
Role of mTOR Signaling Cascade in Epidermal Morphogenesis and Skin Barrier Formation


Abstract
:Simple Summary
Abstract
1. Introduction
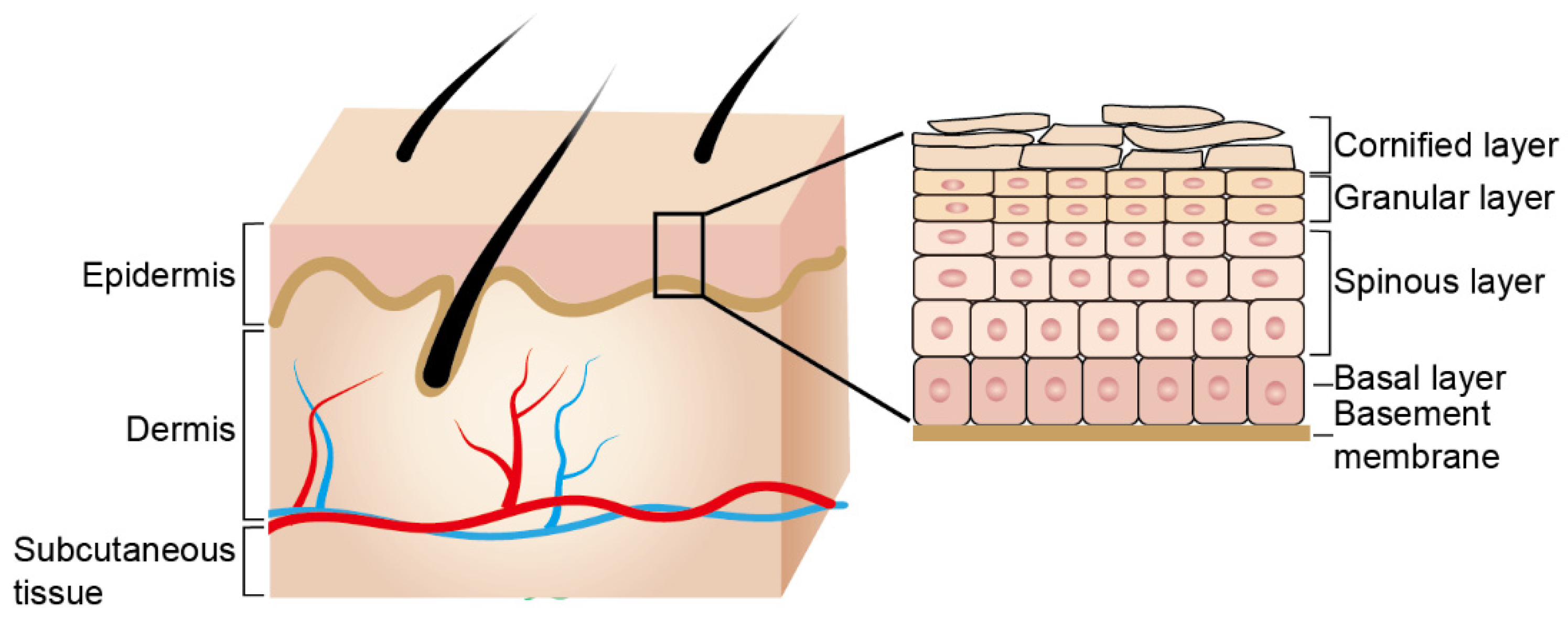
2. Epidermal Barrier Formation
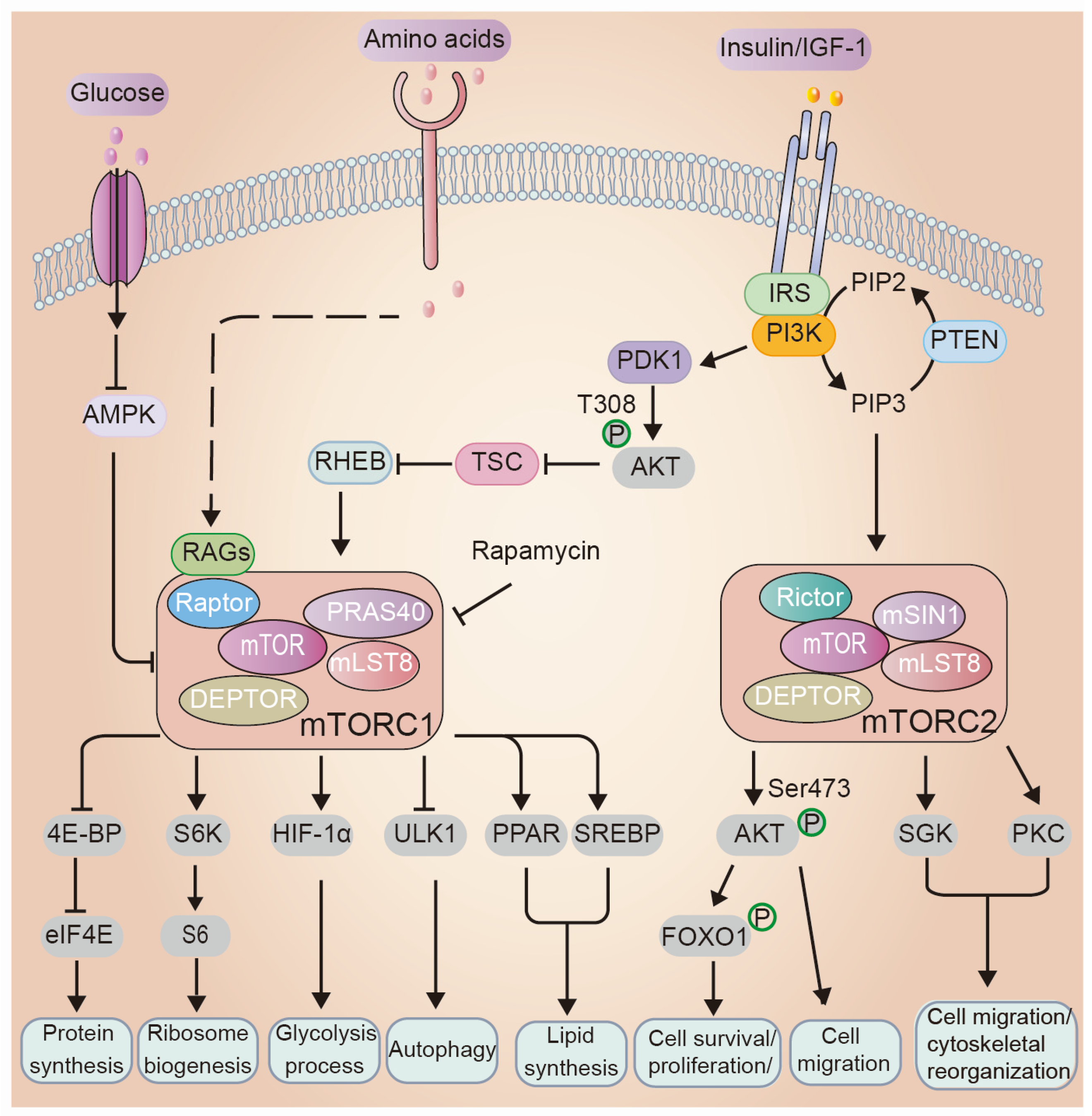
3. Overview of the mTOR Signaling Network: Key Players and Mechanisms
3.1. Regulations of mTOR Signaling
3.2. Cellular Processes Regulated by mTOR
4. mTOR Signaling in Epidermal Morphogenesis and Barrier Formation
4.1. The Role of mTOR Upstream Regulators in Epidermal Morphogenesis and Barrier Formation
4.2. PI3K/AKT/mTOR Signaling Cascade in Epidermal Morphogenesis and Barrier Formation
4.3. mTOR Downstream Mediates in Epidermal Morphogenesis and Skin Barrier Function
4.3.1. Protein Synthesis
4.3.2. Autophagy
4.3.3. PKC
4.3.4. Transcriptional Regulation
4.3.5. Lipid Metabolism
5. Concluding Remarks and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gonzales, K.A.U.; Fuchs, E. Skin and Its Regenerative Powers: An Alliance between Stem Cells and Their Niche. Dev. Cell 2017, 43, 387–401. [Google Scholar] [CrossRef] [Green Version]
- Kubo, A.; Nagao, K.; Amagai, M. Epidermal barrier dysfunction and cutaneous sensitization in atopic diseases. J. Clin. Investig. 2012, 122, 440–447. [Google Scholar] [CrossRef] [Green Version]
- Koster, M.I.; Roop, D.R. Genetic pathways required for epidermal morphogenesis. Eur. J. Cell Biol. 2004, 83, 625–629. [Google Scholar] [CrossRef]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR signaling in growth and metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef] [Green Version]
- Clark, R.A.; Pavlis, M. Dysregulation of the mTOR pathway secondary to mutations or a hostile microenvironment contributes to cancer and poor wound healing. J. Investig. Dermatol. 2009, 129, 529–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naeem, A.S.; Tommasi, C.; Cole, C.; Brown, S.J.; Zhu, Y.; Way, B.; Owen, S.A.W.; Moffatt, M.; Cookson, W.O.; Harper, J.I.; et al. A mechanistic target of rapamycin complex 1/2 (mTORC1)/V-Akt murine thymoma viral oncogene homolog 1 (AKT1)/cathepsin H axis controls filaggrin expression and processing in skin, a novel mechanism for skin barrier disruption in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2017, 139, 1228–1241. [Google Scholar]
- Cibrian, D.; de la Fuente, H.; Sánchez-Madrid, F. Metabolic Pathways That Control Skin Homeostasis and Inflammation. Trends Mol. Med. 2020, 26, 975–986. [Google Scholar] [CrossRef]
- Mercurio, L.; Albanesi, C.; Madonna, S. Recent Updates on the Involvement of PI3K/AKT/mTOR Molecular Cascade in the Pathogenesis of Hyperproliferative Skin Disorders. Front. Med. 2021, 8, 665647. [Google Scholar] [CrossRef]
- Gudjonsson, J.E.; Johnston, A.; Dyson, M.; Valdimarsson, H.; Elder, J.T. Mouse models of psoriasis. J. Investig. Dermatol. 2007, 127, 1292–1308. [Google Scholar]
- Koster, M.I.; Roop, D.R. Mechanisms regulating epithelial stratification. Annu. Rev. Cell Dev. Biol. 2007, 23, 93–113. [Google Scholar] [CrossRef] [PubMed]
- Candi, E.; Schmidt, R.; Melino, G. The cornified envelope: A model of cell death in the skin. Nat. Rev. Mol. Cell Biol. 2005, 6, 328–340. [Google Scholar] [CrossRef]
- Heitman, J.; Movva, N.R.; Hall, M.N. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science 1991, 253, 905–909. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.J.; Albers, M.W.; Shin, T.B.; Ichikawa, K.; Keith, C.T.; Lane, W.S.; Schreiber, S.L. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature 1994, 369, 756–758. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Rudge, D.G.; Koos, J.D.; Vaidialingam, B.; Yang, H.J.; Pavletich, N.P. mTOR kinase structure, mechanism and regulation. Nature 2013, 497, 217–223. [Google Scholar] [CrossRef] [Green Version]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef]
- Oldham, S.; Hafen, E. Insulin/IGF and target of rapamycin signaling: A TOR de force in growth control. Trends Cell Biol. 2003, 13, 79–85. [Google Scholar] [CrossRef]
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.L. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 2002, 4, 648–657. [Google Scholar] [CrossRef]
- Yang, H.; Jiang, X.; Li, B.; Yang, H.J.; Miller, M.; Yang, A.; Dhar, A.; Pavletich, N.P. Mechanisms of mTORC1 activation by RHEB and inhibition by PRAS40. Nature 2017, 552, 368–373. [Google Scholar] [CrossRef]
- González, A.; Hall, M.N. Nutrient sensing and TOR signaling in yeast and mammals. EMBO J. 2017, 36, 397–408. [Google Scholar] [CrossRef] [Green Version]
- Shen, K.; Rogala, K.B.; Chou, H.T.; Huang, R.K.; Yu, Z.; Sabatini, D.M. Cryo-EM Structure of the Human FLCN-FNIP2-Rag-Ragulator Complex. Cell 2019, 179, 1319–1329.e8. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.X.; Guan, K.L. The SIN1-PH Domain Connects mTORC2 to PI3K. Cancer Discov. 2015, 5, 1127–1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.; Gan, W.; Chin, Y.R.; Ogura, K.; Guo, J.; Zhang, J.; Wang, B.; Blenis, J.; Cantley, L.C.; Toker, A.; et al. PtdIns(3,4,5)P3-Dependent Activation of the mTORC2 Kinase Complex. Cancer Discov. 2015, 5, 1194–1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zinzalla, V.; Stracka, D.; Oppliger, W.; Hall, M.N. Activation of mTORC2 by association with the ribosome. Cell 2011, 144, 757–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, W.J.; Wu, C.C.; Kim, S.J.; Facchinetti, V.; Julien, L.A.; Finlan, M.; Roux, P.P.; Su, B.; Jacinto, E. mTORC2 can associate with ribosomes to promote cotranslational phosphorylation and stability of nascent Akt polypeptide. EMBO J. 2010, 29, 3939–3951. [Google Scholar] [CrossRef] [Green Version]
- Harrington, L.S.; Findlay, G.M.; Gray, A.; Tolkacheva, T.; Wigfield, S.; Rebholz, H.; Barnett, J.; Leslie, N.R.; Cheng, S.; Shepherd, P.R.; et al. The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J. Cell Biol. 2004, 166, 213–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Yoon, S.O.; Poulogiannis, G.; Yang, Q.; Ma, X.M.; Villén, J.; Kubica, N.; Hoffman, G.R.; Cantley, L.C.; Gygi, S.P.; et al. Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science 2011, 332, 1322–1326. [Google Scholar] [CrossRef] [Green Version]
- Hara, K.; Yonezawa, K.; Kozlowski, M.T.; Sugimoto, T.; Andrabi, K.; Weng, Q.P.; Kasuga, M.; Nishimoto, I.; Avruch, J. Regulation of eIF-4E BP1 phosphorylation by mTOR. J. Biol. Chem. 1997, 272, 26457–26463. [Google Scholar] [CrossRef] [Green Version]
- Gingras, A.C.; Gygi, S.P.; Raught, B.; Polakiewicz, R.D.; Abraham, R.T.; Hoekstra, M.F.; Aebersold, R.; Sonenberg, N. Regulation of 4E-BP1 phosphorylation: A novel two-step mechanism. Genes Dev. 1999, 13, 1422–1437. [Google Scholar] [CrossRef]
- Thoreen, C.C.; Chantranupong, L.; Keys, H.R.; Wang, T.; Gray, N.S.; Sabatini, D.M. A unifying model for mTORC1-mediated regulation of mRNA translation. Nature 2012, 485, 109–113. [Google Scholar] [CrossRef]
- Peterson, T.R.; Sengupta, S.S.; Harris, T.E.; Carmack, A.E.; Kang, S.A.; Balderas, E.; Guertin, D.A.; Madden, K.L.; Carpenter, A.E.; Finck, B.N.; et al. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell 2011, 146, 408–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.E.; Chen, J. Regulation of peroxisome proliferator-activated receptor-gamma activity by mammalian target of rapamycin and amino acids in adipogenesis. Diabetes 2004, 53, 2748–2756. [Google Scholar] [CrossRef] [Green Version]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef]
- Ben-Sahra, I.; Hoxhaj, G.; Ricoult, S.J.H.; Asara, J.M.; Manning, B.D. mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science 2016, 351, 728–733. [Google Scholar] [CrossRef] [Green Version]
- Ben-Sahra, I.; Howell, J.J.; Asara, J.M.; Manning, B.D. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science 2013, 339, 1323–1328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Düvel, K.; Yecies, J.L.; Menon, S.; Raman, P.; Lipovsky, A.I.; Souza, A.L.; Triantafellow, E.; Ma, Q.; Gorski, R.; Cleaver, S.; et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell 2010, 39, 171–183. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Gomes, A.P.; Wang, X.; Yoon, S.O.; Lee, G.; Nagiec, M.J.; Cho, S.; Chavez, A.; Islam, T.; Yu, Y.; et al. mTORC1 Promotes Metabolic Reprogramming by the Suppression of GSK3-Dependent Foxk1 Phosphorylation. Mol. Cell 2018, 70, 949–960.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.C.; Guan, K.L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puente, C.; Hendrickson, R.C.; Jiang, X. Nutrient-regulated Phosphorylation of ATG13 Inhibits Starvation-induced Autophagy. J. Biol. Chem. 2016, 291, 6026–6035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganley, I.G.; Du Lam, H.; Wang, J.; Ding, X.; Chen, S.; Jiang, X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J. Biol. Chem. 2009, 284, 12297–12305. [Google Scholar] [CrossRef] [Green Version]
- Fu, W.; Hall, M.N. Regulation of mTORC2 Signaling. Genes 2020, 11, 1045. [Google Scholar] [CrossRef]
- Gangloff, Y.G.; Mueller, M.; Dann, S.G.; Svoboda, P.; Sticker, M.; Spetz, J.F.; Um, S.H.; Brown, E.J.; Cereghini, S.; Thomas, G.; et al. Disruption of the mouse mTOR gene leads to early postimplantation lethality and prohibits embryonic stem cell development. Mol. Cell Biol. 2004, 24, 9508–9516. [Google Scholar] [CrossRef] [Green Version]
- Murakami, M.; Ichisaka, T.; Maeda, M.; Oshiro, N.; Hara, K.; Edenhofer, F.; Kiyama, H.; Yonezawa, K.; Yamanaka, S. mTOR is essential for growth and proliferation in early mouse embryos and embryonic stem cells. Mol. Cell Biol. 2004, 24, 6710–6718. [Google Scholar] [CrossRef] [Green Version]
- Tellkamp, F.; Benhadou, F.; Bremer, J.; Gnarra, M.; Knüver, J.; Schaffenrath, S.; Vorhagen, S. Transgenic mouse technology in skin biology: Generation of knockin mice. J. Investig. Dermatol. 2014, 134, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Cornu, M.; Albert, V.; Hall, M.N. mTOR in aging, metabolism, and cancer. Curr. Opin. Genet. Dev. 2013, 23, 53–62. [Google Scholar] [CrossRef]
- Liu, J.P.; Baker, J.; Perkins, A.S.; Robertson, E.J.; Efstratiadis, A. Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r). Cell 1993, 75, 59–72. [Google Scholar] [CrossRef]
- DiGiovanni, J.; Bol, D.K.; Wilker, E.; Beltrán, L.; Carbajal, S.; Moats, S.; Ramirez, A.; Jorcano, J.; Kiguchi, K. Constitutive expression of insulin-like growth factor-1 in epidermal basal cells of transgenic mice leads to spontaneous tumor promotion. Cancer Res. 2000, 60, 1561–1570. [Google Scholar]
- Wertheimer, E.; Spravchikov, N.; Trebicz, M.; Gartsbein, M.; Accili, D.; Avinoah, I.; Nofeh-Moses, S.; Sizyakov, G.; Tennenbaum, T. The regulation of skin proliferation and differentiation in the IR null mouse: Implications for skin complications of diabetes. Endocrinology 2001, 142, 1234–1241. [Google Scholar] [CrossRef]
- Stachelscheid, H.; Ibrahim, H.; Koch, L.; Schmitz, A.; Tscharntke, M.; Wunderlich, F.T.; Scott, J.; Michels, C.; Wickenhauser, C.; Haase, I.; et al. Epidermal insulin/IGF-1 signalling control interfollicular morphogenesis and proliferative potential through Rac activation. EMBO J. 2008, 27, 2091–2101. [Google Scholar] [CrossRef] [Green Version]
- Muraguchi, T.; Nanba, D.; Nishimura, E.K.; Tashiro, T. IGF-1R deficiency in human keratinocytes disrupts epidermal homeostasis and stem cell maintenance. J. Dermatol. Sci. 2019, 94, 298–305. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Zi, Z.; Lee, E.E.; Zhao, J.; Contreras, D.C.; South, A.P.; Abel, E.D.; Chong, B.F.; Vandergriff, T.; Hosler, G.A.; et al. Differential glucose requirement in skin homeostasis and injury identifies a therapeutic target for psoriasis. Nat. Med. 2018, 24, 617–627. [Google Scholar] [CrossRef]
- Boulter, E.; Estrach, S.; Errante, A.; Pons, C.; Cailleteau, L.; Tissot, F.; Meneguzzi, G.; Féral, C.C. CD98hc (SLC3A2) regulation of skin homeostasis wanes with age. J. Exp. Med. 2013, 210, 173–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crane, E.D.; Wong, W.; Zhang, H.; O’Neil, G.; Crane, J.D. AMPK Inhibits mTOR-Driven Keratinocyte Proliferation after Skin Damage and Stress. J. Investig. Dermatol. 2021, 141, 2170–2177.e3. [Google Scholar] [CrossRef] [PubMed]
- Dainichi, T.; Hayden, M.S.; Park, S.G.; Oh, H.; Seeley, J.J.; Grinberg-Bleyer, Y.; Beck, K.M.; Miyachi, Y.; Kabashima, K.; Hashimoto, T.; et al. PDK1 Is a Regulator of Epidermal Differentiation that Activates and Organizes Asymmetric Cell Division. Cell Rep. 2016, 15, 1615–1623. [Google Scholar] [CrossRef] [Green Version]
- Peng, X.D.; Xu, P.Z.; Chen, M.L.; Hahn-Windgassen, A.; Skeen, J.; Jacobs, J.; Sundararajan, D.; Chen, W.S.; Crawford, S.E.; Coleman, K.G.; et al. Dwarfism, impaired skin development, skeletal muscle atrophy, delayed bone development, and impeded adipogenesis in mice lacking Akt1 and Akt2. Genes Dev. 2003, 17, 1352–1365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, H.; Thorvaldsen, J.L.; Chu, Q.; Feng, F.; Birnbaum, M.J. Akt1/PKBalpha is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J. Biol. Chem. 2001, 276, 38349–38352. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.S.; Xu, P.Z.; Gottlob, K.; Chen, M.L.; Sokol, K.; Shiyanova, T.; Roninson, I.; Weng, W.; Suzuki, R.; Tobe, K.; et al. Growth retardation and increased apoptosis in mice with homozygous disruption of the Akt1 gene. Genes Dev. 2001, 15, 2203–2208. [Google Scholar] [CrossRef] [Green Version]
- O’Shaughnessy, R.F.; Welti, J.C.; Cooke, J.C.; Avilion, A.A.; Monks, B.; Birnbaum, M.J.; Byrne, C. AKT-dependent HspB1 (Hsp27) activity in epidermal differentiation. J. Biol. Chem. 2007, 282, 17297–17305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, H.; Mu, J.; Kim, J.K.; Thorvaldsen, J.L.; Chu, Q.; Crenshaw, E.B., 3rd; Kaestner, K.H.; Bartolomei, M.S.; Shulman, G.I.; Birnbaum, M.J. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta). Science 2001, 292, 1728–1731. [Google Scholar] [CrossRef]
- Ding, X.; Bloch, W.; Iden, S.; Rüegg, M.A.; Hall, M.N.; Leptin, M.; Partridge, L.; Eming, S.A. mTORC1 and mTORC2 regulate skin morphogenesis and epidermal barrier formation. Nat. Commun. 2016, 7, 13226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asrani, K.; Sood, A.; Torres, A.; Georgess, D.; Phatak, P.; Kaur, H.; Dubin, A.; Talbot, C.C., Jr.; Elhelu, L.; Ewald, A.J.; et al. mTORC1 loss impairs epidermal adhesion via TGF-β/Rho kinase activation. J. Clin. Investig. 2017, 127, 4001–4017. [Google Scholar] [CrossRef] [Green Version]
- Ding, X.; Willenborg, S.; Bloch, W.; Wickstrom, S.A.; Wagle, P.; Brodesser, S.; Roers, A.; Jais, A.; Bruning, J.C.; Hall, M.N.; et al. Epidermal mammalian target of rapamycin complex 2 controls lipid synthesis and filaggrin processing in epidermal barrier formation. J. Allergy Clin. Immunol. 2020, 145, 283–300.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tassone, B.; Saoncella, S.; Neri, F.; Ala, U.; Brusa, D.; Magnuson, M.A.; Provero, P.; Oliviero, S.; Riganti, C.; Calautti, E. Rictor/mTORC2 deficiency enhances keratinocyte stress tolerance via mitohormesis. Cell Death Differ. 2017, 24, 731–746. [Google Scholar] [CrossRef] [Green Version]
- Soma-Nagae, T.; Nada, S.; Kitagawa, M.; Takahashi, Y.; Mori, S.; Oneyama, C.; Okada, M. The lysosomal signaling anchor p18/LAMTOR1 controls epidermal development by regulating lysosome-mediated catabolic processes. J. Cell Sci. 2013, 126, 3575–3584. [Google Scholar] [CrossRef] [Green Version]
- Hertzler-Schaefer, K.; Mathew, G.; Somani, A.K.; Tholpady, S.; Kadakia, M.P.; Chen, Y.; Spandau, D.F.; Zhang, X. Pten loss induces autocrine FGF signaling to promote skin tumorigenesis. Cell Rep. 2014, 6, 818–826. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, A.; Itami, S.; Ohishi, M.; Hamada, K.; Inoue, T.; Komazawa, N.; Senoo, H.; Sasaki, T.; Takeda, J.; Manabe, M.; et al. Keratinocyte-specific Pten deficiency results in epidermal hyperplasia, accelerated hair follicle morphogenesis and tumor formation. Cancer Res. 2003, 63, 674–681. [Google Scholar] [PubMed]
- Squarize, C.H.; Castilho, R.M.; Bugge, T.H.; Gutkind, J.S. Accelerated wound healing by mTOR activation in genetically defined mouse models. PLoS ONE 2010, 5, e10643. [Google Scholar] [CrossRef]
- Asrani, K.; Murali, S.; Lam, B.; Na, C.H.; Phatak, P.; Sood, A.; Kaur, H.; Khan, Z.; Noë, M.; Anchoori, R.K.; et al. mTORC1 feedback to AKT modulates lysosomal biogenesis through MiT/TFE regulation. J. Clin. Investig. 2019, 129, 5584–5599. [Google Scholar] [CrossRef]
- Shima, H.; Pende, M.; Chen, Y.; Fumagalli, S.; Thomas, G.; Kozma, S.C. Disruption of the p70(s6k)/p85(s6k) gene reveals a small mouse phenotype and a new functional S6 kinase. EMBO J. 1998, 17, 6649–6659. [Google Scholar] [CrossRef] [Green Version]
- Selman, C.; Tullet, J.M.; Wieser, D.; Irvine, E.; Lingard, S.J.; Choudhury, A.I.; Claret, M.; Al-Qassab, H.; Carmignac, D.; Ramadani, F.; et al. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science 2009, 326, 140–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pende, M.; Um, S.H.; Mieulet, V.; Sticker, M.; Goss, V.L.; Mestan, J.; Mueller, M.; Fumagalli, S.; Kozma, S.C.; Thomas, G. S6K1(-/-)/S6K2(-/-) mice exhibit perinatal lethality and rapamycin-sensitive 5’-terminal oligopyrimidine mRNA translation and reveal a mitogen-activated protein kinase-dependent S6 kinase pathway. Mol. Cell Biol. 2004, 24, 3112–3124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Bacquer, O.; Petroulakis, E.; Paglialunga, S.; Poulin, F.; Richard, D.; Cianflone, K.; Sonenberg, N. Elevated sensitivity to diet-induced obesity and insulin resistance in mice lacking 4E-BP1 and 4E-BP2. J. Clin. Investig. 2007, 117, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Le Bacquer, O.; Combe, K.; Montaurier, C.; Salles, J.; Giraudet, C.; Patrac, V.; Domingues-Faria, C.; Guillet, C.; Louche, K.; Boirie, Y.; et al. Muscle metabolic alterations induced by genetic ablation of 4E-BP1 and 4E-BP2 in response to diet-induced obesity. Mol. Nutr. Food Res. 2017, 61, 1700128. [Google Scholar] [CrossRef]
- Pearl, D.; Katsumura, S.; Amiri, M.; Tabatabaei, N.; Zhang, X.; Vinette, V.; Pang, X.; Beug, S.T.; Kim, S.H.; Jones, L.M.; et al. 4E-BP-Dependent Translational Control of Irf8 Mediates Adipose Tissue Macrophage Inflammatory Response. J. Immunol. 2020, 204, 2392–2400. [Google Scholar] [CrossRef] [PubMed]
- Rossiter, H.; König, U.; Barresi, C.; Buchberger, M.; Ghannadan, M.; Zhang, C.F.; Mlitz, V.; Gmeiner, R.; Sukseree, S.; Födinger, D.; et al. Epidermal keratinocytes form a functional skin barrier in the absence of Atg7 dependent autophagy. J. Dermatol. Sci. 2013, 71, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Qiang, L.; Yang, S.; Cui, Y.H.; He, Y.Y. Keratinocyte autophagy enables the activation of keratinocytes and fibroblastsand facilitates wound healing. Autophagy 2021, 17, 2128–2143. [Google Scholar] [CrossRef]
- Cataisson, C.; Joseloff, E.; Murillas, R.; Wang, A.; Atwell, C.; Torgerson, S.; Gerdes, M.; Subleski, J.; Gao, J.L.; Murphy, P.M.; et al. Activation of cutaneous protein kinase C alpha induces keratinocyte apoptosis and intraepidermal inflammation by independent signaling pathways. J. Immunol. 2003, 171, 2703–2713. [Google Scholar] [CrossRef] [Green Version]
- Wheeler, D.L.; Li, Y.; Verma, A.K. Protein kinase C epsilon signals ultraviolet light-induced cutaneous damage and development of squamous cell carcinoma possibly through Induction of specific cytokines in a paracrine mechanism. Photochem. Photobiol. 2005, 81, 9–18. [Google Scholar] [CrossRef]
- Gunschmann, C.; Stachelscheid, H.; Akyuz, M.D.; Schmitz, A.; Missero, C.; Bruning, J.C.; Niessen, C.M. Insulin/IGF-1 controls epidermal morphogenesis via regulation of FoxO-mediated p63 inhibition. Dev. Cell 2013, 26, 176–187. [Google Scholar] [CrossRef] [Green Version]
- Boutin, A.T.; Weidemann, A.; Fu, Z.; Mesropian, L.; Gradin, K.; Jamora, C.; Wiesener, M.; Eckardt, K.U.; Koch, C.J.; Ellies, L.G.; et al. Epidermal sensing of oxygen is essential for systemic hypoxic response. Cell 2008, 133, 223–234. [Google Scholar] [CrossRef] [Green Version]
- Rezvani, H.R.; Ali, N.; Serrano-Sanchez, M.; Dubus, P.; Varon, C.; Ged, C.; Pain, C.; Cario-André, M.; Seneschal, J.; Taïeb, A.; et al. Loss of epidermal hypoxia-inducible factor-1α accelerates epidermal aging and affects re-epithelialization in human and mouse. J. Cell Sci. 2011, 124, 4172–4183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cowburn, A.S.; Alexander, L.E.C.; Southwood, M.; Nizet, V.; Chilvers, E.R.; Johnson, R.S. Epidermal deletion of HIF-2α stimulates wound closure. J. Investig. Dermatol. 2014, 134, 801–808. [Google Scholar] [CrossRef] [Green Version]
- Rounbehler, R.J.; Schneider-Broussard, R.; Conti, C.J.; Johnson, D.G. Myc lacks E2F1’s ability to suppress skin carcinogenesis. Oncogene 2001, 20, 5341–5349. [Google Scholar] [CrossRef] [Green Version]
- Frye, M.; Gardner, C.; Li, E.R.; Arnold, I.; Watt, F.M. Evidence that Myc activation depletes the epidermal stem cell compartment by modulating adhesive interactions with the local microenvironment. Development 2003, 130, 2793–2808. [Google Scholar] [CrossRef] [Green Version]
- Zanet, J.; Pibre, S.; Jacquet, C.; Ramirez, A.; de Alborán, I.M.; Gandarillas, A. Endogenous Myc controls mammalian epidermal cell size, hyperproliferation, endoreplication and stem cell amplification. J. Cell Sci. 2005, 118, 1693–1704. [Google Scholar] [CrossRef] [Green Version]
- Michalik, L.; Desvergne, B.; Tan, N.S.; Basu-Modak, S.; Escher, P.; Rieusset, J.; Peters, J.M.; Kaya, G.; Gonzalez, F.J.; Zakany, J.; et al. Impaired skin wound healing in peroxisome proliferator-activated receptor (PPAR)alpha and PPARbeta mutant mice. J. Cell Biol. 2001, 154, 799–814. [Google Scholar] [CrossRef] [Green Version]
- DeChiara, T.M.; Efstratiadis, A.; Robertson, E.J. A growth-deficiency phenotype in heterozygous mice carrying an insulin-like growth factor II gene disrupted by targeting. Nature 1990, 345, 78–80. [Google Scholar] [CrossRef]
- Blakytny, R.; Jude, E.B.; Martin Gibson, J.; Boulton, A.J.; Ferguson, M.W. Lack of insulin-like growth factor 1 (IGF1) in the basal keratinocyte layer of diabetic skin and diabetic foot ulcers. J. Pathol. 2000, 190, 589–594. [Google Scholar] [CrossRef]
- Lin, M.J.; Lu, M.C.; Chang, H.Y. Sustained Release of Insulin-Like Growth Factor-1 from Bombyx mori L. Silk Fibroin Delivery for Diabetic Wound Therapy. Int. J. Mol. Sci. 2021, 22, 6267. [Google Scholar] [CrossRef]
- Hara, K.; Yonezawa, K.; Weng, Q.P.; Kozlowski, M.T.; Belham, C.; Avruch, J. Amino acid sufficiency and mTOR regulate p70 S6 kinase and eIF-4E BP1 through a common effector mechanism. J. Biol. Chem. 1998, 273, 14484–14494. [Google Scholar] [CrossRef] [Green Version]
- Digomann, D.; Kurth, I.; Tyutyunnykova, A.; Chen, O.; Löck, S.; Gorodetska, I.; Peitzsch, C.; Skvortsova, I.-I.; Negro, G.; Aschenbrenner, B.; et al. The CD98 Heavy Chain Is a Marker and Regulator of Head and Neck Squamous Cell Carcinoma Radiosensitivity. Clin. Cancer Res. 2019, 25, 3152–3163. [Google Scholar] [CrossRef]
- Chamcheu, J.C.; Roy, T.; Uddin, M.B.; Banang-Mbeumi, S.; Chamcheu, R.N.; Walker, A.L.; Liu, Y.Y.; Huang, S. Role and Therapeutic Targeting of the PI3K/Akt/mTOR Signaling Pathway in Skin Cancer: A Review of Current Status and Future Trends on Natural and Synthetic Agents Therapy. Cells 2019, 8, 803. [Google Scholar] [CrossRef] [Green Version]
- Janes, S.M.; Ofstad, T.A.; Campbell, D.H.; Watt, F.M.; Prowse, D.M. Transient activation of FOXN1 in keratinocytes induces a transcriptional programme that promotes terminal differentiation: Contrasting roles of FOXN1 and Akt. J. Cell Sci. 2004, 117, 4157–4168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calautti, E.; Li, J.; Saoncella, S.; Brissette, J.L.; Goetinck, P.F. Phosphoinositide 3-kinase signaling to Akt promotes keratinocyte differentiation versus death. J. Biol. Chem. 2005, 280, 32856–32865. [Google Scholar] [CrossRef] [Green Version]
- Maehama, T.; Dixon, J.E. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 1998, 273, 13375–13378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hildenbrand, C.; Burgdorf, W.H.; Lautenschlager, S. Cowden syndrome-diagnostic skin signs. Dermatology 2001, 202, 362–366. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, T.; Minowa, O.; Sugitani, Y.; Takai, S.; Mitani, H.; Kobayashi, E.; Noda, T.; Hino, O. A germ-line Tsc1 mutation causes tumor development and embryonic lethality that are similar, but not identical to, those caused by Tsc2 mutation in mice. Proc. Natl. Acad. Sci. USA 2001, 98, 8762–8767. [Google Scholar] [CrossRef] [Green Version]
- Tsai, S.; Sitzmann, J.M.; Dastidar, S.G.; Rodriguez, A.A.; Vu, S.L.; McDonald, C.E.; Academia, E.C.; O’Leary, M.N.; Ashe, T.D.; La Spada, A.R.; et al. Muscle-specific 4E-BP1 signaling activation improves metabolic parameters during aging and obesity. J. Clin. Investig. 2015, 125, 2952–2964. [Google Scholar] [CrossRef] [Green Version]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef]
- Deleyto-Seldas, N.; Efeyan, A. The mTOR-Autophagy Axis and the Control of Metabolism. Front. Cell Dev. Biol. 2021, 9, 655731. [Google Scholar] [CrossRef] [PubMed]
- Akinduro, O.; Sully, K.; Patel, A.; Robinson, D.J.; Chikh, A.; McPhail, G.; Braun, K.M.; Philpott, M.P.; Harwood, C.A.; Byrne, C.; et al. Constitutive Autophagy and Nucleophagy during Epidermal Differentiation. J. Investig. Dermatol. 2016, 136, 1460–1470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, D.; Qomaladewi, N.P.; Lee, J.; Park, S.H.; Cho, J.Y. The Role of Autophagy in Skin Fibroblasts, Keratinocytes, Melanocytes, and Epidermal Stem Cells. J. Investig. Dermatol. 2020, 140, 1691–1697. [Google Scholar] [CrossRef]
- Mahanty, S.; Dakappa, S.S.; Shariff, R.; Patel, S.; Swamy, M.M.; Majumdar, A.; Setty, S.R.G. Keratinocyte differentiation promotes ER stress-dependent lysosome biogenesis. Cell Death Dis. 2019, 10, 269. [Google Scholar] [CrossRef] [PubMed]
- Qiang, L.; Sample, A.; Shea, C.R.; Soltani, K.; Macleod, K.F.; He, Y.Y. Autophagy gene ATG7 regulates ultraviolet radiation-induced inflammation and skin tumorigenesis. Autophagy 2017, 13, 2086–2103. [Google Scholar] [CrossRef] [Green Version]
- Ikenoue, T.; Inoki, K.; Yang, Q.; Zhou, X.; Guan, K.L. Essential function of TORC2 in PKC and Akt turn motif phosphorylation, maturation and signalling. EMBO J. 2008, 27, 1919–1931. [Google Scholar] [CrossRef] [Green Version]
- Rosato, B.; Ranieri, D.; Nanni, M.; Torrisi, M.R.; Belleudi, F. Role of FGFR2b expression and signaling in keratinocyte differentiation: Sequential involvement of PKCδ and PKCα. Cell Death Dis. 2018, 9, 565. [Google Scholar] [CrossRef] [Green Version]
- Chew, Y.C.; Adhikary, G.; Xu, W.; Wilson, G.M.; Eckert, R.L. Protein kinase C δ increases Kruppel-like factor 4 protein, which drives involucrin gene transcription in differentiating keratinocytes. J. Biol. Chem. 2013, 288, 17759–17768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jerome-Morais, A.; Rahn, H.R.; Tibudan, S.S.; Denning, M.F. Role for protein kinase C-alpha in keratinocyte growth arrest. J. Investig. Dermatol. 2009, 129, 2365–2375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kloet, D.E.; Burgering, B.M. The PKB/FOXO switch in aging and cancer. Biochim. Biophys. Acta 2011, 1813, 1926–1937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, N.; Rollin, J.; Masse, I.; Lamartine, J.; Gidrol, X. p63 regulates human keratinocyte proliferation via MYC-regulated gene network and differentiation commitment through cell adhesion-related gene network. J. Biol. Chem. 2012, 287, 5627–5638. [Google Scholar] [CrossRef] [Green Version]
- Lechler, T.; Fuchs, E. Asymmetric cell divisions promote stratification and differentiation of mammalian skin. Nature 2005, 437, 275–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kierans, S.J.; Taylor, C.T. Regulation of glycolysis by the hypoxia-inducible factor (HIF): Implications for cellular physiology. J. Physiol. 2021, 599, 23–37. [Google Scholar] [CrossRef]
- Land, S.C.; Tee, A.R. Hypoxia-inducible factor 1alpha is regulated by the mammalian target of rapamycin (mTOR) via an mTOR signaling motif. J. Biol. Chem. 2007, 282, 20534–20543. [Google Scholar]
- Rezvani, H.R.; Ali, N.; Nissen, L.J.; Harfouche, G.; de Verneuil, H.; Taïeb, A.; Mazurier, F. HIF-1α in epidermis: Oxygen sensing, cutaneous angiogenesis, cancer, and non-cancer disorders. J. Investig. Dermatol. 2011, 131, 1793–1805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.H.; Bae, H.C.; Kim, J.; Lee, H.; Ryu, W.I.; Son, E.D.; Lee, T.R.; Jeong, S.H.; Son, S.W. HIF-1α-mediated BMP6 down-regulation leads to hyperproliferation and abnormal differentiation of keratinocytes in vitro. Exp. Dermatol. 2018, 27, 1287–1293. [Google Scholar] [CrossRef] [PubMed]
- Fitsialos, G.; Bourget, I.; Augier, S.; Ginouvès, A.; Rezzonico, R.; Odorisio, T.; Cianfarani, F.; Virolle, T.; Pouysségur, J.; Meneguzzi, G.; et al. HIF1 transcription factor regulates laminin-332 expression and keratinocyte migration. J. Cell Sci. 2008, 121, 2992–3001. [Google Scholar] [CrossRef] [Green Version]
- Florczyk, U.; Czauderna, S.; Stachurska, A.; Tertil, M.; Nowak, W.; Kozakowska, M.; Poellinger, L.; Jozkowicz, A.; Loboda, A.; Dulak, J. Opposite effects of HIF-1α and HIF-2α on the regulation of IL-8 expression in endothelial cells. Free Radic. Biol. Med. 2011, 51, 1882–1892. [Google Scholar] [CrossRef] [Green Version]
- Toschi, A.; Lee, E.; Gadir, N.; Ohh, M.; Foster, D.A. Differential dependence of hypoxia-inducible factors 1 alpha and 2 alpha on mTORC1 and mTORC2. J. Biol. Chem. 2008, 283, 34495–34499. [Google Scholar] [CrossRef] [Green Version]
- Watt, F.M.; Frye, M.; Benitah, S.A. MYC in mammalian epidermis: How can an oncogene stimulate differentiation? Nat. Rev. Cancer 2008, 8, 234–242. [Google Scholar] [CrossRef] [Green Version]
- Kruse, V.; Neess, D.; Færgeman, N.J. The Significance of Epidermal Lipid Metabolism in Whole-Body Physiology. Trends Endocrinol. Metab. 2017, 28, 669–683. [Google Scholar] [CrossRef]
- Caron, A.; Richard, D.; Laplante, M. The Roles of mTOR Complexes in Lipid Metabolism. Annu. Rev. Nutr. 2015, 35, 321–348. [Google Scholar] [CrossRef]
- Porstmann, T.; Santos, C.R.; Griffiths, B.; Cully, M.; Wu, M.; Leevers, S.; Griffiths, J.R.; Chung, Y.L.; Schulze, A. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 2008, 8, 224–236. [Google Scholar] [CrossRef] [Green Version]
- Schmuth, M.; Haqq, C.M.; Cairns, W.J.; Holder, J.C.; Dorsam, S.; Chang, S.; Lau, P.; Fowler, A.J.; Chuang, G.; Moser, A.H.; et al. Peroxisome proliferator-activated receptor (PPAR)-beta/delta stimulates differentiation and lipid accumulation in keratinocytes. J. Investig. Dermatol. 2004, 122, 971–983. [Google Scholar] [CrossRef] [Green Version]
- Hatano, Y.; Man, M.Q.; Uchida, Y.; Crumrine, D.; Mauro, T.M.; Feingold, K.R.; Elias, P.M.; Holleran, W.M. Murine atopic dermatitis responds to peroxisome proliferator-activated receptors alpha and beta/delta (but not gamma) and liver X receptor activators. J. Allergy Clin. Immunol. 2010, 125, 160–169.e5. [Google Scholar] [CrossRef] [Green Version]
- Wallmeyer, L.; Lehnen, D.; Eger, N.; Sochorová, M.; Opálka, L.; Kováčik, A.; Vávrová, K.; Hedtrich, S. Stimulation of PPARα normalizes the skin lipid ratio and improves the skin barrier of normal and filaggrin deficient reconstructed skin. J. Dermatol. Sci. 2015, 80, 102–110. [Google Scholar] [CrossRef]
- Yokoyama, A.; Makishima, M.; Choi, M.; Cho, Y.; Nishida, S.; Hashimoto, Y.; Terui, T. Induction of SREBP-1c mRNA by differentiation and LXR ligand in human keratinocytes. J. Investig. Dermatol. 2009, 129, 1395–1401. [Google Scholar] [CrossRef] [Green Version]
- Porstmann, T.; Griffiths, B.; Chung, Y.L.; Delpuech, O.; Griffiths, J.R.; Downward, J.; Schulze, A. PKB/Akt induces transcription of enzymes involved in cholesterol and fatty acid biosynthesis via activation of SREBP. Oncogene 2005, 24, 6465–6481. [Google Scholar] [CrossRef] [Green Version]
- Eming, S.A.; Murray, P.J.; Pearce, E.J. Metabolic orchestration of the wound healing response. Cell Metab. 2021, 33, 1726–1743. [Google Scholar] [CrossRef]
- Zheng, L.; Zhang, W.; Zhou, Y.; Li, F.; Wei, H.; Peng, J. Recent Advances in Understanding Amino Acid Sensing Mechanisms that Regulate mTORC1. Int. J. Mol. Sci. 2016, 17, 1636. [Google Scholar] [CrossRef] [Green Version]
- Shimobayashi, M.; Hall, M.N. Making new contacts: The mTOR network in metabolism and signalling crosstalk. Nat. Rev. Mol. Cell Biol. 2014, 15, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Evers, T.M.J.; Holt, L.J.; Alberti, S.; Mashaghi, A. Reciprocal regulation of cellular mechanics and metabolism. Nat. Metab. 2021, 3, 456–468. [Google Scholar] [CrossRef] [PubMed]
- Dias Gomes, M.; Iden, S. Orchestration of tissue-scale mechanics and fate decisions by polarity signalling. EMBO J. 2021, 40, e106787. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Gene | Genetic Modification | Epidermal Barrier | Additional Phenotypes | Refs |
|---|---|---|---|---|
| Ligands and receptors | ||||
| Igf1 | Whole body knockout | n.d | Epidermal hypoplasia/neonatal lethality | [47] |
| Epidermal-specific overexpression | n.d | Epidermal hyperplasia/spontaneous tumor formation in aged mice | [48] | |
| Igf2 | Whole body knockout | n.d | No obvious abnormalities | [47] |
| Igf1/Igf2 | Whole body knockout | n.d | Epidermal hypoplasia | [47] |
| Ir | Whole body knockout | + | No obvious skin phenotype | [49] |
| Epidermal-specific knockout | + | No obvious abnormalities/decreased epidermal thickness | [50] | |
| Igf-1r | Whole body knockout | +++ | Translucent skin/thin epidermis/neonatal | [47] |
| Epidermal-specific knockout | ++ | Epidermal hypoplasia/impaired skin barrier | [51] | |
| Ir/Igf-1r | Epidermal-specific knockout | +++ | Epidermal hypoplasia/neonatal lethality | [50] |
| Glut1 | Epidermal-specific knockout | + | No obvious skin phenotype/delayed wound healing | [52] |
| Slc3a2 | Epidermal-specific knockout | Hair growth delay/impaired skin wound healing | [53] | |
| Kinase and scaffold proteins | ||||
| AMPK | Epidermal-Specific knockout | + | Epidermal hyperplasia upon injury and UVB exposure | [54] |
| Pdk1 | Epidermal-Specific knockout | +++ | Epidermal hypoplasia/impaired skin barrier function/neonatal lethality | [55] |
| Akt | AKT1/AKT2 whole body double-knockout | n.d | Defects in skin development/translucent skin/neonatal lethality | [56] |
| AKT1 whole body knockout | + | No obvious abnormalities/stratum corneum defects | [57,58,59] | |
| AKT2 whole body knockout | + | No obvious abnormalities | [60] | |
| Mtor | Epidermal-specific knockout | +++ | Epidermal hypoplasia/impaired skin barrier function/neonatal lethality | [61] |
| Raptor | Epidermal-specific knockout | +++ | Epidermal hypoplasia/impaired skin barrier function/neonatal lethality | [61,62] |
| Rictor | Epidermal-Specific knockout | ++ | Epidermal hypoplasia/impaired skin barrier function | [61,63,64] |
| P18 | Epidermal-specific knockout | +++ | Impaired skin barrier function /neonatal lethality | [65] |
| Pten | Keratinocyte-specific knockout | n.d | Epidermal hyperplasia/tumor formation/enhanced re-epithelization during wound healing | [66,67,68] |
| Tsc1 | Epidermal-specific knockout | n.d | Increased re-epithelization during wound healing | [68] |
| Epidermal hyperplasia/wavy hair and curly whiskers/hair lose | [69] | |||
| Rheb | Epidermal-specific knockout | +++ | Epidermal hypoplasia/impaired skin barrier function | [62] |
| Downstream effectors | ||||
| S6k | S6K1 whole body knockout | n.d | Small body size/increased life span | [70,71] |
| S6K2 whole body knockout | n.d | No obvious phenotypic abnormalities | [70] | |
| S6K1/S6K2 whole body double knockout | n.d | Reduced viability/neonatal death | [72] | |
| 4E-BP | 4E-BP1/2 whole body double-knockout | n.d. | Increased sensitivity to diet-induced obesity | [73,74] |
| 4E-BP1/2/3 whole body triple-knockout | n.d | Increased sensitivity to diet-induced obesity | [75] | |
| Atg7 | Epidermal-specific knockout | + | No obvious skin phenotype/impaired skin wound healing | [76,77] |
| Pkc | PKCa overexpression in epidermis | + | No obvious phenotypic abnormalities/increased sensitivity to TPA | [78] |
| PKC-epsilon over expression in epidermis | + | Mild abnormalities/ more sensitive to TPA | [79] | |
| FoxO1 | Overexpression of nuclear variant in epidermis | +++ | Epidermal hypoplasia/impaired stratification/neonatal lethality | [80] |
| Hif | HIF-1a epidermal-specific knockout | n.d. | Epidermal aging/pruritic inflammation/delayed wound closure | [81,82] |
| HIF-2a epidermal-specific knockout | n.d. | Accelerated wound closure | [83] | |
| Myc | Overexpression in epidermis | n.d. | Epidermal hyperplasia/spontaneous tumor/delayed wound closure | [84,85] |
| Epidermal-specific knockout | +++ | Epidermal hypoplasia /tight and fragile skin/impaired wound healing | [86] | |
| PPAR | PPARα whole body knockout | n.d. | delayed wound healing | [87] |
| Heterozygous PPARβ mutant | n.d. | delayed wound healing | [87] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Eming, S.A.; Ding, X. Role of mTOR Signaling Cascade in Epidermal Morphogenesis and Skin Barrier Formation. Biology 2022, 11, 931. https://doi.org/10.3390/biology11060931
Wang J, Eming SA, Ding X. Role of mTOR Signaling Cascade in Epidermal Morphogenesis and Skin Barrier Formation. Biology. 2022; 11(6):931. https://doi.org/10.3390/biology11060931
Chicago/Turabian StyleWang, Juan, Sabine A. Eming, and Xiaolei Ding. 2022. "Role of mTOR Signaling Cascade in Epidermal Morphogenesis and Skin Barrier Formation" Biology 11, no. 6: 931. https://doi.org/10.3390/biology11060931