2.1. Identification of Billionaire siRNAs in the Rice Inflorescence
In a previous study, we found that the whole-genome methylation level (WGML) in the CHH context was higher in inflorescences than in seedlings mainly because of the up-regulation of RdDM machinery [
11]. However, 24 nt siRNA abundance on the hyper-methylated differentially methylated regions (hyper-DMRs) of inflorescence relative to seedlings was significantly lower in inflorescences than in seedlings (
Figure S1A). The inconsistency between the increase of CHH methylation levels and the decrease in siRNA abundance in inflorescences relative to seedlings led us to analyze 24 nt siRNAs in detail. Using the same cutoff (see Methods), we identified 57,424 24 nt siRNA clusters in seedlings but only 20,200 in inflorescences, indicating that 24 nt siRNAs were reprogramed in inflorescences relative to seedlings. About 90.2% of inflorescence siRNA clusters were also identified in seedlings, and only 1971 clusters were inflorescence-specific (
Figure S1B). On all seedling siRNA clusters (siRNA clusters that were seedling-specific or common to seedlings and inflorescences), the siRNA abundance was higher in seedlings than in inflorescences, but the CHH methylation was lower in seedlings than in inflorescences ([
11];
Figure S1B). By contrast, the 24 nt siRNA abundance and CHH methylation were both higher in inflorescences than in seedlings on the inflorescence-specific siRNA clusters (
Figure S1B).
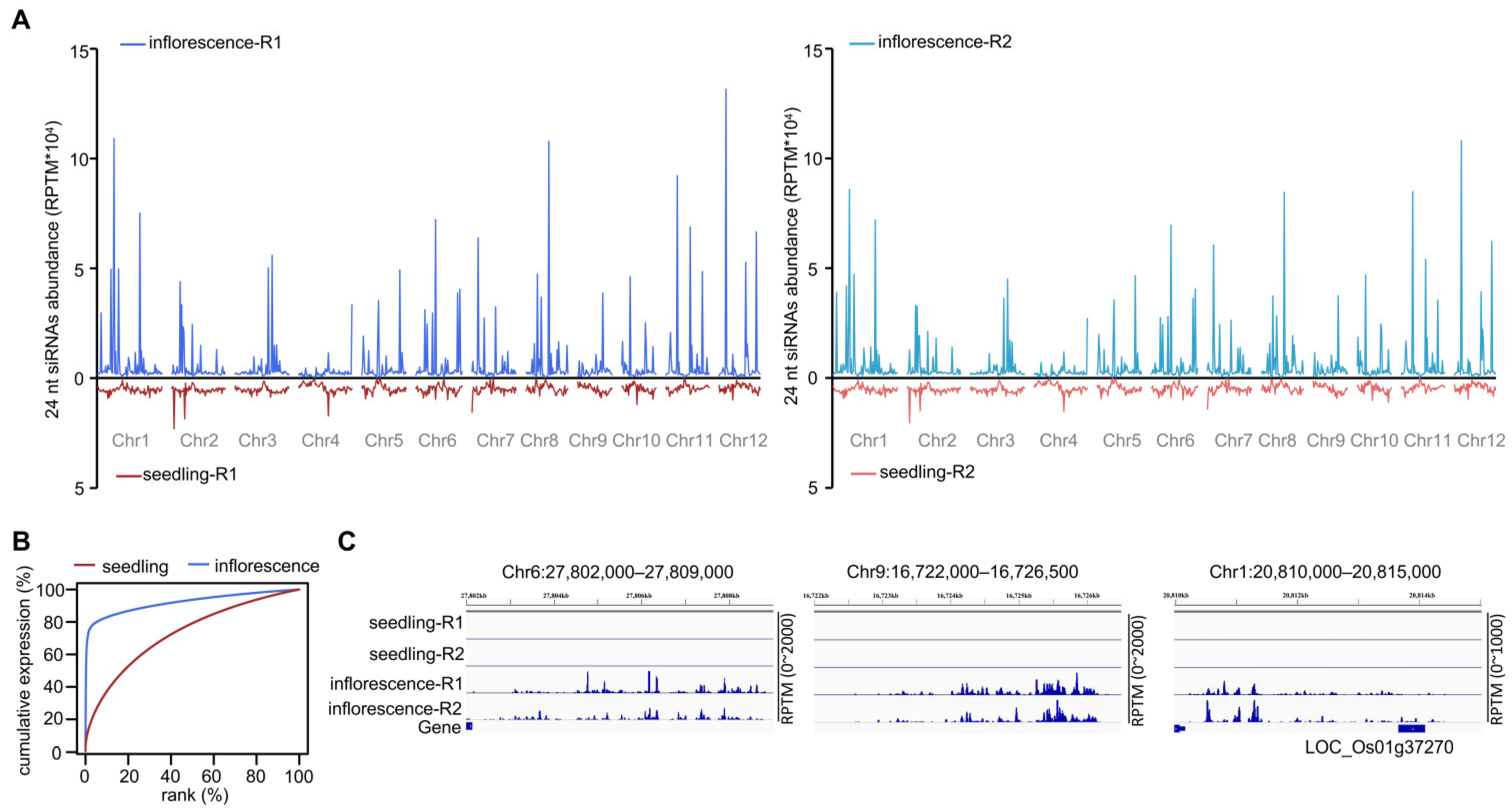
The negative correlation between siRNA abundance and CHH methylation level on hyper-DMRs of inflorescence relative to seedling led us to investigate 24 nt siRNA distribution on the genome in Nipponbare inflorescences and seedlings. siRNA abundance in seedlings was roughly evenly distributed on the genome but was concentrated in certain regions in inflorescences (
Figure 1A). Cumulative expression analysis of 24 nt siRNAs in inflorescences and seedlings revealed that a few regions produced most of the siRNAs in inflorescences (
Figure 1B). We attempted to sort the highly expressed 24 nt siRNA clusters in inflorescences by their abundance. siRNA clusters with reads per ten million (RPTM) > 500, >1000, >1500, >1600, and >2000 accounted for 75.0, 72.7, 70.6, 70.1, and 68.7% of all 24 nt siRNA reads, respectively. To strictly define the highly expressed siRNA clusters, we used RPTM > 2000. We found only 163 siRNA clusters with RPTM > 2000 (
Table S1), which represented <0.81% of all 24 nt siRNA clusters in inflorescences. On these clusters, however, the siRNAs accounted for 68.7% of the 24 nt siRNA reads in inflorescences. In contrast, using the same criterion (RPTM > 2000), we found only 19 highly expressed siRNA clusters in seedlings (
Table S1). On these clusters, the siRNAs accounted for 3.1% of siRNA reads in seedlings. Because of their similarity to the divide between the rich and poor in modern human society, we named these highly expressed siRNA clusters “billionaire-siRNA clusters” (bill-siRNA clusters). The other 24 nt siRNA clusters were accordingly named pauper-siRNA clusters (pau-siRNA clusters). The abundance of 24 nt siRNAs on bill-siRNA clusters was much higher in inflorescences than in seedlings (
Figure 1C).
To investigate the conservation of bill-siRNAs in rice populations, we conducted sRNA sequencing for inflorescences and seedlings in the TP309 variety (
Table S2). As was the case in Nipponbare inflorescences, the 24 nt siRNAs in two replicates of TP309 inflorescences were concentrated in certain regions of the genome (
Figure S2A). Based on cumulative expression analysis, the 24 nt siRNAs were concentrated in a small number of clusters in TP309 inflorescences (
Figure S2B). Using the same criterion that was used for Nipponbare, we identified 129 bill-siRNA clusters in TP309 inflorescences (
Table S1), which represented only 0.5% of the 23,635 siRNA clusters in TP309 inflorescences. On these clusters, however, the siRNAs accounted for 65.2% of all 24 nt siRNA reads. In TP309 seedlings, there were only 12 bill-siRNA clusters (among a total of 46,743 siRNA clusters), which accounted for 2.5% of 24 nt siRNA reads. As was the case with Nipponbare, the abundance of 24 nt siRNAs on bill-siRNA clusters in TP309 was much higher in inflorescences than in seedlings (
Figure S2C).
A Venn diagram showed that 96.1% of bill-siRNA clusters in TP309 inflorescences overlapped with those in Nipponbare inflorescences (
Figure S3A,B), indicating that most of the bill-siRNA clusters are probably conserved in natural populations of rice. There were 39 Nipponbare-specific and 5 TP309-specific bill-siRNA clusters (
Figure S3A,C,D). The siRNA levels on Nipponbare-specific bill-siRNA clusters were significantly higher in Nipponbare inflorescences than in TP309 inflorescences (
Figure S3A,C). In both varieties, the siRNA levels were significantly higher on common bill-siRNA clusters than on variety-specific clusters (
Figure S3E). On common and variety-specific bill-siRNA clusters, SNP and Indel number did not significantly differ between Nipponbare and TP309 (
Figure S3F), indicating that the production of variety-specific bill-siRNAs can probably not be explained by sequence variation.
2.2. Features of Bill-siRNAs
The first base was enriched at adenine for bill-siRNAs in inflorescences (
Figure 2A), which is also true for 24 nt siRNAs in seedlings and for pau-siRNAs in inflorescences, suggesting that both bill-siRNAs and pau-siRNAs are loaded into AGO4 [
13]. We found that bill-siRNA clusters were significantly longer than pau-siRNA clusters in both varieties (
Figure 2B). Even when the siRNA abundance is divided by the cluster length, the siRNA abundance was much higher on bill-siRNA clusters than on pau-siRNA clusters (
Figure 2C). Using previously published ChIP-seq data for various histone modifications in Nipponbare inflorescences [
14], we found that the levels of H3K9me2, a repressive histone mark, were significantly higher on bill-siRNA clusters than on pau-siRNA clusters (
Figure 2D). In contrast, the levels of H3K27me3, another repressive histone mark, were significantly lower on bill-siRNA clusters than on pau-siRNA clusters (
Figure 2D), suggesting that bill-siRNAs tend to locate on stable heterochromatin but not on temporary heterochromatin.
In addition, active histone marks (H3K4me1, H3K4me3, and H3K27ac) were less enriched on bill-siRNA clusters than on pau-siRNA clusters (
Figure 2D). Consequently, RNA polymerase II was less occupied on bill-siRNA clusters than on pau-siRNA clusters (
Figure 2D). Eventually, the expression levels were significantly lower for bill-siRNA cluster-adjacent genes than for pau-siRNA cluster-adjacent genes (
Figure 2E).
Only 12.6% and 15.9% of the genes that overlapped with bill-siRNA clusters exhibit differential expression (fold change > 2, FDR < 0.05) between seedlings and inflorescences in Nipponbare and TP309, respectively (
Figure S4A,B), suggesting that bill-siRNA might have little effect on gene transcription of adjacent genes.
2.3. Accumulation of Bill-siRNAs Is Dependent on OsRDR2 and Is Required for CHH Methylation
On bill-siRNA clusters, siRNA abundance was substantially higher in inflorescences than in seedlings (
Figure 3A). On pau-siRNA clusters, in contrast, siRNA abundance was significantly lower in inflorescences than in seedlings (
Figure 3A). The predominant production of siRNAs on bill-siRNA clusters is the reason for the lower siRNA abundance on pau-siRNA clusters in inflorescences than in seedlings. The siRNA on both bill-siRNA and pau-siRNA clusters was depleted in both inflorescences and seedlings of
osrdr2 (
Figure 3A).
Because Pol IV- and RDR2-generated siRNAs are the hallmarks of RdDM [
4,
15], we assessed CHH methylation on siRNA clusters. On both bill-siRNA and pau-siRNA clusters, the CHH methylation levels were significantly higher in inflorescences than in seedlings (
Figure 3B), which is consistent with our previous finding that CHH methylation is globally higher in reproductive organs than in seedlings [
11]. The CHH methylation on bill-siRNA and pau-siRNA clusters is significantly lower in
osrdr2 inflorescences than in wild-type (WT; Nipponbare) inflorescences (
Figure 3B), demonstrating that
OsRDR2-dependent bill-siRNAs as well as
OsRDR2-dependent pau-siRNAs are required for DNA methylation in inflorescences. Even though siRNA abundance is much higher on bill-siRNA clusters than on pau-siRNA clusters, the CHH methylation levels are lower on bill-siRNA clusters than on pau-siRNA clusters in inflorescences (
Figure 3B), indicating that siRNA abundance is not positively correlated with the levels of DNA methylation. We selected two bill-siRNA clusters (
Figure 3C) and subjected them to Chop-PCR. The Chop-PCR assay demonstrated that CHH methylation is higher in inflorescences than in seedlings, and that the high CHH methylation in inflorescence is dependent on
OsRDR2 (
Figure 3D).
2.4. Bill-siRNAs in Inflorescences Are Mainly Derived from the Ovary
The rice inflorescence is composed of branch, lemma/palea (bract), ovary, and stamen. To clarify the source of bill-siRNAs in the inflorescence, we conducted sRNA sequencing for those tissues at development stage 12 of the anther (
Table S2). Cumulative expression analysis indicated that the accumulation of siRNAs in the ovary was very similar to that in the inflorescence (
Figure 4A and
Figure S5). The siRNAs tend to be concentrated on a small number of loci in the branch, bract, and stamen, but were less concentrated in those tissues than in the inflorescence (
Figure 4A). The siRNAs in the ovary, however, were even more concentrated than in the inflorescence (
Figure 4A and
Figure S5). Using the same criterion that we used for the inflorescence, we identified 158 bill-siRNA clusters in the ovary, 83 in the bract, 46 in the branch, and 75 in the stamen (
Table S1).
On bill-siRNA clusters, siRNA abundance is similar in the ovary and the inflorescence, but is significantly higher in the ovary than in the bract, branch, or stamen (
Figure 4B). In addition, 75.5% of bill-siRNA clusters in the inflorescence are shared by the bill-siRNA clusters in the ovary. In contrast, a much lower percentage of bill-siRNA clusters in the inflorescence are overlapped with bill-siRNA clusters in the bract, branch, or stamen (39.9, 14.1, or 28.8%, respectively;
Figure 4C), suggesting that the majority of bill-siRNAs in the inflorescence are derived from the ovary.
Among the five tissues, there are a total of 233 bill-siRNA clusters. Among these 233 siRNA clusters, 24 nt siRNA abundance is highest in the ovary (
Figure 4D), and is significantly lower in the bract, branch, and stamen than in the inflorescence or ovary (
Figure 4D). Of the bill-siRNA clusters in the bract, branch, and stamen, 85.92, 56.82, and 64.79%, respectively, are overlapped with those in the ovary (
Figure 4E). Together, these results indicate that most of the bill-siRNAs in the inflorescence are produced in the ovary. On the 233 bill-siRNA clusters in the five tissues, siRNA accumulation was significantly reduced in the
osrdr2 mutant (
Figure 4D), suggesting that the biosynthesis of bill-siRNAs is dependent on
OsRDR2. On one representative bill-siRNA cluster, northern blotting confirmed that
OsRDR2 is required for bill-siRNA production in the inflorescence and the ovary (
Figure 4F).
On pau-siRNA clusters of the ovary and stamen, siRNA accumulation was significantly lower in
osrdr2 than in the WT (
Figure S6A,B), suggesting that the biosynthesis of both bill-siRNAs and pau-siRNAs is dependent on
OsRDR2 in the ovary and stamen. The CHH methylation level on bill-siRNA clusters in the ovary and stamen was substantially lower in
osrdr2 than in the WT, suggesting that bill-siRNAs are functional in directing de novo DNA methylation in the ovary and stamen (
Figure S6C,D).
As in the inflorescence, the first bases of bill-siRNAs in the ovary, bract, branch, and stamen are enriched on adenine (
Figure S7A). The bill-siRNA clusters were significantly longer than the pau-siRNA clusters in the same tissues (
Figure S7B). The expression levels of the genes that overlapped with bill-siRNA clusters are significantly lower than those that overlapped with pau-siRNA clusters in both the ovary and stamen (
Figure S7C,D). Only 14.2% of the genes that overlapped with bill-siRNA clusters in the ovary show differential expression between the ovary and seedling (fold change > 2, FDR < 0.05), and 1.9% of the genes that overlapped with ovary bill-siRNA clusters in the ovary change their transcriptional levels when bill-siRNAs are eliminated in the
osrdr2 ovary (
Figure S7E,F). Moreover, 16.4% of the genes that overlapped with bill-siRNA clusters in the stamen exhibit differential expression between stamen and seedlings, and only 1.8% of them change their transcriptional levels when bill-siRNAs are eliminated in the
osrdr2 stamen (
Figure S7G,H), indicating that bill-siRNAs have little role in regulating adjacent gene expression.
2.5. Bill-siRNAs in Gametes
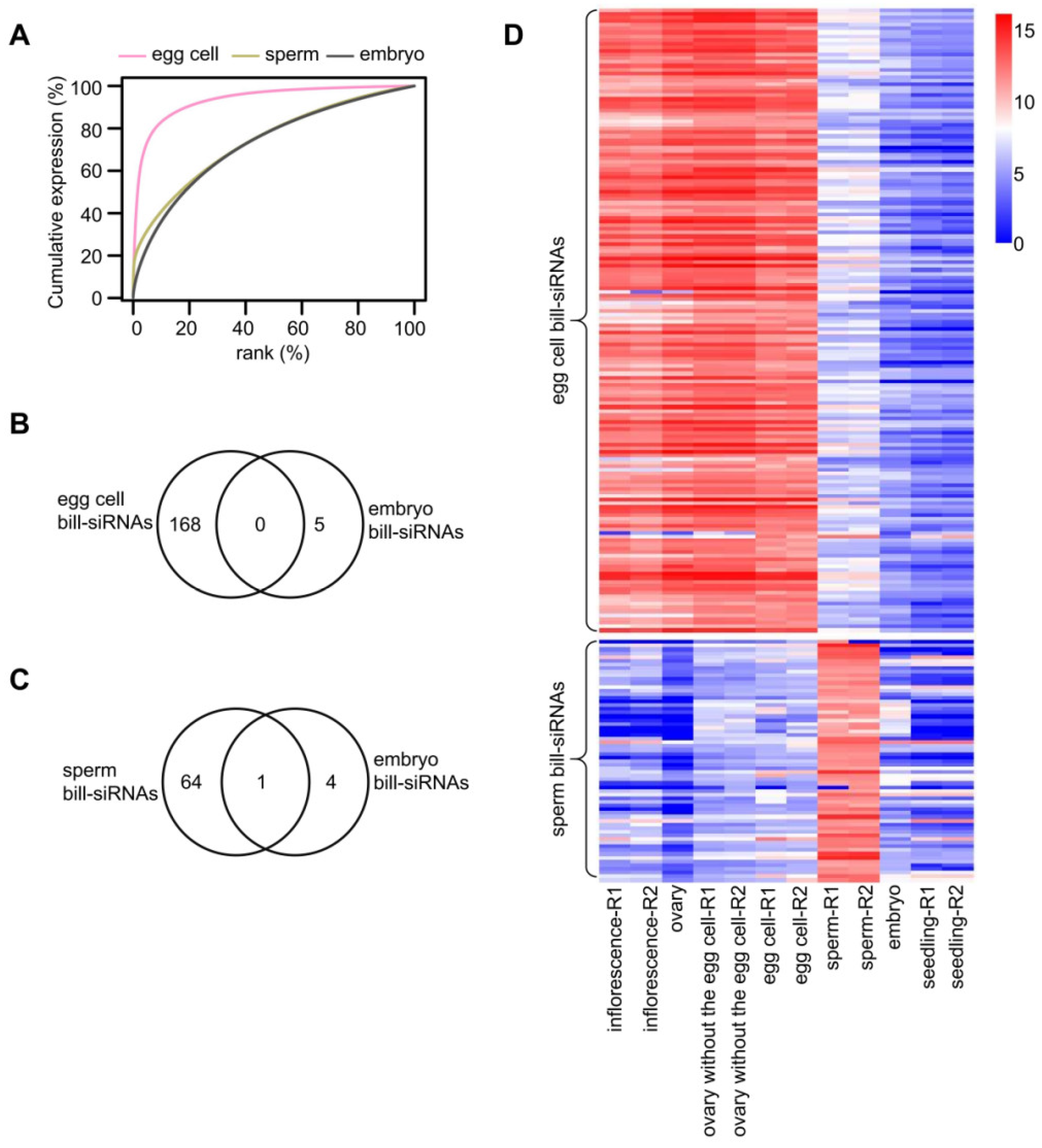
Because bill-siRNAs are abundant in the ovary, we considered the possibility that bill-siRNAs are highly expressed in the egg cell. To test this possibility, we analyzed sRNAs of the egg cell and of the ovary without the egg cell [
16]. The genomic distribution of siRNAs was similar in the ovary with egg, the egg cell alone, and the ovary without the egg cell (
Figure S8A,B). A cumulative expression plot showed that the concentration of siRNAs in the egg cell and the ovary without the egg cell was comparable to that in the ovary with the egg cell (
Figure S8C). We identified 168 and 185 bill-siRNA clusters in the egg cell and in the ovary without the egg cell, respectively. Among the 168 bill-siRNA clusters in the egg cell, 160 (95.2%) are overlapped with those in the ovary (
Figure S8D). Among 185 bill-siRNA clusters in the ovary without the egg cell, 147 (79.5%) are overlapped with those in the ovary with the egg cell (
Figure S8E). Among 168 bill-siRNA clusters in the egg cell, 165 (98.2%) are overlapped with those in the ovary without the egg cell (
Figure S9). These results indicated that the bill-siRNAs are equally produced in the egg cell and other cells of the ovary.
As is the case in the ovary and inflorescence, bill-siRNA clusters are longer than pau-siRNA clusters in the egg cell and in the ovary without the egg cell (
Figure S10A). The first base of bill-siRNAs in the egg cell and in the ovary without the egg cell is enriched for A (
Figure S10B), which is similar to the bill-siRNAs in the inflorescence and ovary.
We used sRNAs of the sperm and vegetative cell [
16] to identify potential bill-siRNAs in the male gamete. Cumulative expression plots revealed that siRNA distribution in the sperm and vegetative cell is similar to siRNA distribution in the stamen (
Figure S11A). Using the same criterion, we identified 65 and 18 bill-siRNA clusters in the sperm and vegetative cell, respectively. Among the 18 bill-siRNA clusters of the vegetative cell, 15 are overlapped with those of the sperm (
Figure S11B). On bill-siRNA clusters in the sperm cell, siRNA abundance is significantly higher in the sperm cell than in the vegetative cell or stamen (
Figure S11B). In addition, the majority of bill-siRNAs of sperm were specific to the sperm relative to the stamen (
Figure S11C). By contrast, a majority of bill-siRNAs of the vegetative cell are shared by the stamen (
Figure S11D). As in other tissues, bill-siRNA clusters were longer than pau-siRNA clusters in the sperm and vegetative cell (
Figure S11E). The first base of bill-siRNAs in the sperm and vegetative is enriched for A (
Figure S11F).
2.7. The Bill-siRNAs of the Gametes Perish but New Bill-siRNAs Are Produced in the Endosperm
Given that sRNA-seq data in the central cell were lacking, and that the siRNAs largely overlapped in the ovary, egg cell, and ovary without the egg cell, we used the sRNA data of the ovary without the egg cell as representative of the central cell. A cumulative expression plot shows that siRNA concentration in the endosperm is similar to that in the ovary without the egg cell, which differs from the pattern of siRNA in the sperm (
Figure 6A). Among 238 bill-siRNA clusters in the endosperm, only one overlapped with that in the ovary without the egg cell (
Figure 6B). On this overlapped siRNA cluster, however, the siRNA distribution was quite different in the endosperm than in the ovary (
Figure S13A). In addition, only two bill-siRNA clusters of the endosperm were shared by the sperm (
Figure 6C and
Figure S13B). A heatmap demonstrated that the bill-siRNAs of the endosperm differed from those of the sperm or of the ovary without the egg cell (
Figure 6D). Overall, the bill-siRNAs of the male and female gamete perished in the endosperm, and new bill-siRNAs were produced in the endosperm.
siRNAs were also concentrated on a few loci in the endosperm of TP309 (
Figure S14A). Among the 279 bill-siRNA clusters in the TP309 endosperm, 218 were shared by Nipponbare (
Figure 7A). The bill-siRNA clusters of the TP309 endosperm were significantly longer than the pau-siRNA clusters of the endosperm in both Nipponbare and TP309 (
Figure S14B). The first base of the bill-siRNAs in the endosperm in both Nipponbare and TP309 was enriched at A (
Figure S14C). The percentage of A was lower in the bill-siRNAs of the endosperm than in those of the ovary (
Figures S14C and S7A). The methylation levels of CG and CHG were significantly lower on bill-siRNA clusters than on pau-siRNA clusters (
Figure 7B). The methylation levels of CHH, however, were significantly higher on bill-siRNA clusters than on pau-siRNA clusters (
Figure 7B). For the repressive epigenetic mark H3K27me3 in the endosperm, the enrichment was significantly higher on bill-siRNA clusters than on pau-siRNA clusters (
Figure 7C). The biosynthesis of bill-siRNAs in the endosperm was partially dependent on
FEM1/
OsRDR2 and Pol IV because the abundance of siRNAs was significantly decreased in
fem1-1 (
osrdr2-1) and
pol iv (
osnprd1a-1/osnrpd1b-1) mutants in the TP309 background (
Figure 7D).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







