
Off-Target Effects of Antidepressants on Vascular Function and Structure

 , , and
, , and
Abstract
:1. Introduction
2. Markers of Subclinical Arterial Disease and Their Clinical Relevance in Depression
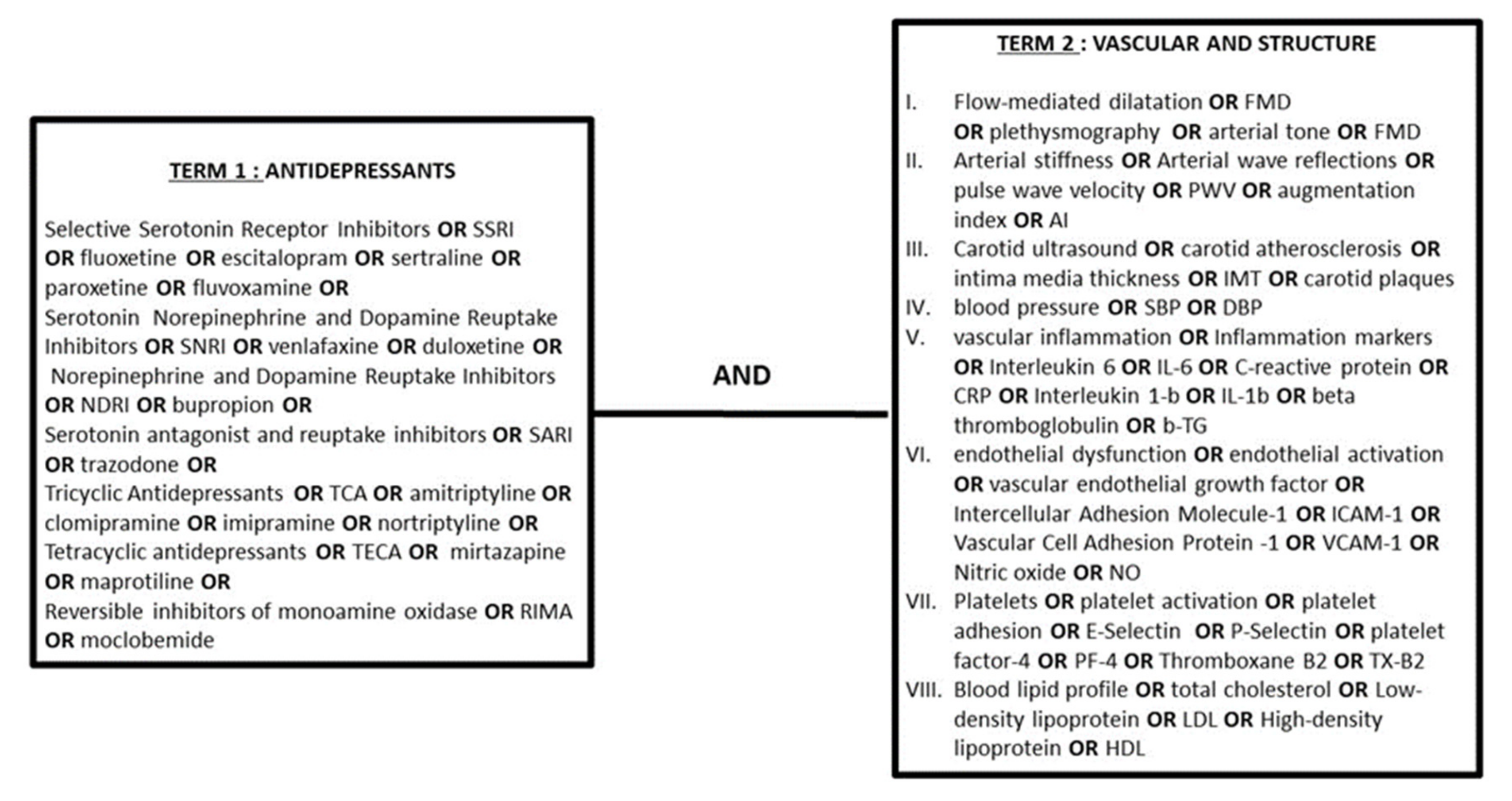
2.1. Search Strategy
2.2. Selection Criteria for Studies in This Review
2.3. Markers of Arterial Wave Reflections and Central Hemodynamics
2.4. Markers of Macrovascular and Microvascular Endothelial Function
2.5. Markers of Carotid Subclinical Atherosclerosis
3. The Effect of Antidepressant Treatment on Arteriosclerotic Processes: Experimental Data

{kind=link}
| Study | Drug/Dose | Treatment Duration | Study Model | Outcome |
|---|---|---|---|---|
| Matchkov et al., 2015 [32] | Escitalopram 5 mg/kg/d | 3 weeks | Male Wistar rats exposed to CMS | ↑ Endothelial function in small arteries via ↓ COX-2 dependent relaxation and ↑ endothelium-dependent hyperpolarization-like pathways |
| Unis et al., 2014 [33] | Escitalopram Dose NA | 6 weeks | Male albino Wistar rats on HFD | ↓ Atherosclerotic changes, together with a significant ↓ in VCAM-1 expression in abdominal aortic endothelium and ↓ TG, TC, and LDL |
| Lopez-Vilchez et al., 2016 [34] | Escitalopram 28 mg/d | 24 weeks | HUVEC exposed to MD patients’ whole blood serum | ↓ ICAM-1 and oxidative stress (with ↑ presence of eNOS and ↓ ROS production) |
| Bruno V D Marques et al., 2017 [35] | Fluoxetine 5 mg/kg/d | 28 h | Wistar rats | ↓ Aortic relaxation to a single restraint test in rat offspring |
| Janaina A Simplicio et al., 2015 [36] | Fluoxetine 10 mg/kg/d | 21 days | Male Wistar rats | ↑ Thoracic aorta vasoconstriction by phenylephrine, ↑ BP and PGF2a, and ↓ nNOS |
| Camila A Pereira et al., 2015 [37] | Fluoxetine 10 mg/kg/d | 21 days | Wistar rats | ↑ Endothelium-dependent and -independent relaxation of mesenteric resistance arteries via ↑ eNOS activity, NO generation, and KCa channel activation |
| Dan Dan-Han et al., 2012 [38] | Fluoxetine 10 mg/d | 21 days | Adult male Sprague Dawley rats | ↓ ROS generation, pulmonary artery pressure, and HIF-1 and VEGF production |
| Mohamed Habib et al., 2015 [39] | Fluoxetine 10 mg/kg/d | 21 days | Male Wistar rats diabetes induced, under CMS | ↓ Aortic expression of IL-1β and TNF-a; ↓ BP; ↓TG, TC, and LDL |
| Isingrini et al., 2011 [40] | Fluoxetine 10 mg/kg/d | 5 weeks | Male DBA/2 J mice subjected to CMS | ↔ MMP-9, PAI-1, VCAM-1, and ICAM-1 expression |
| M. Rami et al., 2018 [41] | Fluoxetine 18 mg/kg/d | 16 weeks | Apo-E-deficient mice | ↑ Atherosclerotic lesions of carotid arteries, ↔ TC |
| Isingrini et al., 2012 [42] | Fluoxetine 10 mg/kg/d | 20 weeks | BALB/c mice subjected to CMS | ↓ CMS detrimental effect on NO-related endothelial-dependent relaxation of aortic rings |
| Tsai et al., 2014 [43] | Fluoxetine/Bupropion/Imipramine/Moclobemide/Venlafaxine/Mirtazapine 10−8–10−5 M | - | LPS-activated THP-1 human monocytes | Fluoxetine and bupropion ↓ LPS-induced IP-10 expression |
| Domokos Gero et al., 2013 [44] | Paroxetine 10 mg/kg/d | 28 days | BEnd.3 murine cells, EA hy926 human endothelial cells, and Male Sprague Dawley rats induced with diabetes | ↑ Acetylcholine-induced rat aortic relaxation, ↓ mitochondrial ROS production in endothelial cells |
| Laleh Rafiee et al., 2016 [45] | Fluvoxamine 10−8 M–10−6 M | - | LPS-stimulated human endothelial cells | ↓ ICAM-1, VCAM, COX2, and iNOS expression |
| Lekakis et al., 2010 [46] | Fluvoxamine/Sertraline 10−7 M–10−4 M | 3 months | HAEC | ↓ U937 cell adhesion to TNFa-stimulated HAECs, ↓ VCAM-1 and ICAM-1 expression |
| Silverstein Metzler et al., 2017 [47] | Sertraline 20 mg/kg | 18 months | Female cynomolgus monkeys | ↑ CAA measured via histomorphometry |
| Shively et al., 2015 [48] | Sertraline 20 mg/kg | 18 months | Female cynomolgus monkeys | ↑ CAA measured via histomorphometry, ↔ plasma lipids |
| Maes et al., 1999 [49] | Sertraline 10−6, 10−8/Clomipramine 10−6, 10−9/ Trazodone 10−6, 10−8 | - | Whole blood of healthy human subjects (9) | All ↓ IFNγ, clomipramine, and sertraline; ↑ IL-10 |
| J M Vila et al., 1999 [50] | Sertraline/Nortriptyline/Amitriptyline 3 × 10−7–10−4 m | - | Human mesenteric arteries | Sertraline, amitriptyline, and nortriptyline ↓ human artery contraction |
| Joost P van Melle et al., 2004 [51] | Sertraline 0.1–300 μmol/L | - | Pre-contracted rat aortae, HIMA | ↑ Endothelial-independent vascular dilation in pre-contracted vessels |
| Prabhat Singh et al., 2016 [52] | Venlafaxine dose NA | - | Adult male Wistar rats | ↑ Endothelial function, assessed by means of a BIOPAC system |
| S Ribback et al., 2012 [53] | Venlafaxine/Fluoxetine/Tranylcypromine/Amitriptyline (0.05–500 μM) | - | Rat aortic ring dilation after precontraction with phenylephrine | ↑ Aortic relaxation in all except for venlafaxine, which promoted contraction |
| Qinghua LV et al., 2014 [54] | Venlafaxine 10−8, 10−5 M | 20 min | HBMEC | Protection against MGO (methylglyoxal)-mediated endothelial cell injury |
| Hoda I Bahr et al., 2019 [55] | Duloxetine 15–30 mg/kg | 13 weeks | Male Swiss albino mice induced with diabetes | ↓ VEGF, ↓ iNOS expression |
| Brustolim et al., 2006 [56] | Buproprion 100 mg/kg | 90 min | LPS-induced male 6-week-old BALB/c mice | ↓ Serum TNF-α, IL1-β, IFN-γ, and NO; ↑ in IL-10 |
| Mai Ahmed et al., 2014 [57] | Buproprion 50 kg | 4 weeks | Wistar male rats on HFD | ↓ Serum TNFa, with no effect seen on aortic IMT or aortic response to acetylcholine, ↔ TG |
| Labib et al., 2019 [58] | Imipramine 20 mg/kg/day | 2 weeks | Male Wistar rats exposed to CMS and HFD | ↔ Imipramine on aortic histological abnormalities, and level of CEPCs and VEGFR-2 |
| Ismail et al., 2014 [59] | Imipramine 20 mg | 3 weeks | Male Wistar rats | ↓ Endothelium-dependent relaxation of the thoracic aorta, ↓ TNF-a expression with imipramine |
| Rodica Lighezan et al., 2016 [60] | Moclobemide/Clorgyline/Selegiline 10 μmol/L | 30 min | HIMA (human internal mammary arteries) | ↑ Endothelium-dependent relaxation |
| Laleh Rafiee et al., 2016 [61] | Maprotiline 108, 10−6 | - | LPS-stimulated HUVEC | ↓ VCAM-1 and ICAM-1 expression |
3.1. Serotonin Reuptake Inhibitors (SSRIs)
3.1.1. Fluoxetine
3.1.2. Escitalopram
3.1.3. Sertraline
3.1.4. Paroxetine
3.1.5. Fluvoxamine
3.1.6. The Effect of SSRIs on Lipid Metabolism
3.2. Serotonin and Norepinephrine Reuptake Inhibitor (SNRIs)
3.2.1. Venlafaxine
3.2.2. Duloxetine
3.3. Norepinephrine–Dopamine Reuptake Inhibitors (NDRIs)
Bupropion
3.4. Serotonin Antagonist and Reuptake Inhibitors (SARI)
3.5. Tricyclic Antidepressants (TCAs)
3.5.1. Amitriptyline
3.5.2. Clomipramine
3.5.3. Imipramine
3.5.4. Nortriptyline
3.6. Tetracyclic Antidepressants (TECAs)
3.6.1. Mirtazapine and Maprotiline
3.6.2. Reversible Inhibitors of Monoamine Oxidase A (RIMAs)
4. The Effect of Antidepressant Treatment on Arteriosclerotic Processes: Human Molecular Data
| Type of Dysfunction | Study | RX (mg) | Duration | Patient Population | Design | Marker |
|---|---|---|---|---|---|---|
| Autonomous Nervous System | Shores et al., 2000 [72] | Sertraline 50 mg/placebo | 2 days | 12 healthy controls | OPC | ↓ NE |
| Barton et al., 2007 [73] | SSRI | 12 weeks | 39 MDD-76 healthy controls | OPC | ↓ SNS activity | |
| Inflammation | Eller et al., 2008 [74] | Escitalopram 10–20 mg/day | 12 weeks | 100 MDD | RCT | ↔ sIL-2R, IL-8, TNF-α |
| Blumenthal, 2012 [75] | Sertraline 50–200 mg/d | 16 weeks | 101 elevated depressive symptoms and ACS | RCT | ↔ PF4, CRP, βTG, IL-6 | |
| Pizzi et al., 2009 [76] | Sertraline 70 ± 39 mg and placebo | 20 weeks | 95 CHD and depression (47 sertraline and 48 placebo) | RCT | ↓ CRP, IL-6 | |
| Endothelial injury/dysfunction | Nathalie Lara, 2003 [77] | Paroxetine 10 mg/d and 20 mg/d | 9 weeks | 18 healthy controls | OPC | ↑ NO |
| M Deuschle, 2015 [78] | Venlafaxine (46 mg)/mirtazapine (215 mg) | 4 weeks | 86 MDD | RCT | ↔ VEGF | |
| Wendy Chrapko, 2006 [79] | Paroxetine 10 mg/d–20 mg/d | 9 weeks | 12 MDD and 12 healthy controls | PC | ↑ NO plasma levels, ↔ eNOS activitiy | |
| Dawood et al., 2016 [80] | SSRI | 95 days | 33 with MDD | OPC | ↔ ICAM-1, VCAM-1, P-selectin, NE | |
| Lopez-Vilchez et al., 2016 [34] | Escitaloprame, average 28 mg/day | 24 weeks | 12 MDD and 12 healthy controls | OPC | ↓ CECs, VWF, VCAM-1; ↑ EPCs | |
| Lekakis et al., 2010 [46] | Sertraline 50 mg/placebo | 3 months | 25 MDD-CHF | RCT | ↓ VCAM-1, ICAM-1 | |
| Platelet activation | N Hergovich, 2000 [81] | Paroxetine 20 mg/d | 14 days | 16 healthy controls | RCT | ↓ intraplatelet serotonin levels, platelet plug formation and responsiveness to thrombin receptor activating peptide |
| Hantsoo et al., 2014 [82] | SSRI (scitalopram 20 mg/sertraline 20 mg/fluoxetine 50 mg) | 4 weeks | 28 MDD/PMDD/PPD | OPC | ↔ platelet aggregation, ↓ platelet NO | |
| Serebruany et al., 2005 [83] | Sertraline | 16 weeks | 55 MDD | RCT | ↓ PF4, PECAM-1, β-TG, P-selectin, TxB2, E-selectin, 6-keto-PGF1a; ↑ VCAM-1 | |
| Serebruany et al., 2003 [84] | Sertraline | 24 weeks | 64 ACS | RCT | ↓ Platelet factor 4, βTG, PECAM-1, P-selectin, TxB2, 6-keto-PGF1a, VCAM-1, and E-selectin | |
| Blood Lipid Profile | Paslakis et al., 2011 [85] | Paroxetine | 4 weeks | 35 MDD and 35 healthy controls | RCT | ↓ Lpa |
| Perry et al., 1990 [86] | Trazodone | 6 weeks | 36 MDD | RCT | ↓ TC | |
| Hummel et al., 2011 [87] | Venlafaxine, mirtazapine | 4 weeks | 65 MDD and 31 healthy controls | RCT | ↓ LDL/HDL ratio in responders |
4.1. Inflammation Markers
4.1.1. SSRIs
4.1.2. Non-SSRIs
4.2. Markers of Endothelial Dysfunction
4.2.1. SSRIs
4.2.2. Non-SSRIs
4.3. Platelet Activation
4.4. Blood Lipid Profile
4.4.1. SSRIs
4.4.2. Non SSRIs
5. The Effect of Antidepressant Treatment on Arteriosclerotic Processes: Clinical Evidence
5.1. Blood Pressure and Arterial Wave Reflections
5.1.1. SSRIs
| Study | Depression | CVD | N | Age | Women | Rx | Res | Tx (Weeks) | Study | En. F. | A. S. | B. P. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Derby et al., 2007 [110] | No | No | 70 | 19–74 | 100% | SNRI (Duloxetine) | 3 | RCT | ↑ | |||
| Diaper et al., 2013 [111] | No | No | 54 | 23 ± 5 | 46% | SNRI (Venlafaxine) | 3 | RCT | ↑ | |||
| Martins et al. 2009 [112] | No | No | 24 | 41 ± 7 | 50% | NDRI (Bupropion) | 1 | RCT | ↑ | |||
| Thase et al., 2008 [113] | No | No | 300 | 44 ± 13 | 39% | NDRI (Bupropion) | 4 | RCT | ↔ | |||
| Dawood et al., 2016 [114] | Yes | No | 31 | 44 ± 2 | 61% | SSRI (Various) | 24 | OPC | FMD ↔ | |||
| Hantsoo et al., 2014 [82] | Yes | No | 27 | 38 ± 8 | 100% | SSRI (Various) | 4 | OPC | FMD ↔ | |||
| Kokras et al., 2019 [102] | Yes | No | 37 | 51 ± 13 | 30% | SSRI (Citalopram) | 24 | 26 | OPC | FMD ↑ | PWV ↓, AI ↓ | ↓ |
| Oulis et al., 2010 [115] | Yes | No | 40 | 57 ± 10 | 100% | SSRI (Various) | 12 | 6 | OPC | PWV ↓ | ||
| Peixoto et al., 2018 [103] | Yes | No | 30 | 57 ± 6 | 76% | SSRI (Escitalopram) | 8 | RCT | ↔ | |||
| Scuteri et al., 2013 [104] | Yes | No | 21 | 77 ± 4 | 76% | SNRI (Duloxetine) | 52 | RCT | PWV ↑ | ↑ | ||
| Scuteri et al., 2013 [104] | Yes | No | 27 | 77 ± 5 | 85% | SSRI (Escitalopram) | 52 | RCT | PWV - | ↑ | ||
| Tudoran et al., 2019 [116] | Yes | No | 128 | 48 ± 6 | 60% | SSRI (Sertraline) | 26 | OPC | IMT↓ | PWV ↓ | ||
| Blumenthal et al., 2012 [75] | Yes | Yes | 64 | 63 ± 11 | 30% | SSRI (Sertraline) | 18 | RCT | FMD ↔ | |||
| Pizzi et al., 2009 [76] | Yes | Yes | 95 | 57 ± 8 | 50% | SSRI (Sertraline) | 20 | RCT | FMD ↑ |
5.1.2. SNRIs
5.1.3. TCAs
5.1.4. Other Antidepressants
5.2. Arterial Stiffness
5.2.1. SSRIs
5.2.2. SNRIs
5.2.3. Other Antidepressants
5.3. Endothelial Function
5.3.1. SSRIs
| Study | Depression | CVD | N | Age | Women | Rx | Res. | Weeks | Study | En. F. | A. S. | B. P. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sherwood et.al, 2005 [71] | Yes | Yes | 143 | 63 ± 10 | 31% | Various | - | - | RCS | FMD ↑ | - | ↑ SBP, DBP (TCAs) |
| Broadley et al., 2002 [129] | Yes | No | 22 | 18–55 | 30% | Various | - | - | CC | FMD ↓ | - | |
| Delaney et al., 2010 [122] | Yes | No | 622 | 45–84 | 41% | Various | - | 85 | LS | - | ||
| Licht et al., 2009 [121] | Yes | No | 20.718 | 40 ± 12 | 69% | Various | - | - | RCS | - | ||
| Crookes et al., 2018 [130] | Yes | No | 11.183 | 16–29 | 47% | Various | - | 672 | CC | - | ↑ SBP, DBP (NS working | |
| Paranthaman et al., 2012 [131] | Yes | No | 25 | 72 ± 5 | 15% | Various | 9 | - | RCS | ↓ lMT Res > NRes | - | |
| Camacho et al., 2016 [132] | Yes | No | 324 | 62.1 | 56% | Various | - | - | RCS | ↔ IMT | - | ↑ SBP, DBP |
5.3.2. Other Antidepressants
5.4. Atherosclerosis of the Carotid Arteries
5.4.1. SSRIs
5.4.2. Other Antidepressants
6. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- World Health Organization. The Global Burden of Disease: 2004 Update; World Health Organization: Geneva, Switzerland, 2008. [Google Scholar]
- Bahls, M.; Lorenz, M.W.; Dörr, M.; Gao, L.; Kitagawa, K.; Tuomainen, T.P.; Agewall, S.; Berenson, G.; Catapano, A.L.; Norata, G.D.; et al. Progression of conventional cardiovascular risk factors and vascular disease risk in individuals: Insights from the PROG-IMT consortium. Eur. J. Prev. Cardiol. 2020, 27, 234–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhar, A.K.; Barton, D.A. Depression and the Link with Cardiovascular Disease. Front. Psychiatry 2016, 7, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ditmars, H.L.; Logue, M.W.; Toomey, R.; McKenzie, R.E.; Franz, C.E.; Panizzon, M.S.; Reynolds, C.A.; Cuthbert, K.N.; Vandiver, R.; Gustavson, D.E.; et al. Associations between depression and cardiometabolic health: A 27-year longitudinal study. In Psychological Medicine; Cambridge University Press: Cambridge, UK, 2021; pp. 1–11. [Google Scholar] [CrossRef]
- Scalco, A.Z.; Scalco, M.Z.; Azul, J.B.; Lotufo Neto, F. Hypertension and depression. Clinics 2005, 60, 241–250. [Google Scholar] [CrossRef] [Green Version]
- Hare, D.L.; Toukhsati, S.R.; Johansson, P.; Jaarsma, T. Depression and cardiovascular disease: A clinical review. Eur. Heart J. 2014, 35, 1365–1372. [Google Scholar] [CrossRef] [Green Version]
- Tiemeier, H.; Breteler, M.M.; van Popele, N.M.; Hofman, A.; Witteman, J.C. Late-life depression is associated with arterial stiffness: A population-based study. J. Am. Geriatr. Soc. 2003, 51, 1105–1110. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.B.; Youngblood, M.E.; Catellier, D.; Veith, R.C.; Carney, R.M.; Burg, M.M.; Kaufmann, P.G.; Shuster, J.; Mellman, T.; Blumenthal, J.A.; et al. Effects of antidepressant medication on morbidity and mortality in depressed patients after myocardial infarction. Arch. Gen. Psychiatry 2005, 62, 792–798. [Google Scholar] [CrossRef] [Green Version]
- Almuwaqqat, Z.; Jokhadar, M.; Norby, F.L.; Lutsey, P.L.; O’Neal, W.T.; Seyerle, A.; Soliman, E.Z.; Chen, L.Y.; Bremner, J.D.; Vaccarino, V.; et al. Association of Antidepressant Medication Type with the Incidence of Cardiovascular Disease in the ARIC Study. J. Am. Heart Assoc. 2019, 8, e012503. [Google Scholar] [CrossRef]
- Hamer, M.; Batty, G.D.; Seldenrijk, A.; Kivimaki, M. Antidepressant medication use and future risk of cardiovascular disease: The Scottish Health Survey. Eur. Heart J. 2010, 32, 437–442. [Google Scholar] [CrossRef] [Green Version]
- Lichtman, J.H.; Froelicher, E.S.; Blumenthal, J.A.; Carney, R.M.; Doering, L.V.; Frasure-Smith, N.; Freedland, K.E.; Jaffe, A.S.; Leifheit-Limson, E.C.; Sheps, D.S.; et al. Depression as a risk factor for poor prognosis among patients with acute coronary syndrome: Systematic review and recommendations: A scientific statement from the American Heart Association. Circulation 2014, 129, 1350–1369. [Google Scholar] [CrossRef] [Green Version]
- Liberati, A.; Altman, D.G.; Tetzlaff, J.; Mulrow, C.; Gøtzsche, P.C.; Ioannidis, J.P.; Clarke, M.; Devereaux, P.J.; Kleijnen, J.; Moher, D. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate healthcare interventions: Explanation and elaboration. BMJ 2009, 339, b2700. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Sun, D.; Wang, B.; Li, Y.; Ma, Y. The relationship of depressive symptoms and functional and structural markers of subclinical atherosclerosis: A systematic review and meta-analysis. Eur. J. Prev. Cardiol. 2018, 25, 706–716. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-Y.; Hu, H.-Y.; Chou, Y.-J.; Huang, N.; Chou, Y.-C.; Li, C.-P. High Blood Pressure and All-Cause and Cardiovascular Disease Mortalities in Community-Dwelling Older Adults. Medicine 2015, 94, e2160. [Google Scholar] [CrossRef] [PubMed]
- McEniery, C.M.; Cockcroft, J.R.; Roman, M.J.; Franklin, S.S.; Wilkinson, I.B. Central blood pressure: Current evidence and clinical importance. Eur. Heart J. 2014, 35, 1719–1725. [Google Scholar] [CrossRef] [Green Version]
- Weber, T.; Auer, J.; O’Rourke, M.F.; Kvas, E.; Lassnig, E.; Berent, R.; Eber, B. Arterial stiffness, wave reflections, and the risk of coronary artery disease. Circulation 2004, 109, 184–189. [Google Scholar] [CrossRef] [Green Version]
- Mayet, J.; Hughes, A. Cardiac and vascular pathophysiology in hypertension. Heart 2003, 89, 1104–1109. [Google Scholar] [CrossRef]
- Nwabuo, C.C.; Vasan, R.S. Pathophysiology of Hypertensive Heart Disease: Beyond Left Ventricular Hypertrophy. Curr. Hypertens. Rep. 2020, 22, 11. [Google Scholar] [CrossRef]
- Diaz, A.; Tringler, M.; Wray, S.; Ramirez, A.J.; Cabrera Fischer, E.I. The effects of age on pulse wave velocity in untreated hypertension. J. Clin. Hypertens. 2018, 20, 258–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomiyama, H.; Odaira, M.; Kimura, K.; Matsumoto, C.; Shiina, K.; Eguchi, K.; Miyashita, H.; Shimada, K.; Yamashina, A. Differences in Effects of Age and Blood Pressure on Augmentation Index. Am. J. Hypertens. 2014, 27, 1479–1485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greco, A.; Maloberti, A.; Varrenti, M.; Lucente, D.; Pessina, L.; Peretti, A.; Franzosi, C.; Mondellini, G.; Palazzini, M.; Laurent, S.; et al. Pulse wave velocity and depression in hypertensive patients. J. Hypertens. 2017, 35, e44. [Google Scholar] [CrossRef]
- Seldenrijk, A.; van Hout, H.P.; van Marwijk, H.W.; de Groot, E.; Gort, J.; Rustemeijer, C.; Diamant, M.; Penninx, B.W. Depression, anxiety, and arterial stiffness. Biol. Psychiatry 2011, 69, 795–803. [Google Scholar] [CrossRef]
- Peng, L.; Bi, S.; Liu, X.; Long, T.; Zhao, Y.; Li, F.; Yang, T.; Zhang, C. Association between depressive symptoms and arterial stiffness: A cross-sectional study in the general Chinese population. BMJ Open 2020, 10, e033408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Arora, R.C.; Hiebert, B.M.; Lerner, B.; Szwajcer, A.; McDonald, K.; Rigatto, C.; Komenda, P.; Sood, M.M.; Tangri, N. Non-invasive endothelial function testing and the risk of adverse outcomes: A systematic review and meta-analysis. Eur. Heart J. Cardiovasc. Imaging 2014, 15, 736–746. [Google Scholar] [CrossRef] [Green Version]
- Cooper, D.C.; Tomfohr, L.M.; Milic, M.S.; Natarajan, L.; Bardwell, W.A.; Ziegler, M.G.; Dimsdale, J.E. Depressed mood and flow-mediated dilation: A systematic review and meta-analysis. Psychosom. Med. 2011, 73, 360–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pirro, M.; Lorenz, M.W.; Gao, L.; Ziegelbauer, K.; Norata, G.D.; Empana, J.P.; Schmidtmann, I.; Lin, H.-J.; McLachlan, S.; Bokemark, L.; et al. Predictive value for cardiovascular events of common carotid intima media thickness and its rate of change in individuals at high cardiovascular risk–Results from the PROG-IMT collaboration. PLoS ONE 2018, 13, e0191172. [Google Scholar] [CrossRef] [Green Version]
- Lorenz, M.W.; Markus, H.S.; Bots, M.L.; Rosvall, M.; Sitzer, M. Prediction of clinical cardiovascular events with carotid intima-media thickness: A systematic review and meta-analysis. Circulation 2007, 115, 459–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Den Ruijter, H.M.; Peters, S.A.; Anderson, T.J.; Britton, A.R.; Dekker, J.M.; Eijkemans, M.J.; Engstrom, G.; Evans, G.W.; de Graaf, J.; Grobbee, D.E.; et al. Common carotid intima-media thickness measurements in cardiovascular risk prediction: A meta-analysis. JAMA 2012, 308, 796–803. [Google Scholar] [CrossRef] [PubMed]
- Nambi, V.; Chambless, L.; Folsom, A.R.; He, M.; Hu, Y.; Mosley, T.; Volcik, K.; Boerwinkle, E.; Ballantyne, C.M. Carotid intima-media thickness and presence or absence of plaque improves prediction of coronary heart disease risk: The ARIC (Atherosclerosis Risk in Communities) study. J. Am. Coll. Cardiol. 2010, 55, 1600–1607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Störk, S.; van den Beld, A.W.; von Schacky, C.; Angermann, C.E.; Lamberts, S.W.; Grobbee, D.E.; Bots, M.L. Carotid artery plaque burden, stiffness, and mortality risk in elderly men: A prospective, population-based cohort study. Circulation 2004, 110, 344–348. [Google Scholar] [CrossRef] [Green Version]
- Stewart, J.C.; Janicki, D.L.; Muldoon, M.F.; Sutton-Tyrrell, K.; Kamarck, T.W. Negative emotions and 3-year progression of subclinical atherosclerosis. Arch. Gen. Psychiatry 2007, 64, 225–233. [Google Scholar] [CrossRef]
- Matchkov, V.V.; Kravtsova, V.V.; Wiborg, O.; Aalkjaer, C.; Bouzinova, E.V. Chronic selective serotonin reuptake inhibition modulates endothelial dysfunction and oxidative state in rat chronic mild stress model of depression. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2015, 309, R814–R823. [Google Scholar] [CrossRef] [Green Version]
- Unis, A.; Abdelbary, A.; Hamza, M. Comparison of the effects of escitalopram and atorvastatin on diet-induced atherosclerosis in rats. Can. J. Physiol. Pharmacol. 2014, 92, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Vilchez, I.; Diaz-Ricart, M.; Navarro, V.; Torramade, S.; Zamorano-Leon, J.; Lopez-Farre, A.; Galan, A.M.; Gasto, C.; Escolar, G. Endothelial damage in major depression patients is modulated by SSRI treatment, as demonstrated by circulating biomarkers and an in vitro cell model. Transl. Psychiatry 2016, 6, e886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marques, B.V.D.; Higashi, C.M.; Novi, D.R.D.S.; Zanluqui, N.G.; Gregório, T.F.; Pinge-Filho, P.; Gerardin, D.C.C.; Pelosi, G.G.; Moreira, E.G.; Ceravolo, G.S. Intrauterine and lactation exposure to fluoxetine blunted in the offspring the aortic adaptive response induced by acute restraint stress. Eur. J. Pharmacol. 2017, 813, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Simplicio, J.A.; Resstel, L.B.; Tirapelli, D.P.; D’Orléans-Juste, P.; Tirapelli, C.R. Contribution of oxidative stress and prostanoids in endothelial dysfunction induced by chronic fluoxetine treatment. Vasc. Pharmacol. 2015, 73, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Pereira, C.A.; Ferreira, N.S.; Mestriner, F.L.; Antunes-Rodrigues, J.; Evora, P.R.; Resstel, L.B.; Carneiro, F.S.; Tostes, R.C. Chronic fluoxetine treatment increases NO bioavailability and calcium-sensitive potassium channels activation in rat mesenteric resistance arteries. Eur. J. Pharmacol. 2015, 765, 375–383. [Google Scholar] [CrossRef] [Green Version]
- Han, D.D.; Wang, Y.; Zhang, X.H.; Liu, J.R.; Wang, H.L. Fluoxetine protects against monocrotaline-induced pulmonary arterial remodeling by inhibition of hypoxia-inducible factor-1α and vascular endothelial growth factor. Can. J. Physiol. Pharmacol. 2012, 90, 445–454. [Google Scholar] [CrossRef]
- Habib, M.; Shaker, S.; El-Gayar, N.; Aboul-Fotouh, S. The effects of antidepressants “fluoxetine and imipramine” on vascular abnormalities and Toll like receptor-4 expression in diabetic and non-diabetic rats exposed to chronic stress. PLoS ONE 2015, 10, e0120559. [Google Scholar] [CrossRef]
- Isingrini, E.; Belzung, C.; d’Audiffret, A.; Camus, V. Early and late-onset effect of chronic stress on vascular function in mice: A possible model of the impact of depression on vascular disease in aging. Am. J. Geriatr. Psychiatry 2011, 19, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Rami, M.; Guillamat-Prats, R.; Rinne, P.; Salvermoser, M.; Ring, L.; Bianchini, M.; Blanchet, X.; Megens, R.T.A.; Döring, Y.; Walzog, B.; et al. Chronic Intake of the Selective Serotonin Reuptake Inhibitor Fluoxetine Enhances Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1007–1019. [Google Scholar] [CrossRef] [Green Version]
- Isingrini, E.; Belzung, C.; Freslon, J.L.; Machet, M.C.; Camus, V. Fluoxetine effect on aortic nitric oxide-dependent vasorelaxation in the unpredictable chronic mild stress model of depression in mice. Psychosom. Med. 2012, 74, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.-H.; Kuo, C.-H.; Yang, P.; Cheng, K.-H.; Wang, P.-W.; Chen, C.-C.; Hung, C.-H. Effects of antidepressants on IP-10 production in LPS-activated THP-1 human monocytes. Int. J. Mol. Sci. 2014, 15, 13223–13235. [Google Scholar] [CrossRef] [Green Version]
- Gerö, D.; Szoleczky, P.; Suzuki, K.; Módis, K.; Oláh, G.; Coletta, C.; Szabo, C. Cell-based screening identifies paroxetine as an inhibitor of diabetic endothelial dysfunction. Diabetes 2013, 62, 953–964. [Google Scholar] [CrossRef] [Green Version]
- Rafiee, L.; Hajhashemi, V.; Javanmard, S.H. Fluvoxamine inhibits some inflammatory genes expression in LPS/stimulated human endothelial cells, U937 macrophages, and carrageenan-induced paw edema in rat. Iran. J. Basic Med. Sci. 2016, 19, 977–984. [Google Scholar]
- Lekakis, J.; Ikonomidis, I.; Papoutsi, Z.; Moutsatsou, P.; Nikolaou, M.; Parissis, J.; Kremastinos, D.T. Selective serotonin re-uptake inhibitors decrease the cytokine-induced endothelial adhesion molecule expression, the endothelial adhesiveness to monocytes and the circulating levels of vascular adhesion molecules. Int. J. Cardiol. 2010, 139, 150–158. [Google Scholar] [CrossRef]
- Silverstein-Metzler, M.G.; Justice, J.N.; Appt, S.E.; Groban, L.; Kitzman, D.W.; Carr, J.J.; Register, T.C.; Shively, C.A. Long-term sertraline treatment and depression effects on carotid artery atherosclerosis in premenopausal female primates. Menopause 2017, 24, 1175–1184. [Google Scholar] [CrossRef]
- Shively, C.A.; Register, T.C.; Appt, S.E.; Clarkson, T.B. Effects of long-term sertraline treatment and depression on coronary artery atherosclerosis in premenopausal female primates. Psychosom. Med. 2015, 77, 267–278. [Google Scholar] [CrossRef] [Green Version]
- Maes, M.; Song, C.; Lin, A.H.; Bonaccorso, S.; Kenis, G.; De Jongh, R.; Bosmans, E.; Scharpé, S. Negative immunoregulatory effects of antidepressants: Inhibition of interferon-gamma and stimulation of interleukin-10 secretion. Neuropsychopharmacology 1999, 20, 370–379. [Google Scholar] [CrossRef]
- Vila, J.M.; Medina, P.; Segarra, G.; Lluch, P.; Pallardó, F.; Flor, B.; Lluch, S. Relaxant effects of antidepressants on human isolated mesenteric arteries. Br. J. Clin. Pharmacol. 1999, 48, 223–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Melle, J.P.; Buikema, H.; van den Berg, M.P.; van Buiten, A.; van Veldhuisen, D.J.; Boonstra, P.W.; van Gilst, W.H. Sertraline causes strong coronary vasodilation: Possible relevance for cardioprotection by selective serotonin reuptake inhibitors. Cardiovasc. Drugs Ther. 2004, 18, 441–447. [Google Scholar] [CrossRef]
- Singh, P.; Sharma, B. Selective Serotonin-norepinephrine Re-uptake Inhibition Limits Renovas-cular-hypertension Induced Cognitive Impairment, Endothelial Dysfunction, and Oxidative Stress Injury. Curr. Neurovasc. Res. 2016, 13. [Google Scholar] [CrossRef]
- Ribback, S.; Pavlovic, D.; Herbst, D.; Nedeljkov-Jancic, R.; Wendt, M.; Nedeljkov, V.; Bleich, S.; Frieling, H. Effects of amitriptyline, fluoxetine, tranylcypromine and venlafaxine on rat vascular smooth muscle in vitro–the role of the endothelium. J. Physiol. Pharmacol. 2012, 63, 119–125. [Google Scholar]
- Lv, Q.; Gu, C.; Chen, C. Venlafaxine protects methylglyoxal-induced apoptosis in the cultured human brain microvascular endothelial cells. Neurosci. Lett. 2014, 569, 99–103. [Google Scholar] [CrossRef]
- Bahr, H.I.; Abdelghany, A.A.; Galhom, R.A.; Barakat, B.M.; Arafa, E.-S.A.; Fawzy, M.S. Duloxetine protects against experimental diabetic retinopathy in mice through retinal GFAP downregulation and modulation of neurotrophic factors. Exp. Eye Res. 2019, 186, 107742. [Google Scholar] [CrossRef] [PubMed]
- Brustolim, D.; Ribeiro-dos-Santos, R.; Kast, R.E.; Altschuler, E.L.; Soares, M.B. A new chapter opens in anti-inflammatory treatments: The antidepressant bupropion lowers production of tumor necrosis factor-alpha and interferon-gamma in mice. Int. Immunopharmacol. 2006, 6, 903–907. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, M.; El-Bakly, W.M.; Zaki, A.M.; abd Alzez, L.F.; El Serafi, O. Bupropion effects on high-fat diet-induced steatohepatitis and endothelial dysfunction in rats: Role of tumour necrosis factor-alpha. J. Pharm. Pharmacol. 2014, 66, 793–801. [Google Scholar] [CrossRef]
- Labib, J.M.W.; Aboul-Fotouh, S.; Habib, M.Z. Pentoxifylline ameliorates chronic stress/high-fat diet-induced vascular wall disease: The role of circulating endothelial progenitor cells. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2019, 392, 669–683. [Google Scholar] [CrossRef]
- Ismail, B.; Aboul-Fotouh, S.; Mansour, A.A.; Shehata, H.H.; Salman, M.I.; Ibrahim, E.A.; Hassan, O.A.; Abdel-tawab, A.M. Behavioural, metabolic, and endothelial effects of the TNF-α suppressor thalidomide on rats subjected to chronic mild stress and fed an atherogenic diet. Can. J. Physiol. Pharmacol. 2014, 92, 375–385. [Google Scholar] [CrossRef]
- Lighezan, R.; Sturza, A.; Duicu, O.M.; Ceausu, R.A.; Vaduva, A.; Gaspar, M.; Feier, H.; Vaida, M.; Ivan, V.; Lighezan, D.; et al. Monoamine oxidase inhibition improves vascular function in mammary arteries from nondiabetic and diabetic patients with coronary heart disease. Can. J. Physiol. Pharmacol. 2016, 94, 1040–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rafiee, L.; Hajhashemi, V.; Javanmard, S.H. Maprotiline inhibits LPS-induced expression of adhesion molecules (ICAM-1 and VCAM-1) in human endothelial cells. Res. Pharm. Sci. 2016, 11, 138–144. [Google Scholar] [PubMed]
- Sangkuhl, K.; Klein, T.E.; Altman, R.B. Selective serotonin reuptake inhibitors pathway. Pharm. Genom. 2009, 19, 907–909. [Google Scholar] [CrossRef] [Green Version]
- Halaris, A.; Meresh, E.; Fareed, J.; Hoppensteadt, D.; Kimmons, S.; Sinacore, J. 2. Tumor Necrosis Factor alpha as a biomarker in major depressive disorder. Brain Behav. Immun. 2012, 26, S1. [Google Scholar] [CrossRef]
- Groves, J.T.; Wang, C.C. Nitric oxide synthase: Models and mechanisms. Curr. Opin. Chem. Biol. 2000, 4, 687–695. [Google Scholar] [CrossRef]
- Casino, P.R.; Kilcoyne, C.M.; Quyyumi, A.A.; Hoeg, J.M.; Panza, J.A. The role of nitric oxide in endothelium-dependent vasodilation of hypercholesterolemic patients. Circulation 1993, 88, 2541–2547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakogiannis, C.; Sachse, M.; Stamatelopoulos, K.; Stellos, K. Platelet-derived chemokines in inflammation and atherosclerosis. Cytokine 2019, 122, 154157. [Google Scholar] [CrossRef]
- Ouyang, W.; Rutz, S.; Crellin, N.K.; Valdez, P.A.; Hymowitz, S.G. Regulation and functions of the IL-10 family of cytokines in inflammation and disease. Annu. Rev. Immunol. 2011, 29, 71–109. [Google Scholar] [CrossRef]
- Lambert, O.; Bourin, M. SNRIs: Mechanism of action and clinical features. Expert Rev. Neurother. 2002, 2, 849–858. [Google Scholar] [CrossRef]
- Hwang, J.; Zheng, L.T.; Ock, J.; Lee, M.G.; Kim, S.H.; Lee, H.W.; Lee, W.H.; Park, H.C.; Suk, K. Inhibition of glial inflammatory activation and neurotoxicity by tricyclic antidepressants. Neuropharmacology 2008, 55, 826–834. [Google Scholar] [CrossRef]
- Sturza, A.; Leisegang, M.S.; Babelova, A.; Schröder, K.; Benkhoff, S.; Loot, A.E.; Fleming, I.; Schulz, R.; Muntean, D.M.; Brandes, R.P. Monoamine oxidases are mediators of endothelial dysfunction in the mouse aorta. Hypertension 2013, 62, 140–146. [Google Scholar] [CrossRef] [Green Version]
- Sherwood, A.; Hinderliter, A.L.; Watkins, L.L.; Waugh, R.A.; Blumenthal, J.A. Impaired endothelial function in coronary heart disease patients with depressive symptomatology. J. Am. Coll. Cardiol. 2005, 46, 656–659. [Google Scholar] [CrossRef] [Green Version]
- Shores, M.M.; Pascualy, M.; Lewis, N.L.; Flatness, D.; Veith, R.C. Short-term sertraline treatment suppresses sympathetic nervous system activity in healthy human subjects. Psychoneuroendocrinology 2001, 26, 433–439. [Google Scholar] [CrossRef]
- Barton, D.A.; Dawood, T.; Lambert, E.A.; Esler, M.D.; Haikerwal, D.; Brenchley, C.; Socratous, F.; Kaye, D.M.; Schlaich, M.P.; Hickie, I.; et al. Sympathetic activity in major depressive disorder: Identifying those at increased cardiac risk? J. Hypertens. 2007, 25, 2117–2124. [Google Scholar] [CrossRef] [PubMed]
- Eller, T.; Vasar, V.; Shlik, J.; Maron, E. Pro-inflammatory cytokines and treatment response to escitalopram in major depressive disorder. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2008, 32, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Blumenthal, J.A.; Sherwood, A.; Babyak, M.A.; Watkins, L.L.; Smith, P.J.; Hoffman, B.M.; O’Hayer, C.V.; Mabe, S.; Johnson, J.; Doraiswamy, P.M.; et al. Exercise and pharmacological treatment of depressive symptoms in patients with coronary heart disease: Results from the UPBEAT (Understanding the Prognostic Benefits of Exercise and Antidepressant Therapy) study. J. Am. Coll. Cardiol. 2012, 60, 1053–1063. [Google Scholar] [CrossRef] [Green Version]
- Pizzi, C.; Mancini, S.; Angeloni, L.; Fontana, F.; Manzoli, L.; Costa, G.M. Effects of selective serotonin reuptake inhibitor therapy on endothelial function and inflammatory markers in patients with coronary heart disease. Clin. Pharmacol. Ther. 2009, 86, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Lara, N.; Archer, S.L.; Baker, G.B.; Le Mellédo, J.M. Paroxetine-induced increase in metabolic end products of nitric oxide. J. Clin. Psychopharmacol. 2003, 23, 408–412. [Google Scholar] [CrossRef]
- Deuschle, M.; Gilles, M.; Scharnholz, B.; Kahl, K.G. Antidepressant Treatment with Venlafaxine and Mirtazapine: No Effect on Serum Concentration of Vascular Endothelial Growth Factor (VEGF). Pharmacopsychiatry 2015, 48, 292–293. [Google Scholar] [CrossRef]
- Chrapko, W.; Jurasz, P.; Radomski, M.W.; Archer, S.L.; Newman, S.C.; Baker, G.; Lara, N.; Le Mellédo, J.M. Alteration of decreased plasma NO metabolites and platelet NO synthase activity by paroxetine in depressed patients. Neuropsychopharmacology 2006, 31, 1286–1293. [Google Scholar] [CrossRef] [Green Version]
- Dawood, T.; Barton, D.A.; Lambert, E.A.; Eikelis, N.; Lambert, G.W. Examining Endothelial Function and Platelet Reactivity in Patients with Depression before and after SSRI Therapy. Front. Psychiatry 2016, 7, 18. [Google Scholar] [CrossRef] [Green Version]
- Hergovich, N.; Aigner, M.; Eichler, H.G.; Entlicher, J.; Drucker, C.; Jilma, B. Paroxetine decreases platelet serotonin storage and platelet function in human beings. Clin. Pharmacol. Ther. 2000, 68, 435–442. [Google Scholar] [CrossRef]
- Hantsoo, L.; Czarkowski, K.A.; Child, J.; Howes, C.; Epperson, C.N. Selective serotonin reuptake inhibitors and endothelial function in women. J. Women’s Health 2014, 23, 613–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serebruany, V.L.; Suckow, R.F.; Cooper, T.B.; O’Connor, C.M.; Malinin, A.I.; Krishnan, K.R.; van Zyl, L.T.; Lekht, V.; Glassman, A.H. Relationship between release of platelet/endothelial biomarkers and plasma levels of sertraline and N-desmethylsertraline in acute coronary syndrome patients receiving SSRI treatment for depression. Am. J. Psychiatry 2005, 162, 1165–1170. [Google Scholar] [CrossRef]
- Serebruany, V.L.; Glassman, A.H.; Malinin, A.I.; Nemeroff, C.B.; Musselman, D.L.; van Zyl, L.T.; Finkel, M.S.; Krishnan, K.R.; Gaffney, M.; Harrison, W.; et al. Platelet/endothelial biomarkers in depressed patients treated with the selective serotonin reuptake inhibitor sertraline after acute coronary events: The Sertraline AntiDepressant Heart Attack Randomized Trial (SADHART) Platelet Substudy. Circulation 2003, 108, 939–944. [Google Scholar] [CrossRef] [Green Version]
- Paslakis, G.; Kopf, D.; Westphal, S.; Gilles, M.; Lederbogen, F.; Hamann, B.; Heuser, I.; Deuschle, M. Treatment with paroxetine, but not amitriptyline, lowers levels of lipoprotein(a) in patients with major depression. J. Psychopharmacol. 2011, 25, 1344–1346. [Google Scholar] [CrossRef]
- Perry, P.J.; Garvey, M.J.; Dunner, D.L.; Rush, A.J.; Kyhl, J. A report of trazodone-associated laboratory abnormalities. Ther. Drug Monit. 1990, 12, 517–519. [Google Scholar] [CrossRef] [PubMed]
- Hummel, J.; Westphal, S.; Weber-Hamann, B.; Gilles, M.; Lederbogen, F.; Angermeier, T.; Luley, C.; Deuschle, M.; Kopf, D. Serum Lipoproteins Improve After Successful Pharmacologic Antidepressant Treatment: A Randomized Open-Label Prospective Trial. J. Clin. Psychiatry 2011, 72, 885–891. [Google Scholar] [CrossRef] [PubMed]
- Vogelzangs, N.; Duivis, H.E.; Beekman, A.T.F.; Kluft, C.; Neuteboom, J.; Hoogendijk, W.; Smit, J.H.; de Jonge, P.; Penninx, B.W.J.H. Association of depressive disorders, depression characteristics and antidepressant medication with inflammation. Transl. Psychiatry 2012, 2, e79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hannestad, J.; DellaGioia, N.; Bloch, M. The effect of antidepressant medication treatment on serum levels of inflammatory cytokines: A meta-analysis. Neuropsychopharmacology 2011, 36, 2452–2459. [Google Scholar] [CrossRef] [PubMed]
- Van Reedt Dortland, A.K.; Giltay, E.J.; van Veen, T.; Zitman, F.G.; Penninx, B.W. Metabolic syndrome abnormalities are associated with severity of anxiety and depression and with tricyclic antidepressant use. Acta Psychiatr. Scand. 2010, 122, 30–39. [Google Scholar] [CrossRef]
- Leung, D.W.; Cachianes, G.; Kuang, W.J.; Goeddel, D.V.; Ferrara, N. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science 1989, 246, 1306–1309. [Google Scholar] [CrossRef]
- Kimura, K.; Hashiguchi, T.; Deguchi, T.; Horinouchi, S.; Uto, T.; Oku, H.; Setoyama, S.; Maruyama, I.; Osame, M.; Arimura, K. Serum VEGF—As a prognostic factor of atherosclerosis. Atherosclerosis 2007, 194, 182–188. [Google Scholar] [CrossRef]
- Kim, M.H.; Huo, S.H.; Kim, K.S.; Kim, M.S.; Song, J.S. Study on the platelet factor and beta-thromboglobulin in the patients with ischemic heart disease. Korean J. Intern. Med. 1986, 1, 1–6. [Google Scholar] [CrossRef]
- Laghrissi-Thode, F.; Wagner, W.R.; Pollock, B.G.; Johnson, P.C.; Finkel, M.S. Elevated platelet factor 4 and beta-thromboglobulin plasma levels in depressed patients with ischemic heart disease. Biol. Psychiatry 1997, 42, 290–295. [Google Scholar] [CrossRef]
- Fjukstad, K.K.; Engum, A.; Lydersen, S.; Dieset, I.; Steen, N.E.; Andreassen, O.A.; Spigset, O. Metabolic Abnormalities Related to Treatment with Selective Serotonin Reuptake Inhibitors in Patients with Schizophrenia or Bipolar Disorder. J. Clin. Psychopharmacol. 2016, 36, 615–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, F.; Crain, A.L.; Whitebird, R.R.; Godlevsky, O.V.; O’Connor, P.J. Effects of paroxetine and sertraline on low-density lipoprotein cholesterol: An observational cohort study. CNS Drugs 2009, 23, 857–865. [Google Scholar] [CrossRef]
- Pan, S.-J.; Tan, Y.-L.; Yao, S.-W.; Xin, Y.; Yang, X.; Liu, J.; Xiong, J. Fluoxetine induces lipid metabolism abnormalities by acting on the liver in patients and mice with depression. Acta Pharmacol. Sin. 2018, 39, 1463–1472. [Google Scholar] [CrossRef]
- Kesim, M.; Tiryaki, A.; Kadioglu, M.; Muci, E.; Kalyoncu, N.I.; Yaris, E. The effects of sertraline on blood lipids, glucose, insulin and HBA1C levels: A prospective clinical trial on depressive patients. J. Res. Med. Sci. Off. J. Isfahan Univ. Med. Sci. 2011, 16, 1525–1531. [Google Scholar]
- McIntyre, R.S.; Soczynska, J.K.; Konarski, J.Z.; Kennedy, S.H. The effect of antidepressants on lipid homeostasis: A cardiac safety concern? Expert Opin. Drug Saf. 2006, 5, 523–537. [Google Scholar] [CrossRef]
- Rabe-Jabłońska, J.; Poprawska, I. Levels of serum total cholesterol and LDL-cholesterol in patients with major depression in acute period and remission. Med. Sci. Monit. 2000, 6, 539–547. [Google Scholar]
- Nicholas, L.M.; Ford, A.L.; Esposito, S.M.; Ekstrom, R.D.; Golden, R.N. The effects of mirtazapine on plasma lipid profiles in healthy subjects. J. Clin. Psychiatry 2003, 64, 883–889. [Google Scholar] [CrossRef] [PubMed]
- Kokras, N.; Papadopoulou, E.; Georgiopoulos, G.; Dalla, C.; Petropoulos, I.; Kontogiannis, C.; Laina, A.; Bampatsias, D.; Stellos, K.; Kouzoupis, A.V.; et al. The effect of treatment response on endothelial function and arterial stiffness in depression. A prospective study. J. Affect. Disord. 2019, 252, 190–200. [Google Scholar] [CrossRef] [PubMed]
- Peixoto, M.F.; Cesaretti, M.; Hood, S.D.; Tavares, A. Effects of SSRI medication on heart rate and blood pressure in individuals with hypertension and depression. Clin. Exp. Hypertens. 2019, 41, 428–433. [Google Scholar] [CrossRef]
- Scuteri, A.; Modestino, A.; Fedullo, F.; Assisi, A.P.; Gianni, W. Depression treatment selectively modifies arterial stiffness in older participants. J. Gerontology. Ser. A Biol. Sci. Med. Sci. 2013, 68, 719–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, Z.; Wang, L.; Wen, X.; Liu, Y.; Fan, Y.; Liu, Z. A meta-analysis of effects of selective serotonin reuptake inhibitors on blood pressure in depression treatment: Outcomes from placebo and serotonin and noradrenaline reuptake inhibitor controlled trials. Neuropsychiatr. Dis. Treat. 2017, 13, 2781–2796. [Google Scholar] [CrossRef] [Green Version]
- Thase, M.E.; Larsen, K.G.; Reines, E.; Kennedy, S.H. The cardiovascular safety profile of escitalopram. Eur. Neuropsychopharmacol. 2013, 23, 1391–1400. [Google Scholar] [CrossRef]
- Laurent, S.; Sharman, J.; Boutouyrie, P. Central versus peripheral blood pressure: Finding a solution. J. Hypertens. 2016, 34, 1497–1499. [Google Scholar] [CrossRef]
- Laurent, S.; Cockcroft, J.; Van Bortel, L.; Boutouyrie, P.; Giannattasio, C.; Hayoz, D.; Pannier, B.; Vlachopoulos, C.; Wilkinson, I.; Struijker-Boudier, H. Expert consensus document on arterial stiffness: Methodological issues and clinical applications. Eur. Heart J. 2006, 27, 2588–2605. [Google Scholar] [CrossRef] [Green Version]
- Pucci, G.; Battista, F.; Schillaci, G. Effects of antihypertensive drugs on central blood pressure: New evidence, more challenges. Hypertens. Res. 2014, 37, 10–12. [Google Scholar] [CrossRef]
- Derby, M.A.; Lu, Z.; Chappell, J.C.; Celedon, R.G.; Callaghan, J.T.; Leibowitz, M.; Ereshefsky, L.; Hoelscher, L.; Mitchell, M.I. The Effects of Supratherapeutic Doses of Duloxetine on Blood Pressure and Pulse Rate. J. Cardiovasc. Pharmacol. 2000, 49, 6. [Google Scholar] [CrossRef]
- Diaper, A.; Rich, A.S.; Wilson, S.J.; Craig, K.; Dourish, C.T.; Dawson, G.R.; Nutt, D.J.; Bailey, J.E. Changes in cardiovascular function after venlafaxine but not pregabalin in healthy volunteers: A double-blind, placebo-controlled study of orthostatic challenge, blood pressure and heart rate. Hum. Psychopharmacol. 2013, 28, 562–575. [Google Scholar] [CrossRef]
- Martins, L.C.; Ferreira-Melo, S.E.; Sabha, M.; Coelho, O.R.; Yugar-Toledo, J.C.; Quinaglia, T.; Moreira, M.M.; Coca, A.; Moreno, H. Acute effects of pharmacotherapies in blood pressure in normotensive moderate smokers. Blood Press. 2009, 18, 255–260. [Google Scholar] [CrossRef]
- Thase, M.E.; Haight, B.R.; Johnson, M.C.; Hunt, T.; Krishen, A.; Fleck, R.J.; Modell, J.G. A randomized, double-blind, placebo-controlled study of the effect of sustained-release bupropion on blood pressure in individuals with mild untreated hypertension. J. Clin. Psychopharmacol. 2008, 28, 302–307. [Google Scholar] [CrossRef] [PubMed]
- Dawood, T.; Schlaich, M.; Brown, A.; Lambert, G. Depression and blood pressure control: All antidepressants are not the same. Hypertension 2009, 54, e1. [Google Scholar] [CrossRef] [Green Version]
- Oulis, P.; Kouzoupis, A.; Kyrkou, K.; Masdrakis, V.G.; Georgiopoulos, G.; Karapoulios, E.; Georgiou, S.; Karakatsanis, N.A.; Lykka, M.; Papadimitriou, G.N.; et al. Reversal of increased arterial stiffness in severely depressed women after 6-week antidepressant treatment. J. Affect. Disord. 2010, 122, 164–166. [Google Scholar] [CrossRef]
- Tudoran, R. Toate Pinzele Sus. Available online: tudoran2019.pdf (accessed on 28 March 2021).
- Thase, M.E.; Tran, P.V.; Wiltse, C.; Pangallo, B.A.; Mallinckrodt, C.; Detke, M.J. Cardiovascular profile of duloxetine, a dual reuptake inhibitor of serotonin and norepinephrine. J. Clin. Psychopharmacol. 2005, 25, 132–140. [Google Scholar] [CrossRef]
- Stahl, S.M.; Grady, M.M.; Moret, C.; Briley, M. SNRIs: Their pharmacology, clinical efficacy, and tolerability in comparison with other classes of antidepressants. CNS Spectr. 2005, 10, 732–747. [Google Scholar] [CrossRef] [PubMed]
- Glassman, A.H.; Bigger, J.T., Jr. Cardiovascular effects of therapeutic doses of tricyclic antidepressants. A review. Arch. Gen. Psychiatry 1981, 38, 815–820. [Google Scholar] [CrossRef]
- Glassman, A.H. Cardiovascular Effects of Tricyclic Antidepressants. Annu. Rev. Med. 1984, 35, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Licht, C.M.; de Geus, E.J.; Seldenrijk, A.; van Hout, H.P.; Zitman, F.G.; van Dyck, R.; Penninx, B.W. Depression is associated with decreased blood pressure, but antidepressant use increases the risk for hypertension. Hypertension 2009, 53, 631–638. [Google Scholar] [CrossRef] [Green Version]
- Delaney, J.A.C.; Oddson, B.E.; Kramer, H.; Shea, S.; Psaty, B.M.; McClelland, R.L. Baseline Depressive Symptoms Are Not Associated with Clinically Important Levels of Incident Hypertension During Two Years of Follow-Up. Hypertension 2010, 55, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Cockhill, L.A.; Remick, R.A. Blood pressure effects of monoamine oxidase inhibitors—The highs and lows. Can. J. Psychiatry 1987, 32, 803–808. [Google Scholar] [CrossRef]
- Baker, J.H.; Vgontzas, A.N.; Fernandez-Mendoza, J.; Kirshnamurthy, V.B.; Gaines, J.; Basta, M.; Criley, C.; Bixler, E.O. 0413 Effects of Trazodone on Blood Pressure: A Longitudinal, Observational Study of Patients Presenting to a Sleep Disorder Clinic. Sleep 2018, 41, A157. [Google Scholar] [CrossRef] [Green Version]
- Montgomery, S.A. Safety of mirtazapine: A review. Int. Clin. Psychopharmacol. 1995, 10 (Suppl. S4), 37–45. [Google Scholar] [CrossRef]
- Nitz, K.; Lacy, M.; Atzler, D. Amino Acids and Their Metabolism in Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 319–330. [Google Scholar] [CrossRef] [Green Version]
- Mäki-Petäjä, K.M.; Barrett, S.M.L.; Evans, S.V.; Cheriyan, J.; McEniery, C.M.; Wilkinson, I.B. The Role of the Autonomic Nervous System in the Regulation of Aortic Stiffness. Hypertension 2016, 68, 1290–1297. [Google Scholar] [CrossRef] [PubMed]
- Wernicke, J.; Lledó, A.; Raskin, J.; Kajdasz, D.K.; Wang, F. An evaluation of the cardiovascular safety profile of duloxetine: Findings from 42 placebo-controlled studies. Drug Saf. 2007, 30, 437–455. [Google Scholar] [CrossRef] [PubMed]
- Broadley, A.J.M.; Korszun, A.; Jones, C.J.H.; Frenneaux, M.P. Arterial endothelial function is impaired in treated depression. Heart 2002, 88, 521–523. [Google Scholar] [CrossRef] [Green Version]
- Crookes, D.M.; Demmer, R.T.; Keyes, K.M.; Koenen, K.S.; Suglia, S.M. Depressive Symptoms, Antidepressant Use, and Hypertension in Young Adulthood. Epidemiology 2018, 29, 547–555. [Google Scholar] [CrossRef]
- Paranthaman, R.; Greenstein, A.; Burns, A.S.; Heagerty, A.M.; Malik, R.A.; Baldwin, R.C. Relationship of endothelial function and atherosclerosis to treatment response in late-life depression. Int. J. Geriatr. Psychiatry 2012, 27, 967–973. [Google Scholar] [CrossRef]
- Camacho, A.; McClelland, R.L.; Delaney, J.A.; Allison, M.A.; Psaty, B.M.; Rifkin, D.E.; Rapp, S.R.; Szklo, M.; Stein, M.B.; Criqui, M.H. Antidepressant Use and Subclinical Measures of Atherosclerosis: The Multi-Ethnic Study of Atherosclerosis. J. Clin. Psychopharmacol. 2016, 36, 340–346. [Google Scholar] [CrossRef] [PubMed]
- Wernicke, J.F.; Prakash, A.; Kajdasz, D.K.; Houston, J. Safety and tolerability of duloxetine treatment of diabetic peripheral neuropathic pain between patients with and without cardiovascular conditions. J. Diabetes Complicat. 2009, 23, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Palmer, S.C.; Natale, P.; Ruospo, M.; Saglimbene, V.M.; Rabindranath, K.S.; Craig, J.C.; Strippoli, G.F. Antidepressants for treating depression in adults with end-stage kidney disease treated with dialysis. Cochrane Database Syst. Rev. 2016, 5, Cd004541. [Google Scholar] [CrossRef] [PubMed]
- Hankey, G.J.; Hackett, M.L.; Almeida, O.P.; Flicker, L.; Mead, G.E.; Dennis, M.S.; Bunce, L. Safety and efficacy of fluoxetine on functional outcome after acute stroke (AFFINITY): A randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2020, 19, 651–660. [Google Scholar] [CrossRef]
- Lundström, E.; Isaksson, E.; Näsman, P.; Wester, P.; Mårtensson, B.; Norrving, B.; Sunnerhagen, K.S. Safety and efficacy of fluoxetine on functional recovery after acute stroke (EFFECTS): A randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2020, 19, 661–669. [Google Scholar] [CrossRef]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef]
- Arnett, D.K.; Blumenthal, R.S.; Albert, M.A.; Buroker, A.B.; Goldberger, Z.D.; Hahn, E.J.; Himmelfarb, C.D.; Khera, A.; Lloyd-Jones, D.; McEvoy, J.W.; et al. 2019 ACC/AHA Guideline on the Primary Prevention of Cardiovascular Disease: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J. Am. Coll. Cardiol. 2019, 74, 1376–1414. [Google Scholar] [CrossRef]
- Patel, K.V.; Pandey, A.; de Lemos, J.A. Conceptual Framework for Addressing Residual Atherosclerotic Cardiovascular Disease Risk in the Era of Precision Medicine. Circulation 2018, 137, 2551–2553. [Google Scholar] [CrossRef]
- Elenkov, I.J.; Chrousos, G.P. Stress hormones, proinflammatory and antiinflammatory cytokines, and autoimmunity. Ann. N. Y. Acad. Sci. 2002, 966, 290–303. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dimoula, A.; Fotellis, D.; Aivalioti, E.; Delialis, D.; Polissidis, A.; Patras, R.; Kokras, N.; Stamatelopoulos, K. Off-Target Effects of Antidepressants on Vascular Function and Structure. Biomedicines 2022, 10, 56. https://doi.org/10.3390/biomedicines10010056
Dimoula A, Fotellis D, Aivalioti E, Delialis D, Polissidis A, Patras R, Kokras N, Stamatelopoulos K. Off-Target Effects of Antidepressants on Vascular Function and Structure. Biomedicines. 2022; 10(1):56. https://doi.org/10.3390/biomedicines10010056
Chicago/Turabian StyleDimoula, Anna, Dimitrios Fotellis, Evmorfia Aivalioti, Dimitrios Delialis, Alexia Polissidis, Raphael Patras, Nikolaos Kokras, and Kimon Stamatelopoulos. 2022. "Off-Target Effects of Antidepressants on Vascular Function and Structure" Biomedicines 10, no. 1: 56. https://doi.org/10.3390/biomedicines10010056