.F.; Benakanakere, M.R. Decitabine inhibits bone resorption in periodontitis by upregulating anti-inflammatory cytokines and suppressing osteoclastogenesis. Biomedicines 2021, 9, 199. [Google Scholar] [CrossRef]
- Cao, X. RANKL-RANK signaling regulates osteoblast differentiation and bone formation. Bone Res. 2018, 6, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gugliucci, A.; Bendayan, M. Reaction of advanced glycation endproducts with renal tissue from normal and streptozotocin-induced diabetic rats: An ultrastructural study using colloidal gold cytochemistry. J. Histochem. Cytochem. 1995, 43, 591–600. [Google Scholar] [CrossRef]
- Yamagishi, S. Role of advanced glycation end products (AGEs) in osteoporosis in diabetes. Curr. Drug Targets 2011, 12, 2096–2102. [Google Scholar] [CrossRef] [PubMed]
- Gugliucci, A. Formation of fructose-mediated advanced glycation end products and their roles in metabolic and inflammatory diseases. Adv. Nutr. 2017, 8, 54–62. [Google Scholar] [CrossRef] [Green Version]
- Goh, S.Y.; Cooper, M.E. The role of advanced glycation end products in progression and complications of diabetes. J. Clin. Endocrinol. Metab. 2008, 93, 1143–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vadakedath, S.; Kandi, V. Role of advanced glycation end products (AGE) in health and disease: An overview. Biochem. Physiol. 2018, 7, 246. [Google Scholar] [CrossRef]
- Yamagishi, S.-I. Role of advanced glycation end products (AGEs) and receptor for AGEs (RAGE) in vascular damage in diabetes. Exp. Gerontol. 2011, 46, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Miyata, T.; Hori, O.; Zhang, J.H.; Yan, S.D.; Ferran, L.; Iida, Y.; Schmidt, A.M. The receptor for advanced glycation end products (RAGE) is a central mediator of the interaction of AGE-β2microglobulin with human mononuclear phagocytes via an oxidant-sensitive pathway: Implications for the pathogenesis of dialysis-related amyloidosis. J. Clin. Investig. 1996, 98, 1088–1094. [Google Scholar] [CrossRef]
- Monnier, V.M.; Taniguchi, N. Advanced glycation in diabetes, aging and age-related diseases: Editorial and dedication. Glycoconj. J. 2016, 33, 483–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baynes, J.W. The role of AGEs in aging: Causation or correlation. Exp. Gerontol. 2001, 36, 1527–1537. [Google Scholar] [CrossRef]
- Horvat, Š.; Jakas, A. Peptide and amino acid glycation: New insights into the maillard reaction. J. Pept. Sci. 2004, 10, 119–137. [Google Scholar] [CrossRef]
- Semba, R.D.; Bandinelli, S.; Sun, K.; Guralnik, J.M.; Ferrucci, L. Plasma carboxymethyl-lysine, an advanced glycation end product, and all-cause and cardiovascular disease mortality in older community-dwelling adults. J. Am. Geriatr. Soc. 2009, 57, 1874–1880. [Google Scholar] [CrossRef] [Green Version]
- Chaudhuri, J.; Bains, Y.; Guha, S.; Kahn, A.; Hall, D.; Bose, N.; Gugliucci, A.; Kapahi, P. The role of advanced glycation end products in aging and metabolic diseases: Bridging association and causality. Cell Metab. 2018, 28, 337–352. [Google Scholar] [CrossRef] [Green Version]
- Quan, W.; Jiao, Y.; Xue, C.; Li, Y.; Liu, G.; He, Z.; Qin, F.; Zeng, M.; Chen, J. The effect of exogenous free Nε-(carboxymethyl)lysine on diabetic-model Goto-Kakizaki rats: Metabolomics analysis in serum and urine. J. Agric. Food Chem. 2021, 69, 783–793. [Google Scholar] [CrossRef]
- Van Deutekom, A.W.; Niessen, H.W.M.; Schalkwijk, C.G.; Heine, R.J.; Simsek, S. Increased Nε-(carboxymethyl)-lysine levels in cerebral blood vessels of diabetic patients and in a (streptozotocin-treated) rat model of diabetes mellitus. Eur. J. Endocrinol. 2008, 158, 655–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drinda, S.; Franke, S.; Canet, C.C.; Petrow, P.; Bräuer, R.; Hüttich, C.; Stein, G.; Hein, G. Identification of the advanced glycation end products Nε-carboxymethyllysine in the synovial tissue of patients with rheumatoid arthritis. Ann. Rheum. Dis. 2002, 61, 488–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, M.; Fujii, K.; Mori, Y.; Marumo, K. Role of collagen enzymatic and glycation induced cross-links as a determinant of bone quality in spontaneously diabetic WBN/Kob rats. Osteoporos. Int. 2006, 17, 1514–1523. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Yagi, M.; Umehara, H.; Yonei, Y. Establishment of a model for evaluating tumor necrosis factor-α production by cultured RAW264.7 in response to glycation stress. Glycative Stress Res. 2014, 1, 1–7. [Google Scholar]
- Sato, K.; Yagi, M.; Takabe, W.; Yonei, Y. Inhibitory effect of plant extract on tumor necrosis factor-α formation from carboxymethyllysine stimulated macrophages. Glycative Stress Res. 2015, 2, 191–196. [Google Scholar]
- Pattanaik, S.S.; Sawant, P.B.; Xavier, K.A.M.; Dube, K.; Srivastava, P.P.; Dhanabalan, V.; Chadha, N.K. Characterization of carotenoprotein from different shrimp shell waste for possible use as supplementary nutritive feed ingredient in animal diets. Aquaculture 2020, 515, 734594. [Google Scholar] [CrossRef]
- Lin, W.C.; Chien, J.T.; Chen, B.H. Determination of carotenoids in spear shrimp shells (Parapenaeopsis hardwickii) by liquid chromatography. J. Agric. Food Chem. 2005, 53, 5144–5149. [Google Scholar] [CrossRef] [PubMed]
- Naguib, Y.M.A. Antioxidant activities of astaxanthin and related carotenoids. J. Agric. Food Chem. 2000, 48, 1150–1154. [Google Scholar] [CrossRef]
- Rao, A.R.; Sindhuja, H.N.; Dharmesh, S.M.; Sankar, K.U.; Sarada, R.; Ravishankar, G.A. Effective inhibition of skin cancer, tyrosinase, and antioxidative properties by astaxanthin and astaxanthin esters from the green alga Haematococcus pluvialis. J. Agric. Food Chem. 2013, 61, 3842–3851. [Google Scholar] [CrossRef] [PubMed]
- Kamath, B.S.; Srikanta, B.M.; Dharmesh, S.M.; Sarada, R.; Ravishankar, G.A. Ulcer preventive and antioxidative properties of astaxanthin from Haematococcus pluvialis. Eur. J. Pharmacol. 2008, 590, 387–395. [Google Scholar] [CrossRef]
- Ohgami, K.; Shiratori, K.; Kotake, S.; Nishida, T.; Mizuki, N.; Yazawa, K.; Ohno, S. Effects of astaxanthin on lipopolysaccharide-induced inflammation in vitro and in vivo. Investig. Ophthalmol. Vis. Sci. 2003, 44, 2694–2701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hama, S.; Takahashi, K.; Inai, Y.; Shiota, K.; Sakamoto, R.; Yamada, A.; Tsuchiya, H.; Kanamura, K.; Yamashita, E.; Kogure, K. Protective effects of topical application of a poorly soluble antioxidant astaxanthin liposomal formulation on ultraviolet-induced skin damage. J. Pharm. Sci. 2012, 101, 2909–2916. [Google Scholar] [CrossRef]
- Pashkow, F.J.; Watumull, D.G.; Campbell, C.L. Astaxanthin: A novel potential treatment for oxidative stress and inflammation in cardiovascular disease. Am. J. Cardiol. 2008, 101, S58–S68. [Google Scholar] [CrossRef]
- Uchiyama, K.; Naito, Y.; Hasegawa, G.; Nakamura, N.; Takahashi, J.; Yoshikawa, T. Astaxanthin protects β-cells against glucose toxicity in diabetic db/db mice. Redox. Rep. 2002, 7, 290–293. [Google Scholar] [CrossRef]
- Iwamoto, T.; Hosoda, K.; Hirano, R.; Kurata, H.; Matsumoto, A.; Miki, W.; Kamiyama, M.; Itakura, H.; Yamamoto, S.; Kondo, K. Inhibition of low-density lipoprotein oxidation by astaxanthin. J. Atheroscler. Thromb. 2000, 7, 216–222. [Google Scholar] [CrossRef] [Green Version]
- Casella, P.; Iovine, A.; Mehariya, S.; Marino, T.; Musmarra, D.; Molino, A. Smart method for carotenoids characterization in haematococcus pluvialis red phase and evaluation of astaxanthin thermal stability. Antioxidants 2020, 9, 422. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.P.; Chen, F. Hydrolysis kinetics of astaxanthin esters and stability of astaxanthin of Haematococcus pluvialis during saponification. J. Agric. Food Chem. 1999, 47, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Ambati, R.R.; Phang, S.-M.; Ravi, S.; Aswathanarayana, R.G. Astaxanthin: Sources, extraction, stability, biological activities and its commercial applications—A review. Mar. Drugs 2014, 12, 128–152. [Google Scholar] [CrossRef]
- Armenta, R.E.; Guerrero-Legarreta, I. Stability studies on astaxanthin extracted from fermented shrimp byproducts. J. Agric. Food Chem. 2009, 57, 6095–6100. [Google Scholar] [CrossRef]
- Villalobos-Castillejos, F.; Cerezal-Mezquita, P.; Hernández-De Jesús, M.L.; Barragán-Huerta, B.E. Production and stability of water-dispersible astaxanthin oleoresin from Phaffia rhodozyma. Int. J. Food Sci. Technol. 2013, 48, 1243–1251. [Google Scholar] [CrossRef]
- Storebakken, T.; Sørensen, M.; Bjerkeng, B.; Harris, J.; Monahan, P.; Hiu, S. Stability of astaxanthin from red yeast, Xanthophyllomyces dendrorhous, during feed processing: Effects of enzymatic cell wall disruption and extrusion temperature. Aquaculture 2004, 231, 489–500. [Google Scholar] [CrossRef]
- Yun, H.H.; Kwang, J.K.; Su, J.K.; Seul, K.M.; Seong, G.H.; Young, J.S.; Sung, T.Y.; Hwang, Y.H.; Kim, K.J.; Kim, S.J.; et al. Suppression effect of astaxanthin on osteoclast formation in vitro and bone loss in vivo. Int. J. Mol. Sci. 2018, 19, 1–17. [Google Scholar]
- Mamun-Or-Rashid, A.N.M.; Takabe, W.; Yonei, Y. Glycated-proteins modulate RANKL-induced osteoclastogenesis in RAW264.7 cells. Glycative Stress Res. 2017, 4, 232–239. [Google Scholar]
- Mamun-Or-Rashid, A.N.M.; Takabe, W.; Yonei, Y. Melatonin and astaxanthin modulate RANKL-induced TRAP activity in RAW264.7 cells in an opposite fashion. Glycative Stress Res. 2019, 6, 135–141. [Google Scholar]
- Mamun-Or-Rashid, A.N.M.; Takabe, W.; Yonei, Y. Melatonin has no direct effect on inflammatory gene expression in CML-HSA stimulated RAW264.7 cells. Glycative Stress Res. 2016, 3, 141–151. [Google Scholar]
- Han, G.; Zuo, J.; Holliday, L.S. Specialized roles for actin in osteoclasts: Unanswered questions and therapeutic opportunities. Biomolecules 2019, 9, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mamun-Or-Rashid, A.N.M.; Takabe, W.; Yagi, M.; Yonei, Y. RANKL regulates RAW264.7 cell osteoclastogenesis in a manner independent of M-CSF, dependent on FBS, media content and cell density. Glycative Stress Res. 2017, 4, 40–52. [Google Scholar]
- Mamun-Or-Rashid, A.N.M.; Takabe, W.; Yagi, M.; Yonei, Y. Glycated-HSA inhibits osteoclastogenesis in RAW264.7 cells depending on the glycating agents via downregulating RANKL-signaling. Glycative Stress Res. 2017, 4, 217–231. [Google Scholar]
- Lam, J.; Takeshita, S.; Barker, J.E.; Kanagawa, O.; Ross, F.P.; Teitelbaum, S.L. TNF-α induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J. Clin. Investig. 2000, 106, 1481–1488. [Google Scholar] [CrossRef] [PubMed]
- Gravallese, E.M.; Harada, Y.; Wang, J.T.; Gorn, A.H.; Thornhill, T.S.; Goldring, S.R. Identification of cell types responsible for bone resorption in rheumatoid arthritis and juvenile rheumatoid arthritis. Am. J. Pathol. 1998, 152, 943. [Google Scholar] [PubMed]
- Walsh, N.C.; Crotti, T.N.; Goldring, S.R.; Gravallese, E.M.; Israel, B. Rheumatic diseases: The effects of inflammation on bone. Immunol. Rev. 2005, 208, 228–251. [Google Scholar] [CrossRef]
- Chou, H.Y.; Ma, D.L.; Leung, C.H.; Chiu, C.C.; Hour, T.C.; Wang, H.M.D. Purified astaxanthin from Haematococcus pluvialis promotes tissue regeneration by reducing oxidative stress and the secretion of collagen in vitro and in vivo. Oxid. Med. Cell. Longev. 2020, 2020, 4946902. [Google Scholar] [CrossRef]
- Suzuki, Y.; Ohgami, K.; Shiratori, K.; Jin, X.H.; Ilieva, I.; Koyama, Y.; Yazawa, K.; Yoshida, K.; Kase, S.; Ohno, S. Suppressive effects of astaxanthin against rat endotoxin-induced uveitis by inhibiting the NF-κB signaling pathway. Exp. Eye Res. 2006, 82, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Iwabayashi, M.; Fujioka, N.; Nomoto, K.; Miyazaki, R.; Takahashi, H.; Hibino, S.; Takahashi, Y.; Nishikawa, K.; Nishida, M.; Yonei, Y. Efficacy and safety of eight-week treatment with astaxanthin in individuals screened for increased oxidative stress burden. Anti-Aging Med. 2009, 6, 15–21. [Google Scholar] [CrossRef] [Green Version]
- Yonei, Y.; Yagi, M.; Nakamura, M.; Parengkuan, L.; Ogura, M.; Taira, T.; Asano, S.; Liu, H.-H. Effects of astaxanthin on intestinal microflora in mice fed a high-fat diet. Anti-Aging Med. 2013, 10, 77–91. [Google Scholar]
- Valenti, M.T.; Perduca, M.; Romanelli, M.G.; Mottes, M.; Dalle Carbonare, L. A potential role for astaxanthin in the treatment of bone diseases (Review). Mol. Med. Rep. 2020, 22, 1695–1701. [Google Scholar] [CrossRef]
- Balci Yuce, H.; Lektemur Alpan, A.; Gevrek, F.; Toker, H. Investigation of the effect of astaxanthin on alveolar bone loss in experimental periodontitis. J. Periodontal Res. 2018, 53, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Hoshi, H.; Monoe, F.; Ohsawa, I.; Ohta, S.; Miyamoto, T. Astaxanthin improves osteopenia caused by aldehyde-stress resulting from Aldh2 mutation due to impaired osteoblastogenesis. Biochem. Biophys. Res. Commun. 2020, 527, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Petrova, N.L.; Petrov, P.K.; Edmonds, M.E.; Shanahan, C.M. Inhibition of TNF-α reverses the pathological resorption pit profile of osteoclasts from patients with acute charcot osteoarthropathy. J. Diabetes Res. 2015, 2015, 917945. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Forward | Reverse |
|---|---|---|
| TNFα | ACC CTC ACA CTC AGA TCA TCT TC | TGG TGG TTT GCT ACG ACG T |
| IL-1β | TGT AAT GAA AGA CGG CAC ACC | TCT TCT TTG GGT ATT GCT TGG |
| IL-6 | ACA ACC ACG GCC TTC CCT ACT T | CAC GAT TTC CCA GAG AAC ATG TG |
| iNOS | CCA AGC CCT CAC CTA CTT CC | CTC TGA GGG CTG ACA CAA GG |
| NFATc1 | GGA GCG GAG AAA CTT TGC G | GTG ACA CTA GGG GAC ACA TAA CT |
| TRAP | GCG ACC ATT GTT AGC CAC ATA CG | CGT TGA TGT CGC ACA GAG GGA T |
| RAGE | ACT ACC GAG TCC GAG TCT ACC | GTA GCT TCC CTC AGA CAC ACA |
| c-Fos | CGG GTT TCA ACG CCG ACT A | TTG GCA CTA GAG ACG GAC AGA |
| CTSK | GAA GAA GAC TCA CCA GAA GCA G | TCC AGG TTA TGG GCA GAG ATT |
| Atp6v0 | ACG GTG ATG TCA CAG CAG ACG T | CCT CTG GAT AGA GCC TGC CGC A |
| GAPDH | AGG TCG GTG TGA ACG GAT TTG | TGT AGA CCA TGT AGT TGA GGT CA |
| MMP9 | CTG GAC AGC CAG ACA CTA AAG | CTC GCG GCA AGT CTT CAG AG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mamun-Or-Rashid, A.N.M.; Lucy, T.T.; Yagi, M.; Yonei, Y. Inhibitory Effects of Astaxanthin on CML-HSA-Induced Inflammatory and RANKL-Induced Osteoclastogenic Gene Expression in RAW 264.7 Cells. Biomedicines 2022, 10, 54. https://doi.org/10.3390/biomedicines10010054
Mamun-Or-Rashid ANM, Lucy TT, Yagi M, Yonei Y. Inhibitory Effects of Astaxanthin on CML-HSA-Induced Inflammatory and RANKL-Induced Osteoclastogenic Gene Expression in RAW 264.7 Cells. Biomedicines. 2022; 10(1):54. https://doi.org/10.3390/biomedicines10010054
Chicago/Turabian StyleMamun-Or-Rashid, A. N. M., Tanzima Tarannum Lucy, Masayuki Yagi, and Yoshikazu Yonei. 2022. "Inhibitory Effects of Astaxanthin on CML-HSA-Induced Inflammatory and RANKL-Induced Osteoclastogenic Gene Expression in RAW 264.7 Cells" Biomedicines 10, no. 1: 54. https://doi.org/10.3390/biomedicines10010054