
Proteomic Profiling and Rhizosphere-Associated Microbial Communities Reveal Adaptive Mechanisms of Dioclea apurensis Kunth in Eastern Amazon’s Rehabilitating Minelands

 , , , and
, , , and
Abstract
:1. Introduction
2. Results
2.1. Physical and Chemical Properties of Canga and Mining Area Soil Substrates
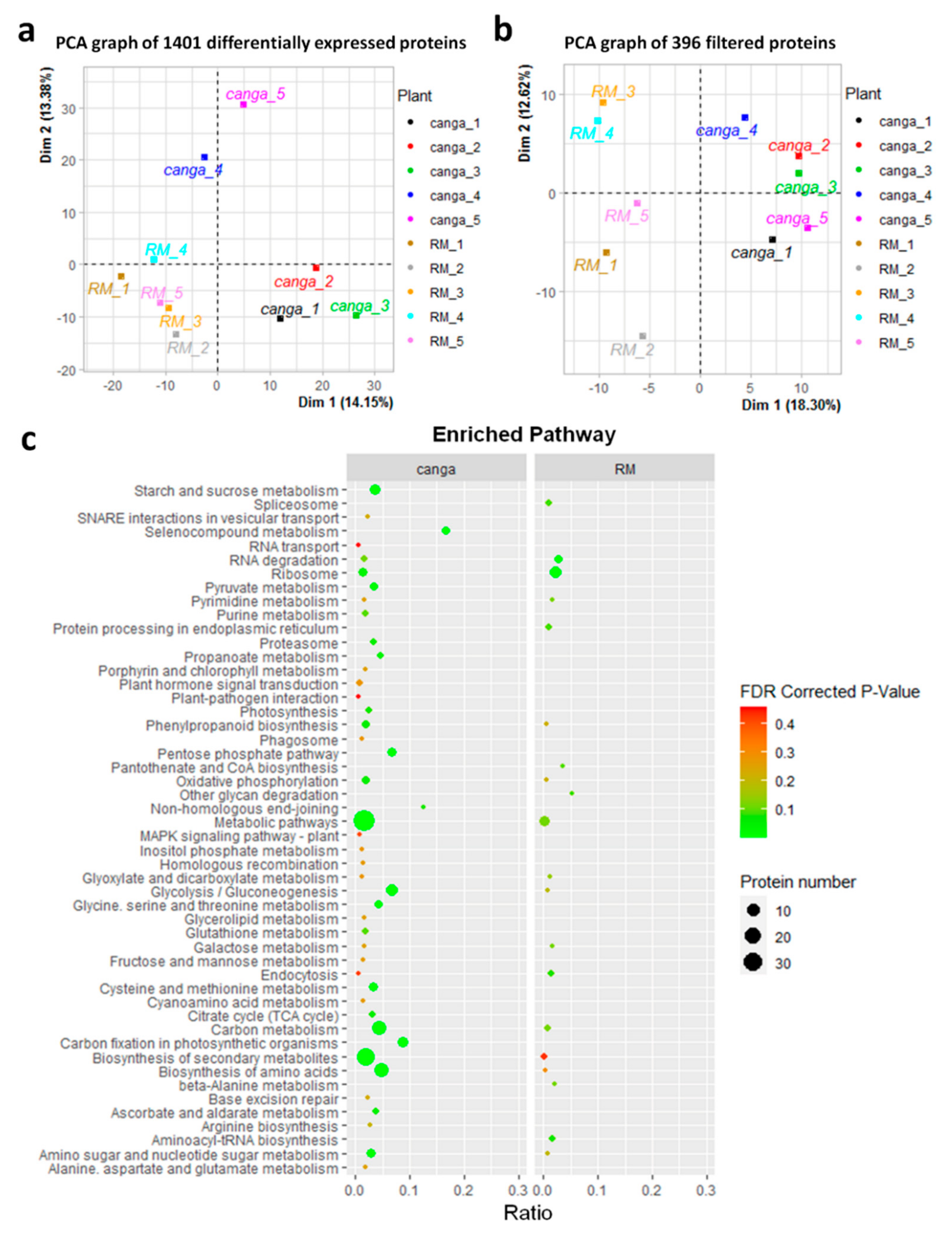
2.2. Protein Profiles, Annotation, and Functional Enrichment
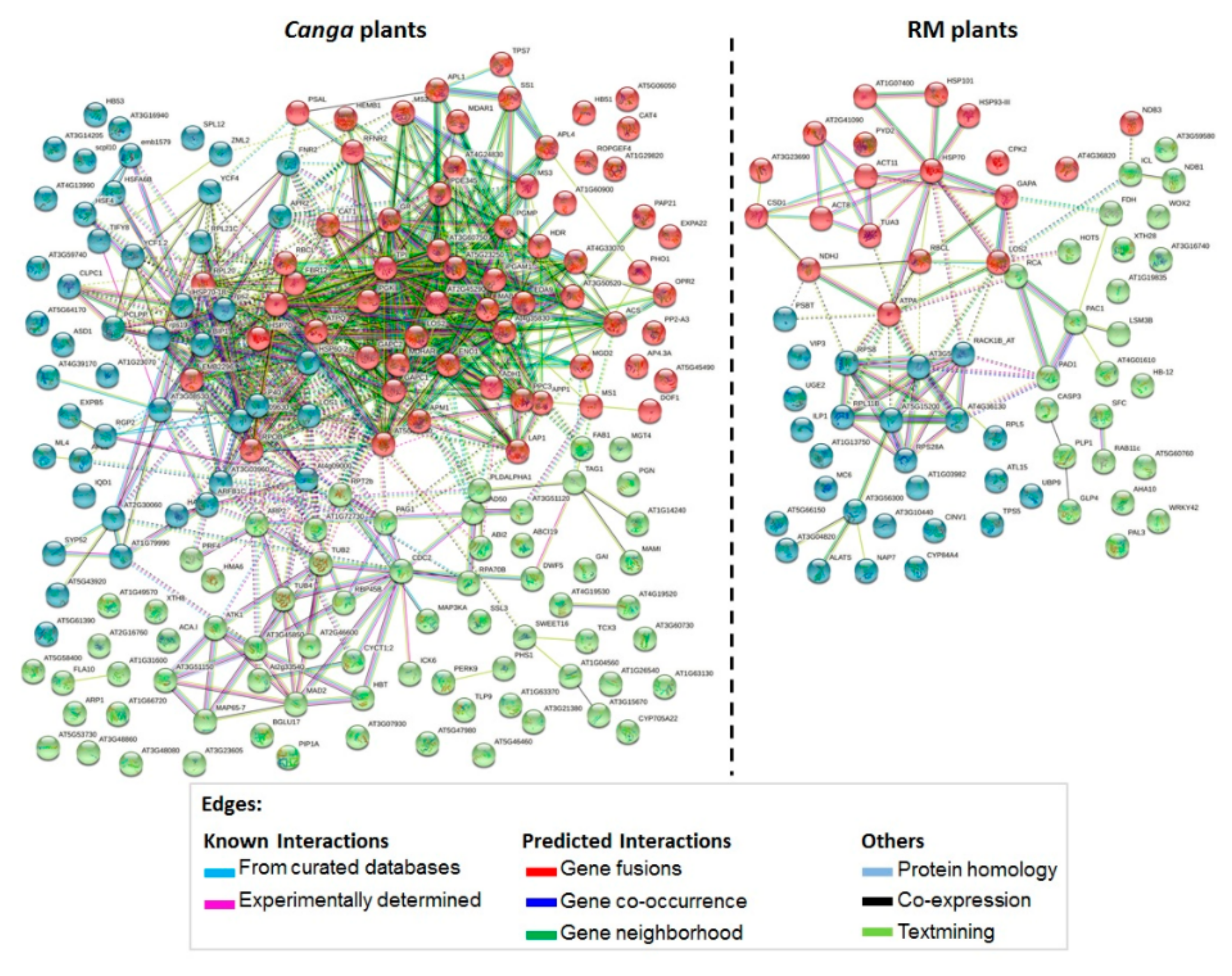
2.3. Protein–Protein Interactions (PPI)
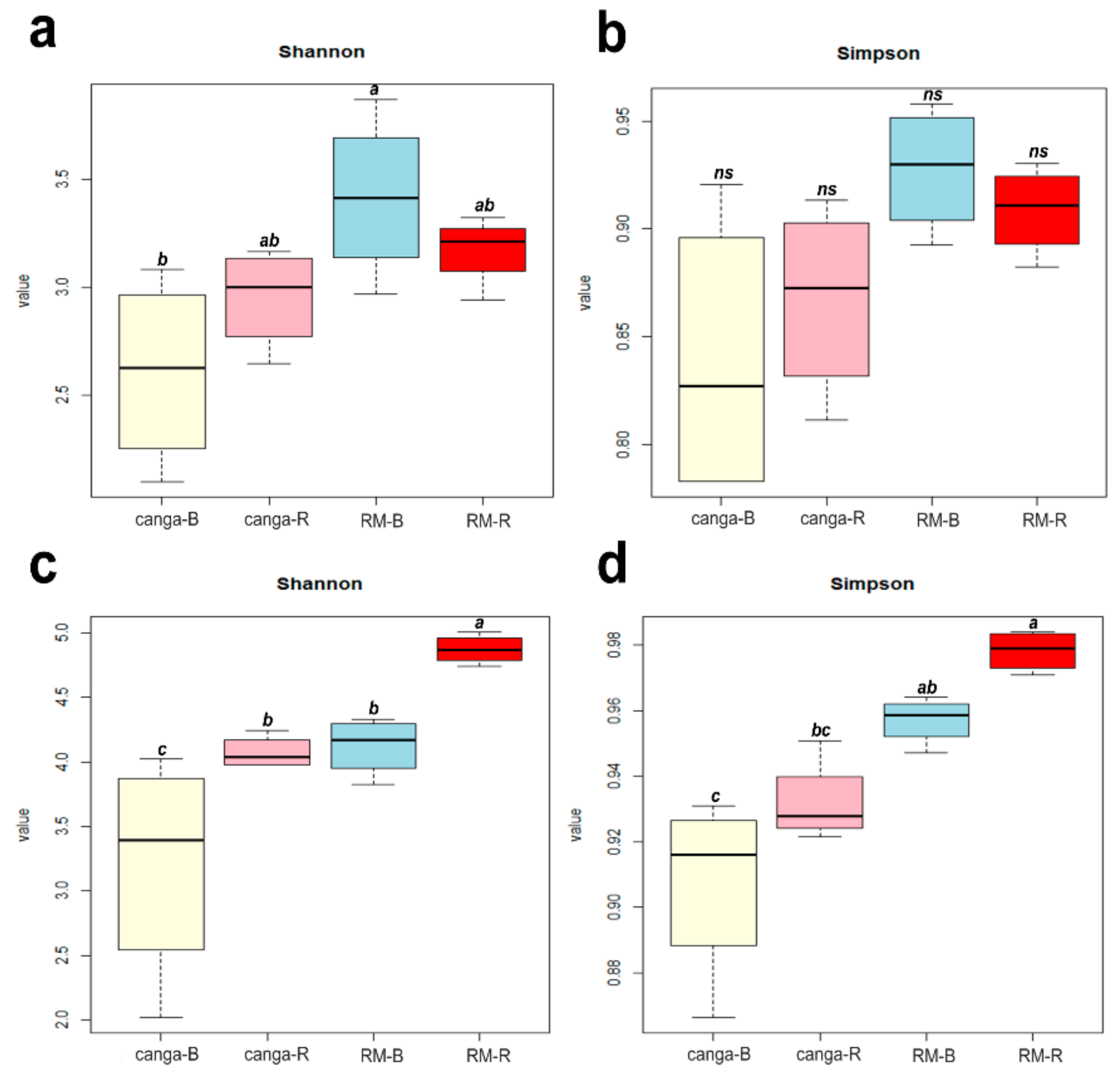
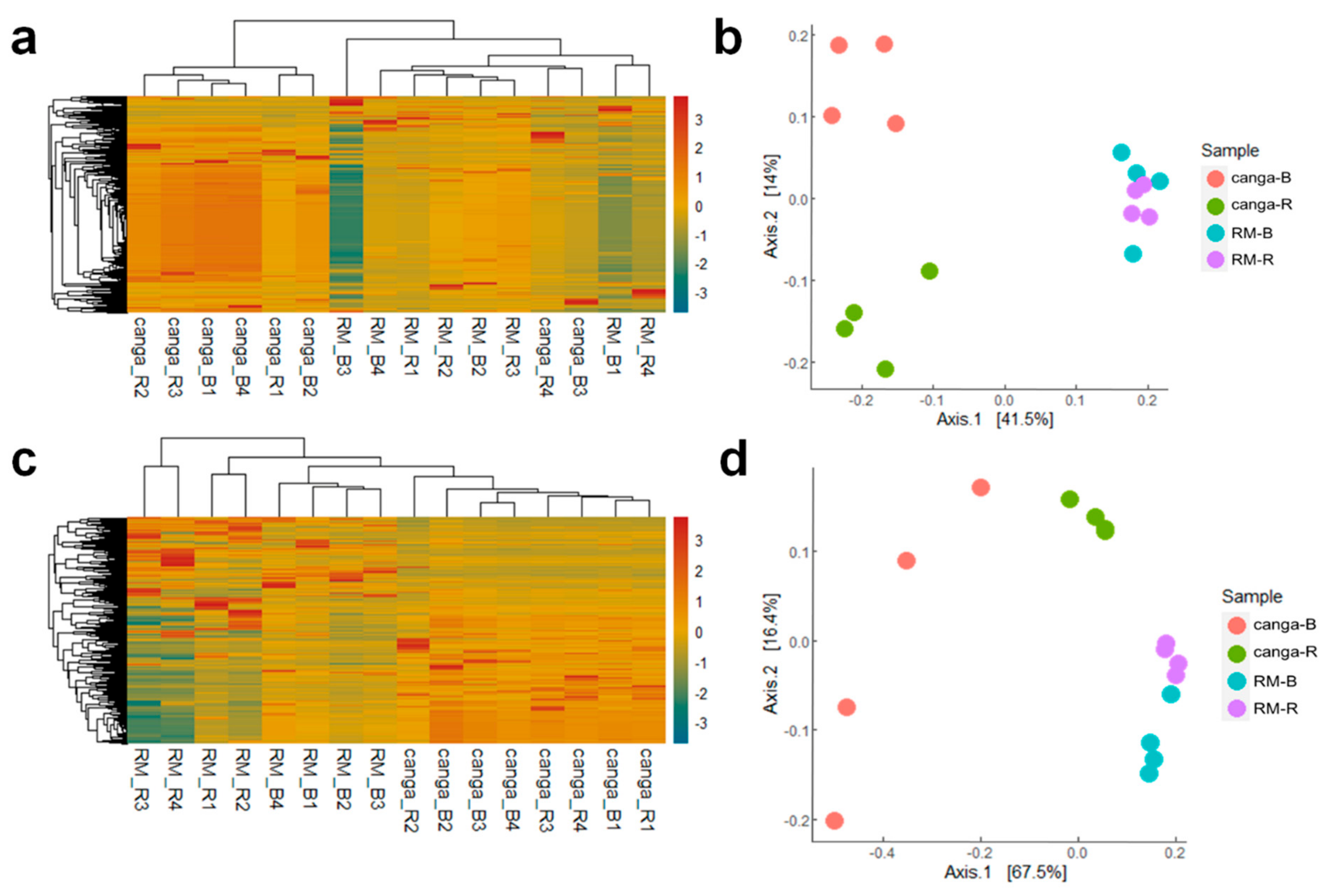
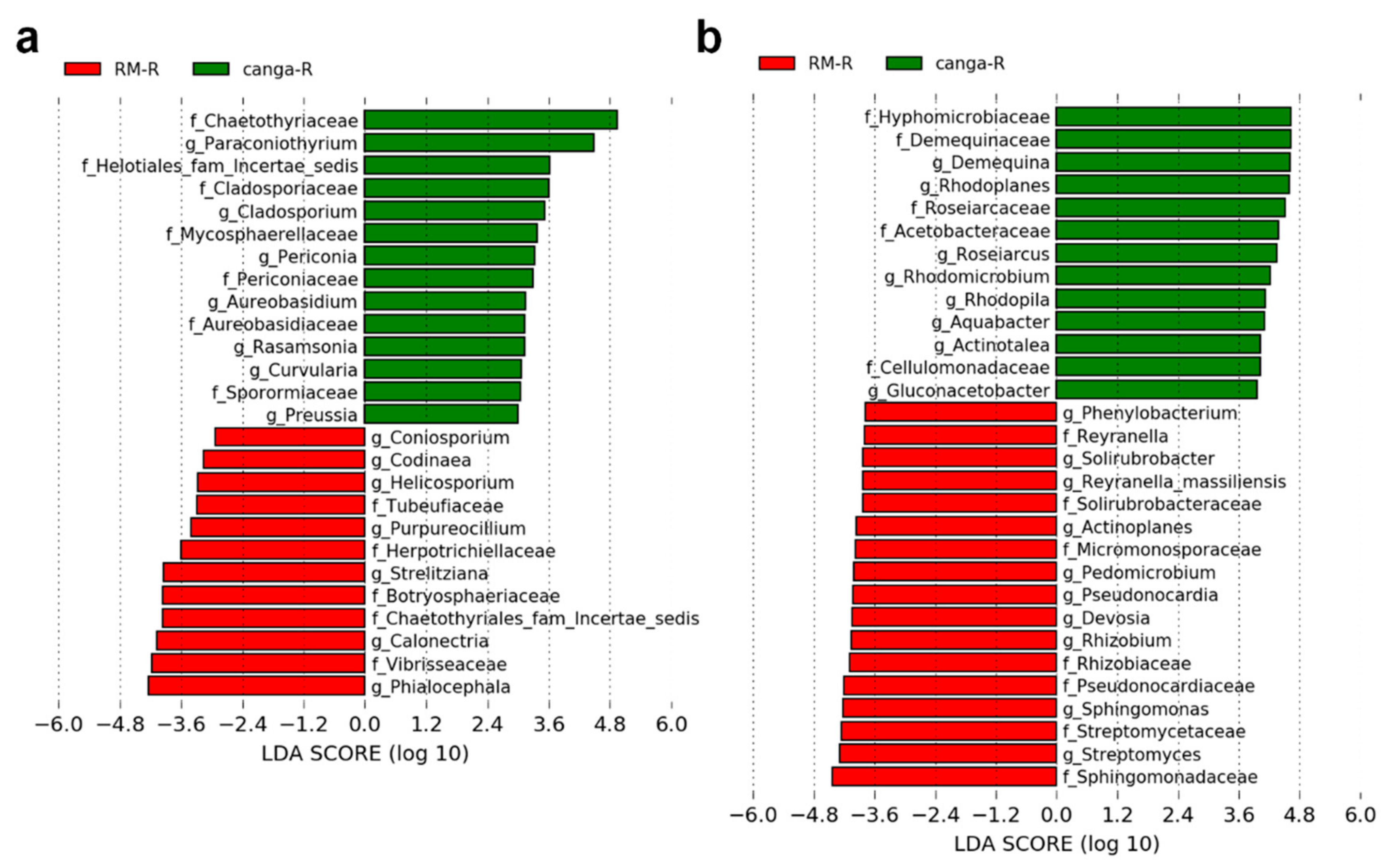
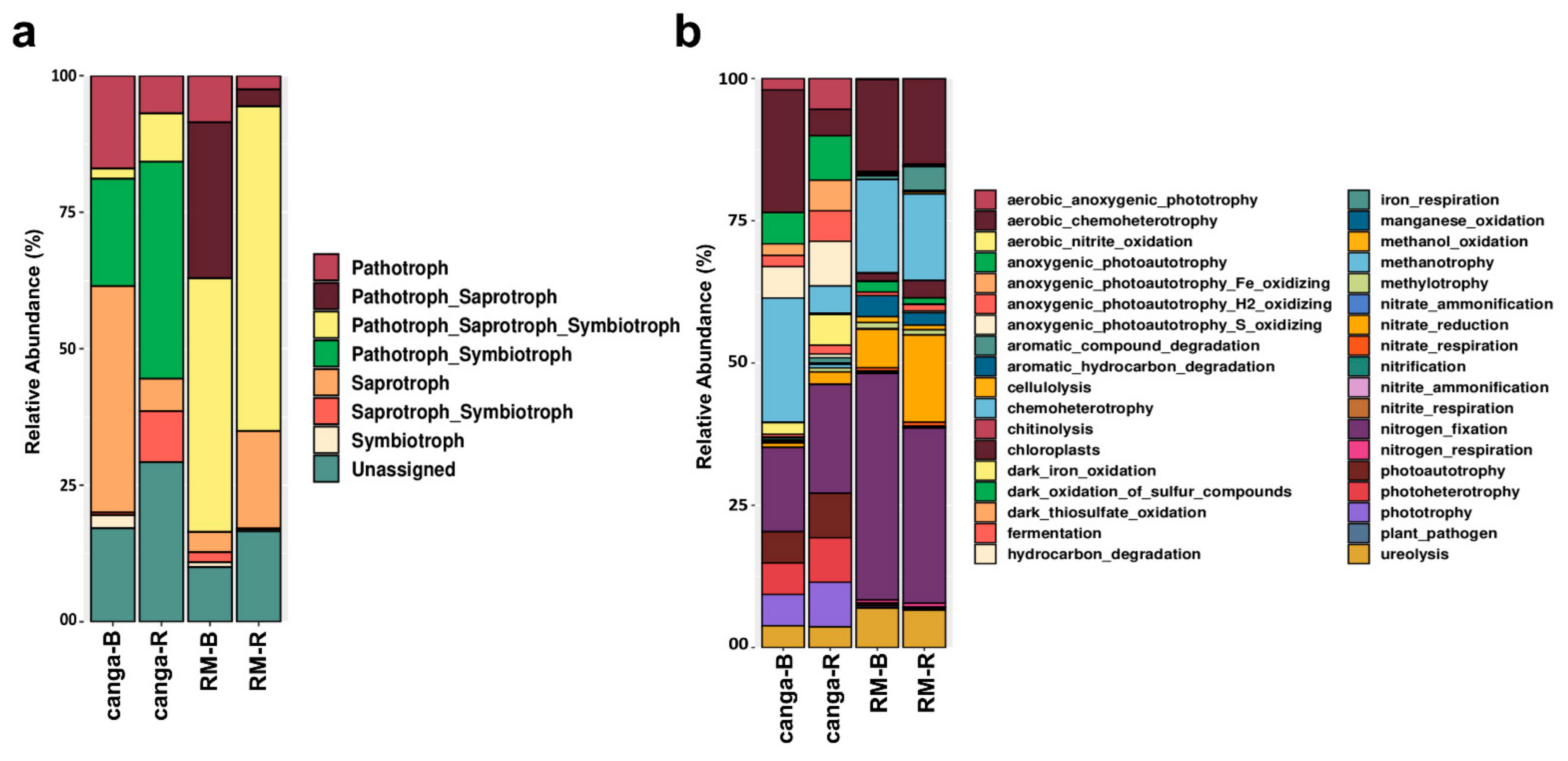
2.4. Microbial Diversity
3. Discussion
3.1. Physical and Chemical Properties of Canga and Mining Area Soil Substrates
3.2. Proteins Involved in the Response to Water Deficit
3.3. Proteins Involved in the Response to Metal Stress
3.4. Proteins Involved in the Response to P-Starvation
3.5. Proteins Involved in Symbiosis
3.6. Rhizosphere-Associated Microbial Communities
4. Materials and Methods
4.1. Soil Substrate Sampling and Chemical Analyses
4.2. Root Sampling and Protein Isolation
4.3. Proteome Analysis
4.4. Microbial Diversity
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- dos Santos, R.S.P.; Milanez, B. The Global Production Network for iron ore: Materiality, corporate strategies, and social contestation in Brazil. Extr. Ind. Soc. 2015, 2, 756–765. [Google Scholar] [CrossRef]
- Jacobi, C.M.; Do Carmo, F.F.; Vincent, R.C.; Stehmann, J.R. Plant communities on ironstone outcrops: A diverse and endangered Brazilian ecosystem. Biodivers. Conserv. 2007, 16, 2185–2200. [Google Scholar] [CrossRef]
- Skirycz, A.; Castilho, A.; Chaparro, C.; Carvalho, N.; Tzotzos, G.; Siqueira, J.O. Canga biodiversity, a matter of mining. Front. Plant Sci. 2014, 5, 653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann, J.; da Silva Cravo, M.; de Macêdo, J.L.V.; Moreira, A.; Schroth, G. Phosphorus management for perennial crops in central Amazonian upland soils. Plant Soil 2001, 237, 309–319. [Google Scholar] [CrossRef]
- Gastauer, M.; Souza Filho, P.W.M.; Ramos, S.J.; Caldeira, C.F.; Silva, J.R.; Siqueira, J.O.; Neto, A.E.F. Mine land rehabilitation in Brazil: Goals and techniques in the context of legal requirements. Ambio 2019, 48, 74–88. [Google Scholar] [CrossRef]
- Carvalho, C.S.; Forester, B.R.; Mitre, S.K.; Alves, R.; Imperatriz-Fonseca, V.L.; Ramos, S.J.; Resende-Moreira, L.C.; Siqueira, J.O.; Trevelin, L.C.; Caldeira, C.F. Combining genotype, phenotype, and environmental data to delineate site-adjusted provenance strategies for ecological restoration. Mol. Ecol. Resour. 2021, 21, 44–58. [Google Scholar] [CrossRef]
- Ramos, S.J.; Gastauer, M.; Mitre, S.K.; Caldeira, C.F.; Silva, J.R.; Neto, A.E.F.; Oliveira, G.; Souza Filho, P.W.; Siqueira, J.O. Plant growth and nutrient use efficiency of two native Fabaceae species for mineland revegetation in the eastern Amazon. J. For. Res. 2020, 31, 2287–2293. [Google Scholar] [CrossRef]
- Costa, P.H.d.O.; Nascimento, S.V.d.; Herrera, H.; Gastauer, M.; Ramos, S.J.; Caldeira, C.F.; Oliveira, G.; Valadares, R.B.d.S. Non-Specific Interactions of Rhizospheric Microbial Communities Support the Establishment of Mimosa acutistipula var. ferrea in an Amazon Rehabilitating Mineland. Processes 2021, 9, 2079. [Google Scholar] [CrossRef]
- Gastauer, M.; de Medeiros Sarmento, P.S.; Santos, V.C.A.; Caldeira, C.F.; Ramos, S.J.; Teodoro, G.S.; Siqueira, J.O. Vegetative functional traits guide plant species selection for initial mineland rehabilitation. Ecol. Eng. 2020, 148, 105763. [Google Scholar] [CrossRef]
- Queiroz, L.P.d. Leguminosas da Caatinga; Universidad Estadual de Feira de Santana: Feira de Santana, Brazil, 2009. [Google Scholar]
- Ramos, S.J.; Caldeira, C.F.; Gastauer, M.; Costa, D.L.P.; Neto, A.E.F.; de Souza, F.B.M.; Souza-Filho, P.W.M.; Siqueira, J.O. Native leguminous plants for mineland revegetation in the eastern Amazon: Seed characteristics and germination. New For. 2019, 50, 859–872. [Google Scholar] [CrossRef]
- Giannini, T.C.; Giulietti, A.M.; Harley, R.M.; Viana, P.L.; Jaffe, R.; Alves, R.; Pinto, C.E.; Mota, N.F.; Caldeira, C.F., Jr.; Imperatriz-Fonseca, V.L. Selecting plant species for practical restoration of degraded lands using a multiple-trait approach. Austral Ecol. 2017, 42, 510–521. [Google Scholar] [CrossRef]
- Aggangan, N.S.; Anarna, J.A.; Cadiz, N.M. Tree legume–microbial symbiosis and other soil amendments as rehabilitation strategies in mine tailings in the Philippines. Philipp. J. Sci. 2019, 148, 481–491. [Google Scholar]
- Jasper, D.A. Beneficial soil microorganisms of the jarrah forest and their recovery in bauxite mine restoration in Southwestern Australia. Restor. Ecol. 2007, 15, S74–S84. [Google Scholar] [CrossRef]
- de Oliveira-Longatti, S.M.; Marra, L.M.; Lima Soares, B.; Bomfeti, C.A.; Da Silva, K.; Avelar Ferreira, P.A.; de Souza Moreira, F.M. Bacteria isolated from soils of the western Amazon and from rehabilitated bauxite-mining areas have potential as plant growth promoters. World J. Microbiol. Biotechnol. 2014, 30, 1239–1250. [Google Scholar] [CrossRef]
- Oliveira Silva, A.; Azarias Guimarães, A.; da Costa, A.M.; Louzada Rodrigues, T.; de Soares Carvalho, T.; Reis Sales, F.; de Souza Moreira, F.M. Plant growth-promoting rhizobacterial communities from an area under the influence of iron mining and from the adjacent phytophysiognomies which have high genetic diversity. Land Degrad. Dev. 2020, 31, 2237–2254. [Google Scholar] [CrossRef]
- Herrera, H.; Fuentes, A.; Ortiz, J.; Soto, J.; da Silva Valadares, R.B.; Salas-Eljatib, C.; Arriagada, C. Root-associated endophytes isolated from juvenile Ulex europaeus L. (Fabaceae) plants colonizing rural areas in South-Central Chile. Plant Soil 2022, 1–13. [Google Scholar] [CrossRef]
- Lugtenberg, B.J.; Malfanova, N.; Kamilova, F.; Berg, G. Plant growth promotion by microbes. Mol. Microb. Ecol. Rhizosphere 2013, 2, 561–573. [Google Scholar]
- Xie, H.; Yang, D.-H.; Yao, H.; Bai, G.; Zhang, Y.-H.; Xiao, B.-G. iTRAQ-based quantitative proteomic analysis reveals proteomic changes in leaves of cultivated tobacco (Nicotiana tabacum) in response to drought stress. Biochem. Biophys. Res. Commun. 2016, 469, 768–775. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, P.; Jaleel, C.A.; Salem, M.A.; Nabi, G.; Sharma, S. Roles of enzymatic and nonenzymatic antioxidants in plants during abiotic stress. Crit. Rev. Biotechnol. 2010, 30, 161–175. [Google Scholar] [CrossRef]
- Li, R.; Wang, J.; Li, S.; Zhang, L.; Qi, C.; Weeda, S.; Zhao, B.; Ren, S.; Guo, Y.-D. Plasma membrane intrinsic proteins SlPIP2; 1, SlPIP2; 7 and SlPIP2; 5 conferring enhanced drought stress tolerance in tomato. Sci. Rep. 2016, 6, 31814. [Google Scholar] [CrossRef] [Green Version]
- Caldeira, C.F.; Jeanguenin, L.; Chaumont, F.; Tardieu, F. Circadian rhythms of hydraulic conductance and growth are enhanced by drought and improve plant performance. Nat. Commun. 2014, 5, 5365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paudel, I.; Gerbi, H.; Zisovich, A.; Sapir, G.; Ben-Dor, S.; Brumfeld, V.; Klein, T. Drought tolerance mechanisms and aquaporin expression of wild vs. cultivated pear tree species in the field. Environ. Exp. Bot. 2019, 167, 103832. [Google Scholar] [CrossRef]
- González-Villagra, J.; Kurepin, L.V.; Reyes-Díaz, M.M. Evaluating the involvement and interaction of abscisic acid and miRNA156 in the induction of anthocyanin biosynthesis in drought-stressed plants. Planta 2017, 246, 299–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baek, D.; Kim, M.C.; Kumar, D.; Park, B.; Cheong, M.S.; Choi, W.; Park, H.C.; Chun, H.J.; Park, H.J.; Lee, S.Y. AtPR5K2, a PR5-like receptor kinase, modulates plant responses to drought stress by phosphorylating protein phosphatase 2Cs. Front. Plant Sci. 2019, 10, 1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Wang, M.; Guo, C.; Wang, N.; Li, X.; Chen, H.; Dong, Y.; Chen, X.; Wang, Z.; Li, H. Cloning and characterization of a novel betaine aldehyde dehydrogenase gene from Suaeda corniculata. Genet. Mol. Res. 2016, 15, gmr.1502784. [Google Scholar] [CrossRef] [PubMed]
- Golestan Hashemi, F.S.; Ismail, M.R.; Rafii, M.Y.; Aslani, F.; Miah, G.; Muharam, F.M. Critical multifunctional role of the betaine aldehyde dehydrogenase gene in plants. Biotechnol. Biotechnol. Equip. 2018, 32, 815–829. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Zhou, Y.; Fan, J.; Fu, Y.; Shen, L.; Yao, Y.; Li, R.; Fu, S.; Duan, R.; Hu, X. SpBADH of the halophyte Sesuvium portulacastrum strongly confers drought tolerance through ROS scavenging in transgenic Arabidopsis. Plant Physiol. Biochem. 2015, 96, 377–387. [Google Scholar] [CrossRef]
- Zhang, L.; Gao, M.; Hu, J.; Zhang, X.; Wang, K.; Ashraf, M. Modulation role of abscisic acid (ABA) on growth, water relations and glycinebetaine metabolism in two maize (Zea mays L.) cultivars under drought stress. Int. J. Mol. Sci. 2012, 13, 3189–3202. [Google Scholar] [CrossRef] [Green Version]
- Satish, L.; Rency, A.S.; Ramesh, M. Spermidine sprays alleviate the water deficit-induced oxidative stress in finger millet (Eleusine coracana L. Gaertn.) plants. 3 Biotech 2018, 8, 63. [Google Scholar] [CrossRef]
- Bai, L.; Zhang, G.; Zhou, Y.; Zhang, Z.; Wang, W.; Du, Y.; Wu, Z.; Song, C.P. Plasma membrane-associated proline-rich extensin-like receptor kinase 4, a novel regulator of Ca2+ signalling, is required for abscisic acid responses in Arabidopsis thaliana. Plant J. 2009, 60, 314–327. [Google Scholar] [CrossRef]
- Tiwari, S.; Lata, C. Heavy metal stress, signaling, and tolerance due to plant-associated microbes: An overview. Front. Plant Sci. 2018, 9, 452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jalmi, S.K.; Bhagat, P.K.; Verma, D.; Noryang, S.; Tayyeba, S.; Singh, K.; Sharma, D.; Sinha, A.K. Traversing the links between heavy metal stress and plant signaling. Front. Plant Sci. 2018, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Li, T.; Wang, J.; Zhao, Z. Diverse strategies conferring extreme cadmium (Cd) tolerance in the dark septate endophyte (DSE), Exophiala pisciphila: Evidence from RNA-seq data. Microbiol. Res. 2015, 170, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Erbasol, I.; Bozdag, G.O.; Koc, A.; Pedas, P.; Karakaya, H.C. Characterization of two genes encoding metal tolerance proteins from Beta vulgaris subspecies maritima that confers manganese tolerance in yeast. Biometals 2013, 26, 795–804. [Google Scholar] [CrossRef] [Green Version]
- Vatansever, R.; Filiz, E.; Eroglu, S. Genome-wide exploration of metal tolerance protein (MTP) genes in common wheat (Triticum aestivum): Insights into metal homeostasis and biofortification. Biometals 2017, 30, 217–235. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Bi, Y.; Zhang, R.; Feng, S. Expression of RcHSP70, heat shock protein 70 gene from Chinese rose, enhances host resistance to abiotic stresses. Sci. Rep. 2020, 10, 2445. [Google Scholar] [CrossRef] [PubMed]
- Kottmann, L.; Wilde, P.; Schittenhelm, S. How do timing, duration, and intensity of drought stress affect the agronomic performance of winter rye? Eur. J. Agron. 2016, 75, 25–32. [Google Scholar] [CrossRef]
- Shevyakova, N.; Eshinimaeva, B.T.; Kuznetsov, V.V. Expression of ferritin gene in Mesembryanthemum crystallinum plants under different supply with iron and different intensity of oxidative stress. Russ. J. Plant Physiol. 2011, 58, 768–775. [Google Scholar] [CrossRef]
- Stein, R.J.; Ricachenevsky, F.K.; Fett, J.P. Differential regulation of the two rice ferritin genes (OsFER1 and OsFER2). Plant Sci. 2009, 177, 563–569. [Google Scholar] [CrossRef]
- Fink, J.R.; Inda, A.V.; Tiecher, T.; Barrón, V. Iron oxides and organic matter on soil phosphorus availability. Cienc. Agrotecnol. 2016, 40, 369–379. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Ding, D.; Li, J.; He, L.; Xu, X.; Zhao, Y.; Yan, B.; Li, Z.; Xu, J. Characterisation of genes involved in galactolipids and sulfolipids metabolism in maize and Arabidopsis and their differential responses to phosphate deficiency. Funct. Plant Biol. 2020, 47, 279–292. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.L.; Yao, H.Y.; Jia, L.H.; Tan, J.F.; Xu, Z.H.; Zheng, W.M.; Xue, H.W. Phospholipase D-derived phosphatidic acid promotes root hair development under phosphorus deficiency by suppressing vacuolar degradation of PIN-FORMED2. New Phytol. 2020, 226, 142. [Google Scholar] [CrossRef]
- Tran, H.T.; Hurley, B.A.; Plaxton, W.C. Feeding hungry plants: The role of purple acid phosphatases in phosphate nutrition. Plant Sci. 2010, 179, 14–27. [Google Scholar] [CrossRef]
- Shimojima, M.; Watanabe, T.; Madoka, Y.; Koizumi, R.; Yamamoto, M.P.; Masuda, K.; Yamada, K.; Masuda, S.; Ohta, H. Differential regulation of two types of monogalactosyldiacylglycerol synthase in membrane lipid remodeling under phosphate-limited conditions in sesame plants. Front. Plant Sci. 2013, 4, 469. [Google Scholar] [CrossRef] [Green Version]
- Shimojima, M.; Madoka, Y.; Fujiwara, R.; Murakawa, M.; Yoshitake, Y.; Ikeda, K.; Koizumi, R.; Endo, K.; Ozaki, K.; Ohta, H. An engineered lipid remodeling system using a galactolipid synthase promoter during phosphate starvation enhances oil accumulation in plants. Front. Plant Sci. 2015, 6, 664. [Google Scholar] [CrossRef] [Green Version]
- Alexova, R.; Millar, A.H. Proteomics of phosphate use and deprivation in plants. Proteomics 2013, 13, 609–623. [Google Scholar] [CrossRef]
- Chevalier, F.; Rossignol, M. Proteomic analysis of Arabidopsis thaliana ecotypes with contrasted root architecture in response to phosphate deficiency. J. Plant Physiol. 2011, 168, 1885–1890. [Google Scholar] [CrossRef] [PubMed]
- Muneer, S.; Jeong, B.R. Proteomic analysis provides new insights in phosphorus homeostasis subjected to pi (inorganic phosphate) starvation in tomato plants (Solanum lycopersicum L.). PLoS ONE 2015, 10, e0134103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Jiao, F.; Wu, Z.; Li, Y.; Wang, X.; He, X.; Zhong, W.; Wu, P. OsPHR2 is involved in phosphate-starvation signaling and excessive phosphate accumulation in shoots of plants. Plant Physiol. 2008, 146, 1673–1686. [Google Scholar] [CrossRef] [Green Version]
- Aleksza, D.; Horváth, G.V.; Sándor, G.; Szabados, L. Proline accumulation is regulated by transcription factors associated with phosphate starvation. Plant Physiol. 2017, 175, 555–567. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, K.; Ogiwara, N.; Kaji, T.; Sugimoto, Y.; Ueno, M.; Sonoda, M.; Matsui, A.; Ishida, J.; Tanaka, M.; Totoki, Y. Transcriptome analysis of soybean (Glycine max) root genes differentially expressed in rhizobial, arbuscular mycorrhizal, and dual symbiosis. J. Plant Res. 2019, 132, 541–568. [Google Scholar] [CrossRef] [PubMed]
- Vadassery, J.; Tripathi, S.; Prasad, R.; Varma, A.; Oelmüller, R. Monodehydroascorbate reductase 2 and dehydroascorbate reductase 5 are crucial for a mutualistic interaction between Piriformospora indica and Arabidopsis. J. Plant Physiol. 2009, 166, 1263–1274. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Yin, J.; Peng, Y.; Xie, J.; Wu, H.; He, D.; Li, X.; Cheng, G. Glutathione is Involved in Detoxification of Peroxide and Root Nodule Symbiosis of Mesorhizobium huakuii. Curr. Microbiol. 2020, 77, 1–10. [Google Scholar] [CrossRef]
- Nunes, J.A.; Schaefer, C.E.; Ferreira Júnior, W.G.; Neri, A.V.; Correa, G.R.; Enright, N.J. Soil-vegetation relationships on a banded ironstone ‘island’, Carajás Plateau, Brazilian Eastern Amazonia. An. Acad. Bras. Ciências 2015, 87, 2097–2110. [Google Scholar] [CrossRef]
- Thavamani, P.; Samkumar, R.A.; Satheesh, V.; Subashchandrabose, S.R.; Ramadass, K.; Naidu, R.; Venkateswarlu, K.; Megharaj, M. Microbes from mined sites: Harnessing their potential for reclamation of derelict mine sites. Environ. Pollut. 2017, 230, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Fuentes, A.; Herrera, H.; Charles, T.C.; Arriagada, C. Fungal and Bacterial Microbiome Associated with the Rhizosphere of Native Plants from the Atacama Desert. Microorganisms 2020, 8, 209. [Google Scholar] [CrossRef] [Green Version]
- Herrera, H.; Novotná, A.; Ortiz, J.; Soto, J.; Arriagada, C. Isolation and identification of plant growth-promoting bacteria from rhizomes of Arachnitis uniflora, a fully mycoheterotrophic plant in southern Chile. Appl. Soil Ecol. 2020, 149, 103512. [Google Scholar] [CrossRef]
- Fowler, W.M.; Fontaine, J.B.; Enright, N.J.; Veber, W.P. Evaluating restoration potential of transferred topsoil. Appl. Veg. Sci. 2015, 18, 379–390. [Google Scholar] [CrossRef] [Green Version]
- Silva, J.R.; Gastauer, M.; Ramos, S.J.; Mitre, S.K.; Neto, A.E.F.; Siqueira, J.O.; Caldeira, C.F. Initial growth of Fabaceae species: Combined effects of topsoil and fertilizer application for mineland revegetation. Flora 2018, 246, 109–117. [Google Scholar] [CrossRef]
- Dastogeer, K.M.; Li, H.; Sivasithamparam, K.; Wylie, S.J. In vitro salt and thermal tolerance of fungal endophytes of Nicotiana spp. growing in arid regions of north-western Australia. Arch. Phytopathol. Plant Prot. 2018, 51, 602–616. [Google Scholar] [CrossRef]
- Dardanelli, M.S.; González, P.S.; Medeot, D.B.; Paulucci, N.S.; Bueno, M.A.; Garcia, M.B. Effects of peanut rhizobia on the growth and symbiotic performance of Arachis hypogaea under abiotic stress. Symbiosis 2009, 47, 175–180. [Google Scholar] [CrossRef]
- Zhan, F.; Li, B.; Jiang, M.; Li, T.; He, Y.; Li, Y.; Wang, Y. Effects of arbuscular mycorrhizal fungi on the growth and heavy metal accumulation of bermudagrass [Cynodon dactylon (L.) Pers.] grown in a lead–zinc mine wasteland. Int. J. Phytoremediat. 2019, 21, 849–856. [Google Scholar] [CrossRef] [PubMed]
- Trindade, F.C.; Ramos, S.J.; Gastauer, M.; Saraiva, A.M.M.; Caldeira, C.F.; Oliveira, G.; da Silva Valadares, R.B. Metaproteomes reveal increased capacity for stress tolerance of soil microbes in ferruginous tropical rocky outcrops. Pedobiologia 2020, 81, 150664. [Google Scholar] [CrossRef]
- Viana, P.L.; Mota, N.F.d.O.; Gil, A.d.S.B.; Salino, A.; Zappi, D.C.; Harley, R.M.; Ilkiu-Borges, A.L.; Secco, R.d.S.; Almeida, T.E.; Watanabe, M.T.C. Flora das cangas da Serra dos Carajás, Pará, Brasil: História, área de estudos e metodologia. Rodriguésia 2016, 67, 1107–1124. [Google Scholar] [CrossRef]
- Kirk, P.L. Kjeldahl method for total nitrogen. Anal. Chem. 1950, 22, 354–358. [Google Scholar] [CrossRef]
- Kettler, T.; Doran, J.W.; Gilbert, T. Simplified method for soil particle-size determination to accompany soil-quality analyses. Soil Sci. Soc. Am. J. 2001, 65, 849–852. [Google Scholar] [CrossRef] [Green Version]
- do Nascimento, S.V.; Magalhaes, M.M.; Cunha, R.L.; de Oliveira Costa, P.H.; de Oliveira Alves, R.C.; de Oliveira, G.C.; da Silva Valadares, R.B. Differential accumulation of proteins in oil palms affected by fatal yellowing disease. PLoS ONE 2018, 13, e0195538. [Google Scholar] [CrossRef] [Green Version]
- Herrera, H.; Valadares, R.; Oliveira, G.; Fuentes, A.; Almonacid, L.; do Nascimento, S.V.; Bashan, Y.; Arriagada, C. Adaptation and tolerance mechanisms developed by mycorrhizal Bipinnula fimbriata plantlets (Orchidaceae) in a heavy metal-polluted ecosystem. Mycorrhiza 2018, 28, 651–663. [Google Scholar] [CrossRef]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Oliveira, R.R.; Silva, R.L.; Nunes, G.L.; Oliveira, G. PIMBA: A PIpeline for MetaBarcoding Analysis. bioRxiv 2021. [Google Scholar] [CrossRef]
- Zhang, J.; Kobert, K.; Flouri, T.; Stamatakis, A. PEAR: A fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 2014, 30, 614–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abarenkov, K.; Henrik Nilsson, R.; Larsson, K.H.; Alexander, I.J.; Eberhardt, U.; Erland, S.; Høiland, K.; Kjøller, R.; Larsson, E.; Pennanen, T. The UNITE database for molecular identification of fungi–recent updates and future perspectives. New Phytol. 2010, 186, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Cole, J.R.; Wang, Q.; Fish, J.A.; Chai, B.; McGarrell, D.M.; Sun, Y.; Brown, C.T.; Porras-Alfaro, A.; Kuske, C.R.; Tiedje, J.M. Ribosomal Database Project: Data and tools for high throughput rRNA analysis. Nucleic Acids Res. 2014, 42, D633–D642. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, N.H.; Song, Z.; Bates, S.T.; Branco, S.; Tedersoo, L.; Menke, J.; Schilling, J.S.; Kennedy, P.G. FUNGuild: An open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecol. 2016, 20, 241–248. [Google Scholar] [CrossRef]
- Louca, S.; Parfrey, L.W.; Doebeli, M. Decoupling function and taxonomy in the global ocean microbiome. Science 2016, 353, 1272–1277. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RM | Canga | |
|---|---|---|
| Clay (g kg−1) | 486.7 ± 37.2 | 287.5 ± 103 |
| Silt (g kg−1) | 158.3 ± 40.8 | 87.5 ± 75 |
| Sand (g kg−1) | 355 ± 68.9 | 625 ± 177 |
| pH H2O | 6.1 ± 0.3 | 4.7 ± 0.6 |
| pH CaCl2 | 5.3 ± 0.2 | 4.2 ± 0.3 |
| Available P (mg dm−3) | 19.5 ± 15.9 | 1 ± 0.9 |
| Total N (dag kg−1) | 0.1 ± 0.06 | 0.4 ± 0.3 |
| Organic matter (dag kg−1) | 1.4 ± 0.6 | 7.7 ± 0.9 |
| Na (mg dm−3) | 4.7 ± 0.6 | 11.4 ± 6.2 |
| K (mg dm−3) | 22.6 ± 6.8 | 22.7 ± 6.8 |
| B (mg dm−3) | 0.1 ± 0.03 | 0.2 ± 0.08 |
| Cu (mg dm−3) | 0.6 ± 0.3 | 1.8 ± 0.5 |
| Fe (mg dm−3) | 11.3 ± 6.3 | 372 ± 92 |
| Mn (mg dm−3) | 59.7 ± 9.4 | 2.8 ± 1.5 |
| Zn (mg dm−3) | 2.1 ± 1.6 | 1.4 ± 0.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nascimento, S.V.d.; Costa, P.H.d.O.; Herrera, H.; Caldeira, C.F.; Gastauer, M.; Ramos, S.J.; Oliveira, G.; Valadares, R.B.d.S. Proteomic Profiling and Rhizosphere-Associated Microbial Communities Reveal Adaptive Mechanisms of Dioclea apurensis Kunth in Eastern Amazon’s Rehabilitating Minelands. Plants 2022, 11, 712. https://doi.org/10.3390/plants11050712
Nascimento SVd, Costa PHdO, Herrera H, Caldeira CF, Gastauer M, Ramos SJ, Oliveira G, Valadares RBdS. Proteomic Profiling and Rhizosphere-Associated Microbial Communities Reveal Adaptive Mechanisms of Dioclea apurensis Kunth in Eastern Amazon’s Rehabilitating Minelands. Plants. 2022; 11(5):712. https://doi.org/10.3390/plants11050712
Chicago/Turabian StyleNascimento, Sidney Vasconcelos do, Paulo Henrique de Oliveira Costa, Hector Herrera, Cecílio Frois Caldeira, Markus Gastauer, Silvio Junio Ramos, Guilherme Oliveira, and Rafael Borges da Silva Valadares. 2022. "Proteomic Profiling and Rhizosphere-Associated Microbial Communities Reveal Adaptive Mechanisms of Dioclea apurensis Kunth in Eastern Amazon’s Rehabilitating Minelands" Plants 11, no. 5: 712. https://doi.org/10.3390/plants11050712